XALKORI
PFIZER
crizotinibe
Inibidor da tirosina-quinase.
Apresentações.
Xalkori® 200 mg ou 250 mg em embalagens contendo 60 cápsulas.
VIA DE ADMINISTRAÇÃO: USO ORAL
USO ADULTO
Composição.
Cada cápsula de Xalkori® 200 mg ou 250 mg contém o equivalente a 200 mg ou 250 mg de crizotinibe, respectivamente. Excipientes: dióxido de silício coloidal, celulose microcristalina, fosfato de cálcio dibásico anidro, amidoglicolato de sódio, estearato de magnésio, cápsula de gelatina dura (gelatina, dióxido de titânio e óxido férrico vermelho) e tinta de impressão preta (goma laca, propilenoglicol, solução concentrada de amônia, hidróxido de potássio e óxido férrico preto).
Indicações.
Xalkori® (crizotinibe) é indicado para o tratamento de câncer de pulmão não pequenas células (CPNPC) avançado que seja positivo para quinase de linfoma anaplásico (ALK).
Resultados de eficácia.
População pediátrica
A segurança e a eficácia do crizotinibe em pacientes pediátricos não foram estabelecidas. Diminuição da formação óssea no crescimento de ossos grandes foi observada em ratos imaturos com 150 mg/kg/dia, seguindo uma dosagem diária por 28 dias (aproximadamente 3 vezes a exposição clínica humana baseada em AUC). Outras toxicidades em potencial com relação à pacientes pediátricos não foram avaliadas em animais jovens.
Estudos clínicos
CPNPC avançado positivo para ALK não tratado previamente -Estudo 1014 randomizado de Fase 3
O uso de crizotinibe como agente único para o tratamento de primeira linha de CPNPC avançado positivo para ALK, em pacientes com ou sem metástases cerebrais, foi investigado em um estudo multicêntrico, multinacional, aberto e randomizado de Fase 3, o Estudo 1014. O objetivo primário deste estudo foi demonstrar que crizotinibe é superior ao tratamento quimioterápico padrão de primeira linha baseado em platina (pemetrexede-cisplatina ou pemetrexede-carboplatina) no prolongamento da Sobrevida Livre de Progressão (SLP), avaliada por revisão radiológica independente (RRI) em pacientes com CPNPC avançado positivo para ALK que não receberam tratamento sistêmico prévio para a doença avançada. Os objetivos secundários foram comparar as medidas da eficácia clínica, incluindo a Taxa de Resposta Objetiva (TRO) avaliada por RRI, Duração da Resposta (DR), Sobrevida Global (SG), Tempo para a Progressão Intracraniana (TPP-IC) avaliada por RRI e Resultados Relatados pelo Paciente (RRP).
A análise populacional completa do Estudo 1014 incluiu 343 pacientes com CPNPC avançado positivo para ALK identificado pela Hibridização in situ por fluorescência (FISH) antes da randomização. Cento e setenta e dois (172) pacientes foram randomizados para o braço crizotinibe (171 pacientes receberam 250 mg de crizotinibe por via oral duas vezes ao dia) e 171 pacientes foram randomizados para o braço quimioterapia (169 pacientes receberam quimioterapia; 91 pacientes foram tratados com pemetrexede/cisplatina e 78 pacientes foram tratados com pemetrexede/carboplatina). A quimioterapia consistiu em pemetrexede 500 mg/m2 em combinação com cisplatina 75 mg/m2 ou carboplatina na dose calculada, para produzir uma área sob a curva concentração-tempo (AUC) de 5 ou 6 mg min/mL. A quimioterapia foi administrada por infusão intravenosa a cada 3 semanas por até 6 ciclos. A duração mediana de tratamento do estudo foi de 47 semanas no braço crizotinibe e 18 semanas no braço quimioterapia. Os pacientes poderiam continuar o tratamento com crizotinibe além do tempo de progressão de doença definida por RECIST (Critério de Avaliação de Resposta em Tumores Sólidos), conforme avaliado por RRI, a critério do investigador, caso o paciente ainda estivesse experimentando benefício clínico. Os pacientes no braço quimioterapia que completaram os 6 ciclos deveriam continuar no estudo sem tratamento adicional, mas fazer avaliações contínuas dos tumores até a progressão da doença definida por RECIST, conforme determinado pela RRI. Os pacientes no braço quimioterapia que tiveram a progressão da doença definida por RECIST e avaliada pela RRI tiveram a opção de receber crizotinibe. Cento e vinte (70%) pacientes receberam crizotinibe após a fase de randomização (109 pacientes por meio do processo de crossover e 11 pacientes como terapia de acompanhamento).
A randomização foi estratificada pelo estado de desempenho ECOG (0 -1 vs 2), raça (asiático versus não asiático) e metástases cerebrais (presentes vs ausente).
Os dados demográficos basais e as características da doença foram semelhantes entre os braços de tratamento crizotinibe e quimioterapia com relação ao gênero (feminino: 61% vs 63%; para crizotinibe vs quimioterapia, respectivamente), idade mediana (52 anos vs 54 anos), raça (caucasiano: 53% vs 50%, e asiático: 45% vs 47%); status de tabagismo (fumantes atuais: 6% vs 3%, ex-fumantes: 33% vs 32% e que nunca fumaram: 62% vs 65%), doença metastática (98% em ambos os braços de tratamento), histologia do tumor (adenocarcinoma: 92% vs 93%), estado de desempenho (ECOG 0 ou 1: 94% vs 95%, e ECOG 2: 6% vs 5%) e metástases cerebrais (presentes 26% vs 28%).
O crizotinibe prolongou significativamente a SLP em comparação com a quimioterapia, conforme avaliado por RRI. Os dados de Sobrevida Global (SG) não estavam maduros no momento da análise da SLP. Os dados de eficácia do Estudo 1014 randomizado Fase 3 estão resumidos na Tabela 1, e a curva de Kaplan-Meier para SLP está ilustrada na Figura 1.
Tabela 1.
Resultados de Eficácia do Estudo 1014 Randomizado de Fase 3 (Análise Populacional Completa) em Pacientes com CPNPC Avançado Positivo para ALK Não Tratado Previamente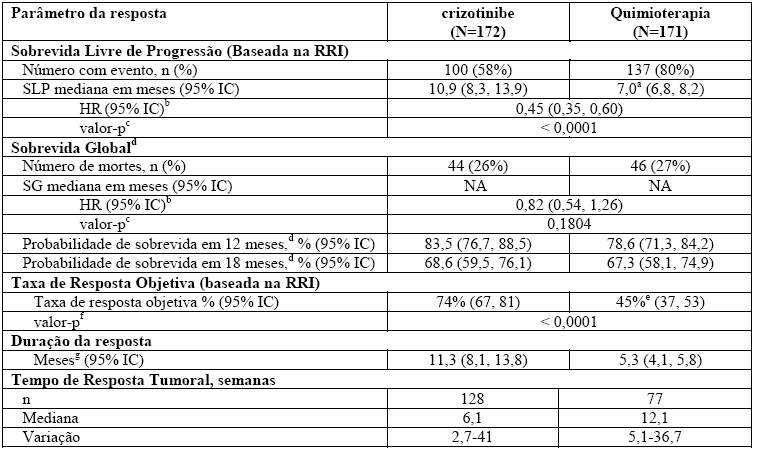
Abreviações: IC = intervalo de confiança; HR = hazard ratio ou taxa de risco; RRI = revisão radiológica independente; NA = não alcançado; SLP = sobrevida livre de progressão; SG = sobrevida global.
a. Tempos da SLP mediana foram 6,9 meses (95% IC: 6,6, 8,3) para pemetrexede/cisplatina (HR = 0,49; valor-p < 0,0001 para crizotinibe em comparação com pemetrexede/cisplatina) e 7,0 meses (95% IC: 5,9; 8,3) para pemetrexede/carboplatina (HR = 0,45; valor-p < 0,0001 para crizotinibe em comparação com pemetrexede/carboplatina).
b. Com base na análise de riscos proporcionais de Cox estratificada.
c. Com base no teste log-rank estratificado (unilateral).
d. A análise da SG não foi ajustada para os efeitos de crossover, que potencialmente geram confusão.
e. TROs foram de 47% (95% IC: 37, 58) para pemetrexede/cisplatina (valor-p < 0,0001 em comparação com crizotinibe) e 44% (95% IC: 32, 55) para pemetrexede/carboplatina (valor-p < 0,0001 em comparação com crizotinibe).
f. Com base no teste de Cochran-Mantel-Haenszel estratificado (bilateral).
g. Estimativa utilizando o método Kaplan-Meier.
Figura 1. Curvas de Kaplan-Meier para a Sobrevida Livre de Progressão (Baseada na RRI) por Braço de Tratamento no Estudo 1014 Randomizado de Fase 3 (Análise Populacional Completa) em pacientes com CPNPC Avançado Positivo para ALK Não Tratado Previamente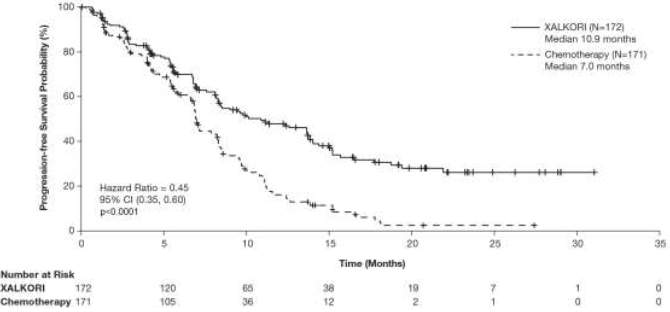
Com base na RRI, um total de 9 (23,1%) dos 39 pacientes no braço crizotinibe e 12 (30,0%) dos 40 pacientes no braço quimioterapia, com metástases cerebrais basais tratadas previamente, apresentaram progressão das lesões intracranianas ou desenvolveram novas lesões intracranianas. Para os pacientes com metástases cerebrais basais tratadas previamente, o TPP intracraniano mediano (TPP-IC) foi de 15,7 meses no braço crizotinibe e de 12,5 meses no braço quimioterapia (HR = 0,45 [95% IC: 0,19; 1,07]; unilateral valor-p = 0,0315). Um total de 16 (12,1%) dos 132 pacientes no braço crizotinibe e 14 (10,7%) dos 131 pacientes no braço quimioterapia, sem metástases cerebrais basais, desenvolveram novas lesões intracranianas. Para pacientes sem metástases cerebrais basais, o TPP-IC mediano não foi alcançado nos braços crizotinibe ou quimioterapia (HR = 0,69 [95% IC: 0,33; 1,45]; unilateral valor-p = 0,1617).
Os sintomas relatados pelo paciente e a Qualidade de Vida (QoL) global foram coletadas com o EORTC QLQC30 e seu módulo de câncer de pulmão (EORTC QLQ-LC13) no valor basal (Dia 1), Dia 7 e Dia 15 do ciclo 1 e no Dia 1 de cada ciclo de tratamento subsequente. Um total de 166 pacientes no braço crizotinibe e 163 pacientes no braço quimioterapia preencheram o EORTC QLQ-C30 e os questionários LC-13 basais e pelo menos uma visita pós-basal.
O tempo até a deterioração (TTD) foi pré-especificado como o tempo desde a randomização até a primeira ocorrência de um aumento de ≥10 pontos na escala basal, nos sintomas de dor (EORTC QLQ-LC13 dor no peito), tosse (EORTC QLQ-LC13 tosse) ou dispneia (EORTC QLQ-LC13 dispneia). O TTD mediano em dor no peito, dispneia ou tosse relatados pelo paciente, como um desfecho composto, foi de 2,1 meses (95% IC: 0,8 mês, 4,2 meses) no braço crizotinibe, comparado com 0,5 mês (95% IC: 0,4 mês, 0,7 mês) no braço quimioterapia. O tratamento com crizotinibe foi associado a um TTD significativamente maior nos sintomas de dor no peito, dispneia ou tosse, em comparação com a quimioterapia (taxa de risco 0,59; 95% IC: 0,45; 0,77; bilateral, baseado no teste log-rank ajustado para Hochberg valor-p = 0,0005).
Figura 2. Plotagem de Kaplan-Meier do Tempo até a Deterioração da Dor (no Peito), Dispneia ou Tosse (desfecho composto) por Braço de Tratamento (Resultados Relatados pelo Paciente na População Avaliável) em Pacientes com CPNPC Avançado Positivo para ALK não Tratado Previamente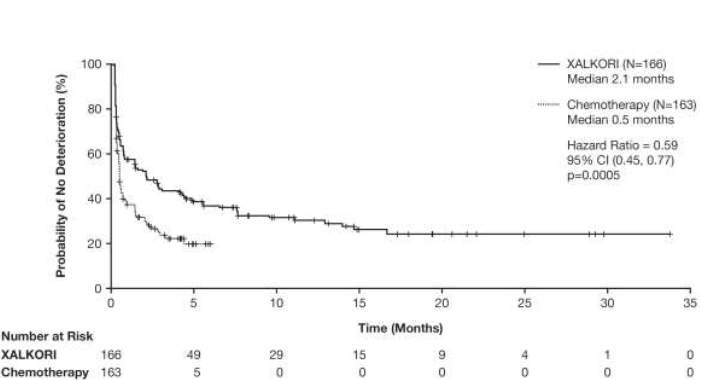
A mudança em relação às escalas basais foi considerada significativamente diferente entre os 2 braços de tratamento, com uma melhoria significativamente maior observada na qualidade global de vida no braço crizotinibe, quando comparado com o braço quimioterapia (diferença geral na mudança em relação às escalas basais 13,8: valor-p < 0,0001).
CPNPC Avançado Positivo para ALK Tratado Previamente -Estudo 1007 Randomizado de Fase 3
O uso de crizotinibe como agente único para o tratamento de CPNPC avançado positivo para ALK com ou sem metástases cerebrais foi investigado em um estudo multicêntrico, multinacional, aberto e randomizado de Fase 3, (Estudo 1007). O objetivo primário deste estudo foi demonstrar que o crizotinibe 250 mg via oral, duas vezes ao dia é superior ao tratamento quimioterápico padrão (pemetrexede 500 mg/m2 ou docetaxel 75 mg/m2) intravenoso (IV) a cada 21 dias, no prolongamento da Sobrevida Livre de Progress en(SLP) em pacientes com CPNPC avançado positivo para ALK que haviam recebido 1 regime prévio de quimioterapia. Foi exigido que os pacientes tivessem CPNPC com ALK positivo, conforme identificado por Hibridização in situ por fluorescência (FISH) antes da randomização. Os pacientes randomizados para a quimioterapia poderiam ser "cruzados" para receber crizotinibe no Estudo 1005 na progressão da doença definida pelo Critério de Avaliação da Resposta em Tumores Sólidos (RECIST), confirmados pela revisão radiológica independente (RRI). O desfecho primário de eficácia foi SLP com eventos de progressão de doença determinados por RRI. Os desfechos secundários incluíram Taxa de Resposta Objetiva (TRO) avaliada por RRI, Duração da Resposta (DR), Sobrevida Global (SG) e Resultados Relatados pelo Paciente (RRP). A análise populacional para o Estudo 1007 incluiu 347 pacientes com CPNPC avançado positivo para ALK. Cento e setenta e três (173) pacientes foram randomizados para o braço do crizotinibe (172 pacientes receberam crizotinibe) e 174 pacientes foram randomizados para o braço quimioterapia (99 pacientes [58%] receberam pemetrexede e 72 pacientes [42%] receberam docetaxel). A randomização foi estratificada pelo estado de desempenho ECOG (0-1, 2), metástase cerebral (presente, ausente) e tratamento prévio com inibidor de tirosina quinase do EGFR (sim, não). A duração mediana de tratamento do estudo foi de 31 semanas no braço crizotinibe comparado com 12 semanas no braço quimioterapia.
Os pacientes poderiam continuar o tratamento, conforme designado, além do tempo de progressão de doença definido por RECIST, conforme avaliado por RRI, a critério do investigador, caso o paciente ainda estivesse experimentando benefício clínico. Cinquenta e oito dos 84 (69%) pacientes tratados com crizotinibe e 17 dos 119 (14%) tratados com quimioterapia continuaram o tratamento por pelo menos 3 semanas após a progressão objetiva da doença.
Os dados demográficos basais e as características da doença para pacientes neste estudo foram semelhantes entre os braços crizotinibe e quimioterapia com relação ao gênero (feminino: 57% vs 55%; para crizotinibe vs quimioterapia, respectivamente), idade mediana (51 anos vs 49 anos), raça (caucasiano: 52% em ambos os braços de tratamento e asiático: 46% vs 45%), status de tabagismo (fumantes atuais: 3% vs 5%, ex-fumantes: 34% vs 31% e que nunca fumaram: 62% vs 64%), doença metastática (95% vs 91%), histologia do tumor (adenocarcinoma: 94% vs 92%), estado de desempenho (ECOG 0 ou 1: 89% vs 91%, ECOG 2: 11% vs 9%), e metástases cerebrais (presentes: 35% em ambos os braços de tratamento).
O crizotinibe prolongou significativamente a SLP em comparação com a quimioterapia, conforme avaliado por RRI. Os dados de Sobrevida Global não estavam maduros no momento da análise da SLP. Os dados de eficácia do Estudo 1007 randomizado Fase 3 estão resumidos na Tabela 2 e a curva de Kaplan-Meier para SLP está ilustrada na Figura 3.
Tabela 2. Resultados de Eficácia do Estudo 1007 Randomizado de Fase 3 (Análise Populacional Completa) em Pacientes com CPNPC Avançado Positivo para ALK Tratados Previamente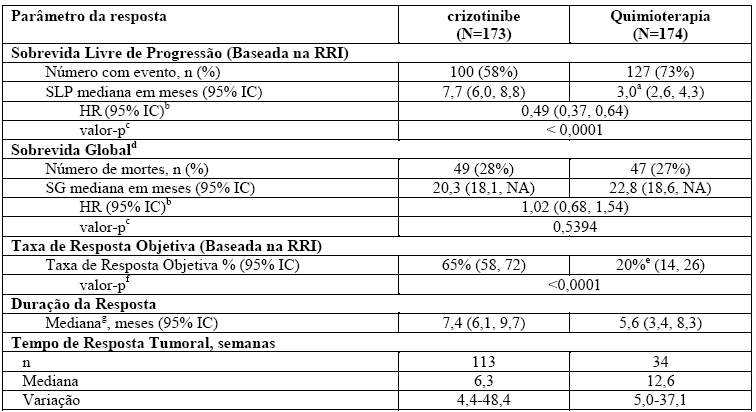
Abreviações: IC = intervalo de confiança; HR = hazard ratio ou taxa de risco; RRI = revisão radiológica independente; NA = não alcançado; SLP = sobrevida livre de progressão; SG = sobrevida global.
a. Tempos da SLP mediana foram 4,2 meses (95% IC: 2,8, 5,7) para pemetrexede (HR = 0,59; valor-p = 0,0004 para crizotinibe em comparação com pemetrexede) e 2,6 meses (95% IC: 1,6; 4,0) para docetaxel (HR = 0,30; valor-p < 0,0001 para crizotinibe em comparação com docetaxel).
b. Com base na análise de riscos proporcionais de Cox estratificada.
c. Com base no teste log-rank estratificado (unilateral).
d. Análise intermediária de SG realizada em 40% do total de eventos exigidos para a análise final.
e. TROs foram de 29% (95% IC: 21, 39) para pemetrexede (valor-p < 0,0001 em comparação com crizotinibe) e 7% (95% IC: 2, 16) para docetaxel (valor-p < 0,0001 em comparação com crizotinibe).
f. Com base no teste de Cochran-Mantel-Haenszel estratificado (bilateral).
g. Estimativa utilizando o método Kaplan-Meier.
Figura 3. Curvas de Kaplan-Meier para Sobrevida Livre de Progressão (Baseada na RRI) por Braço de Tratamento no Estudo 1007 Randomizado de Fase 3 (Análise Populacional Completa) em Pacientes com CPNPC Avançado Positivo para ALK Tratados Previamente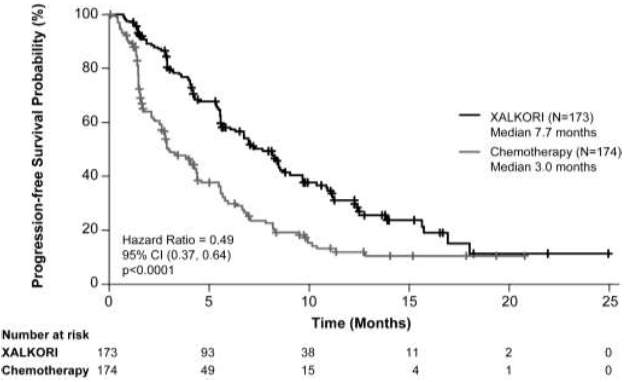
Os sintomas relatados pelo paciente e a qualidade de vida (QoL) global foram coletados usando o EORTC QLQC30 e seu módulo de câncer de pulmão (EORTC QLQ-LC13) no valor basal (Dia 1 do Ciclo 1) e no Dia 1 de cada ciclo de tratamento subsequente. Um total de 162 pacientes no braço crizotinibe e 151 pacientes no braço quimioterapia preencheram o EORTC QLQ-C30 e os questionários LC13 basais e pelo menos uma visita pósbasal.
O tempo até a deterioração (TTD) foi pré-especificado como o tempo desde a randomização até a primeira ocorrência de um aumento de ≥10 pontos na escala basal, nos sintomas de dor (EORTC QLQ-LC13 dor no peito), tosse (EORTC QLQ-LC13 tosse) ou dispneia (EORTC QLQ-LC13 dispneia). O TTD mediano em dor no peito, dispneia ou tosse relatados pelo paciente, como um desfecho composto, foi de 4,5 meses (95% IC: 3,0 meses, 6,9 meses) no braço crizotinibe, comparado com 1,4 meses (95% IC: 1,0 mês, 1,6 meses) no braço quimioterapia. O tratamento com crizotinibe foi associado a um TTD significativamente maior nos sintomas de dor no peito, dispneia ou tosse, em comparação com a quimioterapia (taxa de risco 0,50; 95% IC: 0,37; 0,66; log-rank ajustado para Hochberg valor-p < 0,0001).
A mudança em relação às escalas basais foi considerada significativamente diferente entre os dois braços de tratamento, com uma melhoria significativamente maior observada na qualidade global de vida no braço crizotinibe, quando comparado com o braço quimioterapia (diferença total na mudança em relação às pontuações basais: 9,84, valor-p < 0,0001).
Figura 4. Plotagem de Kaplan-Meier do Tempo até a Deterioração da Dor (no Peito), Dispneia ou Tosse (Desfecho Composto) por Braço de Tratamento (Resultados Relatados pelo Paciente na População Avaliável) em Pacientes com CPNPC Avançado Positivo para ALK Tratados Previamente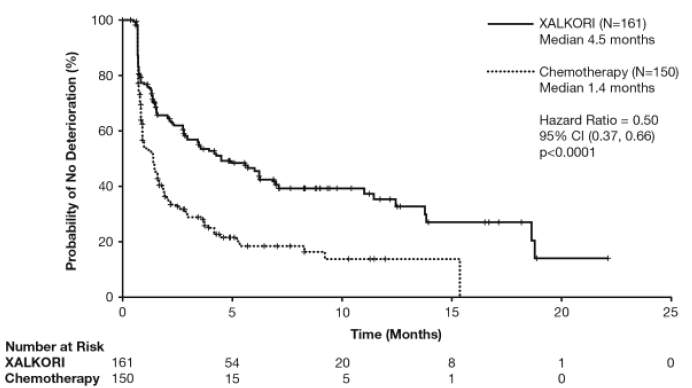
Estudos de braço único em CPNPC Avançado Positivo para ALK
O uso de crizotinibe como agente único no tratamento de CPNPC avançado positivo para ALK, com ou sem metástase cerebral, foi investigado em 2 estudos multicêntricos e multinacionais de braço único (Estudos 1001 e 1005). Os pacientes incluídos nesses estudos haviam recebido terapia sistêmica anterior, com exceção de 16 pacientes no Estudo 1001 e 3 pacientes no Estudo 1005, que não tiveram tratamento sistêmico anterior para doença avançada localmente ou metastática. O desfecho primário da eficácia em ambos os estudos foi o TRO de acordo com o RECIST. O desfecho secundário incluiu o Tempo até a Resposta Tumoral (TRT), DR, SLP e SG. Os pacientes receberam 250 mg de crizotinibe por via oral duas vezes por dia.
No Estudo 1001 (N = 119), as características demográficas foram 50% do sexo feminino; idade mediana de 51 anos; estado de desempenho no ECOG basal de 0 ou 1 (87%) ou 2 (12%), 62% caucasianos e 29% asiáticos; < 1% fumantes atuais, 27% ex-fumantes e 72% nunca fumaram. As características da doença foram de 96% metastáticas, 98% histologia de adenocarcinoma e 13% com nenhuma terapia sistêmica prévia para a doença metastática.
No Estudo 1005 (N = 934), as características demográficas foram 57% do sexo feminino; idade mediana de 52 anos; estado de desempenho no ECOG basal de 0/1 (82%) ou 2/3 (18%), 52% caucasianos e 44% asiáticos; 4% fumantes atuais, 30% ex-fumantes e 66% nunca fumaram. As características da doença foram de 92% metastáticos, 94% histologia de adenocarcinoma.
No Estudo 1001, foi exigido que os pacientes com CPNPC avançado tivessem tumores positivos para ALK antes de entrarem no estudo clínico. O CPNPC positivo para ALK foi identificado usando uma série de ensaios de estudos clínicos locais. Cento e dezenove pacientes com CPNPC avançado positivo para ALK foram incluídos no Estudo 1001 no momento do corte de dados, para as análises de SLP e TRO. A duração mediana do tratamento foi de 32 semanas. Houve 2 respostas completas e 69 respostas parciais para um TRO de 61%. A DR mediana foi de 48 semanas. Cinquenta e cinco por cento das respostas tumorais objetivas foram alcançadas dentro das primeiras 8 semanas de tratamento. Os dados de SG do Estudo 1001 foram atualizados com base em 154 pacientes com CPNPC avançado positivo para ALK. A SG mediana no momento do corte de dados foi de 28,9 meses (95% IC: 21,1; 40,1).
No Estudo 1005, foi exigido que os pacientes com CPNPC avançado tivessem tumores positivos para ALK antes de entrarem no estudo clínico. Para a maioria dos pacientes, CPNPC para ALK positivo foi identificado por FISH. Novecentos e trinta e quatro pacientes com CPNPC avançado positivo para ALK foram tratados com crizotinibe no Estudo em 1005 no momento do corte de dados para as análises de SLP e TRO. A duração mediana de tratamento para esses pacientes foi de 23 semanas. Os pacientes poderiam continuar o tratamento, conforme designado, além do tempo de progressão de doença definido por RECIST a critério do investigador, caso a avaliação do risco/benefício justificasse a continuação do tratamento. Setenta e sete dos 106 pacientes (73%) continuaram o tratamento com crizotinibe por pelo menos 3 semanas após a progressão objetiva de doença.
Setecentos e sessenta e cinco pacientes com CPNPC avançado positivo para ALK do Estudo 1005 estavam avaliáveis para resposta e foram identificados pelo mesmo ensaio FISH utilizado no Estudo 1007 randomizado Fase 3. Houve 8 respostas completas e 357 respostas parciais para um TRO de 48%. A DR mediana foi de 47 semanas. Oitenta e três por cento das respostas de tumor objetivas foram alcançadas dentro das primeiras 12 semanas de tratamento. Os dados de SG do Estudo 1005 foram atualizados com base em 905 pacientes com CPNPC avançado positivo para ALK identificados pelo mesmo ensaio FISH utilizado no Estudo 1007 randomizado de Fase 3. A SG mediana no momento do corte de dados foi de 21,5 meses (95% IC: 19,3, 23,6).
Dados de eficácia dos Estudos 1001 e 1005 são fornecidos na Tabela 3.
Tabela 3. Resultados de Eficácia de CPNPC Avançado Positivo para ALK dos Estudos 1001 e 1005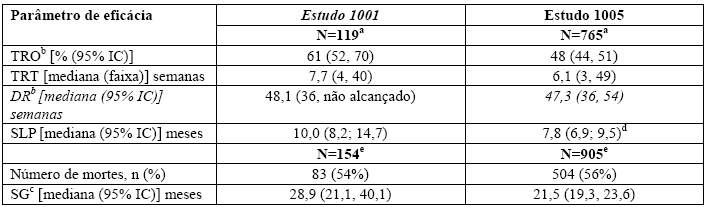
Abreviações: IC = intervalo de confiança: TRO = taxa de resposta objetiva; TRT = tempo até a resposta tumoral; DR = duração da resposta; SLP = sobrevida livre de progressão; SG = sobrevida global a Por datas de corte de dados 15 de setembro de 2010 (Estudo 1001) e 15 de fevereiro de 2012 (Estudo de 1005). b Três pacientes não estavam avaliáveis para resposta no Estudo 1001 e 42 pacientes não estavam avaliáveis para resposta no Estudo 1005. c Estimativa utilizando o método Kaplan-Meier. d Dados de SLP do Estudo 1005 incluíram 807 pacientes na análise populacional de segurança que foram identificados pelo ensaio FISH (por data de corte de dados 15 fevereiro de 2012). e Por data de corte de dados 30 de novembro de 2013
Idosos (vide também item 8. Posologia e Modo de Usar e item 3. Características Farmacológicas -Propriedades Farmacocinéticas)
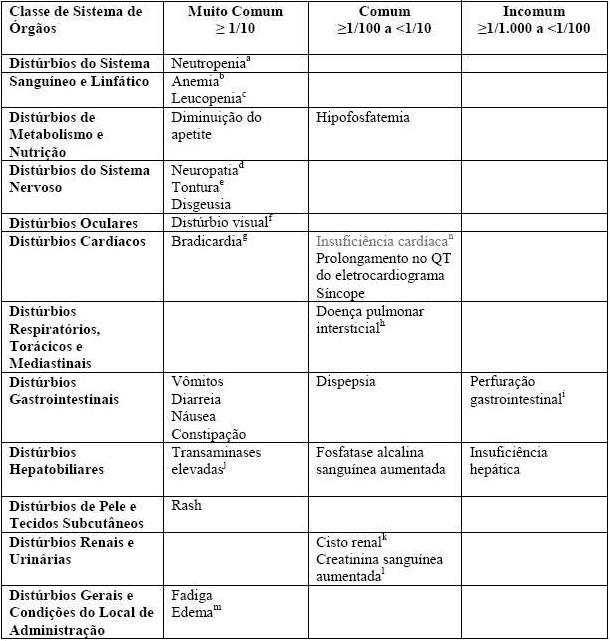
Dos 171 pacientes tratados com crizotinibe no Estudo 1014 randomizado Fase 3, 22 (13%) tinham 65 anos ou mais. Dos 109 pacientes tratados com crizotinibe que foram cruzados para o braço quimioterapia, 26 (24%) tinham 65 anos ou mais. Dos 172 pacientes tratados com crizotinibe no Estudo 1007 randomizado Fase 3, 27 (16%) tinham 65 anos ou mais. Dos 154 pacientes no Estudo 1001, 22 (14%) tinham 65 anos ou mais. Dos 1063 pacientes no Estudo 1005, 173 (16%) tinham 65 anos ou mais. A frequência de reações adversas foi geralmente semelhante para pacientes < 65 anos de idade e pacientes com ³ 65 anos de idade, com a exceção de edema e constipação, que foram relatados com maior frequência no Estudo 1014 entre os pacientes tratados com crizotinibe com ³ 65 anos de idade. Não foram observadas diferenças gerais na eficácia em comparação com pacientes mais jovens.
Referências
Solomon BJ, Mok T, Kim DW et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N Engl J Med 2014; 371:2167-77.
Shaw AT, Kim DW, Nakagawa K et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N Engl J Med 2013; 368: 2385-94.
Crinò L, Kim D-W, Riely GJ et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J. Clin. Oncol., Vol 29, No 15_suppl (May 20 Supplement), 2011: 7514.
Kim D-W, Ahn M-J, Shi Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol 2012; 30: Suppl. abstract. · Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-celllung cancer: updated results from a phase1 study. Lancet Oncol 2012; 13: 1011-9.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
O crizotinibe é uma molécula pequena, inibidor seletivo do receptor de tirosina quinase (RTK) ALK e suas variáveis oncogênicas (por exemplo, eventos de fusão ALK e mutações ALK selecionadas). O crizotinibe é também um inibidor do Receptor de Fator de Crescimento de Hepatócito (HGFR, c-Met) RTK, ROS1 (c-ros), e RTKs RON (Recepteur d'Origine Nantais). O crizotinibe demonstrou uma inibição dependente de concentração de atividade de quinase de ALK, ROS1 e c-Met em ensaios bioquímicos e fosforilação inibida e fenótipos dependentes de quinase modulados em ensaios baseados em célula. O crizotinibe demonstrou uma potente e seletiva atividade inibitória do crescimento e apoptose induzida em linhas de células tumorais que apresentam eventos de fusão ALK (incluindo EML4-ALK e NPM-ALK), eventos de fusão ROS1, ou exibindo amplificação de ALK ou gene locus MET. O crizotinibe demonstrou eficácia antitumor, incluindo atividade antitumoral citorredutora acentuada, em ratos portadores de xenoenxertos de tumores que expressavam proteínas de fusão ALK. A eficácia antitumoral do crizotinibe foi dependente da dose e correlacionada com a inibição farmacodinâmica da fosforilação de proteínas de fusão ALK (incluindo EML4-ALK e NPM-ALK) em tumores in vivo.
Propriedades Farmacocinéticas
Absorção
Após a administração oral de uma única dose em jejum, o crizotinibe é absorvido com tempo mediano para atingir concentrações de pico em 4 a 6 horas. Após 250 mg de crizotinibe duas vezes ao dia, o estado de equilíbrio foi atingido dentro de 15 dias e permaneceu estável com uma proporção de acumulação mediana de 4,8. A biodisponibilidade absoluta do crizotinibe demonstrou ser de 43% (faixa: 32% a 66%) seguindo a administração de uma única dose oral de 250 mg.
Uma alimentação rica em gordura reduziu AUCinf e Cmáx do crizotinibe em aproximadamente 14% quando fornecida uma única dose de 250 mg a voluntários saudáveis. O crizotinibe pode ser administrado com ou sem alimento (vide item 8. Posologia e Modo de Usar).
Distribuição
O volume médio geométrico de distribuição (Vss) do crizotinibe foi 1772 L após a administração de uma dose intravenosa de 50 mg, indicando distribuição extensiva nos tecidos a partir do plasma.
A ligação do crizotinibe às proteínas do plasma humano in vitro é de 91% e independe da concentração do medicamento. Estudos in vitro sugeriram que o crizotinibe é um substrato para glicoproteína-P (GpP). A proporção de concentração plasma para sangue é de aproximadamente 1.
Metabolismo
Estudos in vitro demonstraram que CYP3A4/5 foram as enzimas principais envolvidas na depuração metabólica do crizotinibe. As principais vias metabólicas em seres humanos foram oxidação do anel piperidina a lactama do crizotinibe e O-desalquilação com subsequente conjugação Fase 2 dos metabólitos da O-desalquilação. Estudos in vitro nos microssomos do fígado humano demonstraram que o crizotinibe é um inibidor de CYP2B6 e CYP3A dependente de tempo.
Eliminação
Após doses únicas de crizotinibe, a meia-vida terminal aparente do plasma do crizotinibe foi de 42 horas nos pacientes.
Seguindo a administração de uma única dose de crizotinibe radiomarcada de 250 mg em indivíduos saudáveis, 63% e 22% da dose administrada foi recuperada nas fezes e urina, respectivamente. O crizotinibe inalterado representou aproximadamente 53% e 2,3% da dose administrada nas fezes e urina, respectivamente.
A depuração média aparente (CL/F) do crizotinibe foi inferior no estado de equilíbrio (60 L/h) após 250 mg duas vezes ao dia do que após uma única dose via oral de 250 mg (100 L/h), o que provavelmente foi devido a autoinibição de CYP3A pelo crizotinibe após dosagem múltipla.
Interações medicamentosas
Coadministração de crizotinibe e substratos CYP3A
O crizotinibe foi identificado como um inibidor de CYP3A tanto in vitro como in vivo. Após 28 dias de crizotinibe 250 mg tomado duas vezes ao dia por pacientes com câncer, a AUC de midazolam oral foi de 3,7 vezes (90% IC: 2,63-5,07) em comparação àquela vista quando o midazolam foi administrado sozinho, sugerindo que o crizotinibe é um inibidor moderado de CYP3A (vide item 6. Interações Medicamentosas).
Coadministração de crizotinibe e inibidores de CYP3A
A coadministração de uma única dose oral de 150 mg de crizotinibe na presença de cetoconazol (200 mg duas vezes ao dia), um forte inibidor de CYP3A, resultou em aumentos na exposição sistêmica do crizotinibe, com valores de AUCinf e Cmáx de crizotinibe sendo de aproximadamente 3,2 vezes e 1,4 vezes, respectivamente, àqueles vistos quando o crizotinibe foi administrado sozinho. Entretanto, a magnitude do efeito dos inibidores de CYP3A na exposição de crizotinibe em estado de equilíbrio não foi estabelecida (vide item 6. Interações Medicamentosas).
Coadministração de crizotinibe e indutores de CYP3A
A coadministração de uma única dose de 250 mg de crizotinibe com rifampina (600 mg uma vez ao dia), um forte indutor de CYP3A, resultou em 82% e 69% de diminuição na AUCinf e Cmáx do crizotinibe, respectivamente, comparado a quando o crizotinibe foi tomado sozinho. Entretanto, o efeito dos indutores de CYP3A na exposição de crizotinibe em estado de equilíbrio não foi estabelecido (vide item 6. Interações Medicamentosas).
Coadministração de crizotinibe e agentes que aumentam o pH Gástrico
A solubilidade aquosa do crizotinibe é dependente do pH, com pH baixo (ácido) resultando em maior solubilidade. A administração de uma única dose de 250 mg de crizotinibe após tratamento com 40 mg de esomeprazol uma vez ao dia durante 5 dias resultou em aproximadamente 10% da diminuição da exposição total de crizotinibe (AUCinf) e nenhuma mudança na exposição em pico (Cmáx); a extensão da mudança da exposição total não foi clinicamente significativa. Portanto, o ajuste da dose inicial não é necessário quando o crizotinibe é coadministrado com agentes que aumentam o pH gástrico (como inibidores de bomba de prótons, bloqueadores de H2, ou antiácidos).
Coadministração com outros substratos de CYP
Estudos in vitro indicaram que é improvável a ocorrência de interações clínicas medicamento-medicamento como resultado da inibição mediada por crizotinibe do metabolismo de medicamentos que são substratos para CYP1A2, CYP2C8, CYP2C9, CYP2C19 ou CYP2D6.
O crizotinibe é um inibidor de CYP2B6 in vitro. Contudo, o crizotinibe pode ter potencial para aumentar as concentrações plasmáticas de medicamentos coadministrados que são predominantemente metabolizados por CYP2B6. Estudos in vitro em hepatócitos humanos indicaram que é improvável a ocorrência de interações clínicas medicamento-medicamento como resultado da indução mediada por crizotinibe do metabolismo de medicamentos que são substratos para CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ou CYP3A.
Coadministração com substratos UGT
Estudos in vitro indicaram que é improvável a ocorrência de interações clínicas medicamento-medicamento como resultado da inibição mediada por crizotinibe do metabolismo de medicamentos que são substratos para UGT1A1, UGT1A4, UGT1A6, UGT1A9 ou UGT2B7.
Coadministração com medicamentos que são substratos de transportadores
O crizotinibe é um inibidor da glicoproteína-P (GpP) in vitro. Contudo, o crizotinibe pode ter o potencial para aumentar as concentrações plasmáticas de medicamentos coadministrados que são substratos de GpP.
O crizotinibe é um inibidor de OCT1 e OCT2 in vitro. Portanto, o crizotinibe pode ter o potencial de aumentar as concentrações plasmáticas de medicamentos coadministrados que são substratos de OCT1 ou OCT2.
In vitro, o crizotinibe não inibiu as proteínas de transporte de captação hepática em humanos OATP1B1 ou OATP1B3, ou as proteínas de transporte de captação renal OAT1 ou OAT3 em concentrações clinicamente relevantes. Contudo, as interações clínicas medicamento-medicamento são improváveis de ocorrer como resultado da inibição mediada por crizotinibe da captação hepática ou renal de medicamentos que são substratos para estes transportadores.
Efeito em Outras Proteínas de Transporte
In vitro, o crizotinibe não é um inibidor de BSEP (bomba de exportação do sal da bile) em concentrações clinicamente relevantes.
Farmacocinética em Grupos Especiais de Pacientes Insuficiência Hepática: como o crizotinibe é extensivamente metabolizado no fígado, é provável que a insuficiência hepática aumente as concentrações plasmáticas de crizotinibe. Contudo, o crizotinibe não foi estudado em pacientes com insuficiência hepática. Estudos clínicos que foram realizados excluíram pacientes com ALT ou AST > 2,5 x LSN ou, se devido à malignidade subjacente, > 5,0 x LSN ou com bilirrubina total > 1,5 x LSN (vide item 8. Posologia e Modo de Usar -Tabela 5. Modificação da dose de Xalkori® -Toxidades não-hematológicas e, vide item 5. Advertências e Precauções). A análise farmacocinética da população, usando os dados desses estudos, indicou que os níveis basais de bilirrubina total ou AST não tiveram um efeito clinicamente significativo na farmacocinética do crizotinibe.
Insuficiência Renal: Pacientes com insuficiência renal leve (60 ≤ CLcr < 90 mL/min) e moderada (30 ≤ CLcr < 60 mL/min) foram incluídos nos Estudos 1001 e 1005 de braço único. O efeito da função renal, medida pelo clearance basal de crizotinibe (CLcr) observado em concentrações mínimas no estado equilíbrio (Ctrough, ss) foi avaliado. No Estudo 1001, as médias geométricas ajustadas de Ctrough, ss no plasma de pacientes com insuficiência renal leve (N=35) e moderada (N=8) foram mais altas 5,1% e 11%, respectivamente, do que em pacientes com funções renais normais. No Estudo 1005, as médias geométricas ajustadas Ctrough, ss de crizotinibe em grupos com insuficiência renal leve (N=191) e moderada (N=65) foram mais altas 9,1% e 15%, respectivamente, que em pacientes com funções renais normais. Além disso, a análise da farmacocinética populacional dos Estudos 1001, 1005 e 1007 indicou que o CLcr não teve um efeito clinicamente significativo na farmacocinética do crizotinibe. Devido aos pequenos aumentos na exposição de crizotinibe (5%-15%), nenhum ajuste de dose inicial é recomendado a pacientes com insuficiência renal leve ou moderada. Após uma dose única de 250 mg em indivíduos com insuficiência renal grave (CLcr < 30 mL/min) não necessitando de diálise peritoneal ou hemodiálise, o AUC e o Cmáx do crizotinibe aumentaram em 79% e 34%, respectivamente, em comparação com aqueles com função renal normal. Recomenda-se um ajuste da dose de crizotinibe, ao administrá-lo em pacientes com insuficiência renal grave não submetidos à diálise peritoneal ou hemodiálise (vide item 8. Posologia e Modo de Usar -Modificação da dose e item 5. Advertências e Precauções).
Idade: Com base na análise da farmacocinética populacional dos Estudos 1001, 1005 e 1007, a idade não possui nenhum efeito na farmacocinética do crizotinibe (vide item 8. Posologia e Modo de usar -Modificações da dose e item 3. Características Farmacológicas -Propriedades Farmacodinâmicas).
Peso corporal e gênero: Com base na análise da farmacocinética populacional dos Estudos 1001, 1005 e 1007, não houve efeito clinicamente significativo do peso corporal ou gênero na farmacocinética do crizotinibe.
Etnia: Com base na análise da farmacocinética populacional dos Estudos 1001, 1005 e 1007, o estado de equilíbrio previsto da AUC (95% IC) foi 23%-37% mais alto em pacientes asiáticos (n=523) do que em pacientes não asiáticos (n=691).
Eletrofisiologia cardíaca
O potencial de prolongamento do intervalo QT de crizotinibe foi avaliado em todos os pacientes que receberam 250 mg de crizotinibe duas vezes ao dia. Eletrocardiogramas (ECG) triplicados em série foram coletados após uma única dose e em estado de equilíbrio para avaliar o efeito do crizotinibe nos intervalos QT. Trinta e dois dos 1560 pacientes (2,1%) apresentaram QTcF (QT corrigido pelo mètodo Fridericia) ≥ 500 msec e 76 de 1520 pacientes (5,0%) tiveram um aumento de QTcF da linha de base ≥60 msec por avaliação lida por máquina automatizada de ECG. (vide item 5. Advertências e Precauções).
Um subestudo de ECG, utilizando medições manuais cegas de ECG, foi realizado em 52 pacientes com CPNPC positivo para ALK que receberam crizotinibe 250 mg duas vezes por dia.
Análise da tendência central indicou que um efeito QTc ≥20 ms pode ser excluído. Análise farmacocinética/farmacodinâmica sugeriu uma relação entre a concentração plasmática de crizotinibe e QTc. Além disso, a diminuição da frequência cardíaca foi considerada associada ao aumento nas concentrações plasmáticas de crizotinibe (vide item 5. Advertências e Precauções).
Dados de Segurança Pré-clínicos
Genotoxicidade
O crizotinibe não foi mutagênico in vitro no ensaio de mutação bacteriana reversa (Ames). O crizotinibe foi aneugênico em um ensaio de micronúcleo in vitro em células Ovarianas de Hamsters Chineses e em ensaio de aberração cromossômica de linfócito humano in vitro. Pequenos aumentos de aberrações cromossômicas estruturais em concentrações citotóxicas foram vistas nos linfócitos humanos. Na medula óssea de rato in vivo, aumentos no micronúcleo foram apenas vistos em doses significativamente excedentes à exposição humana esperada. Aumentos no micronúcleo foram observados nos ratos a 250 mg/kg/dia (aproximadamente 4 vezes a AUC na dose recomendada para humanos).
Carcinogenicidade
Estudos de carcinogenicidade com crizotinibe não foram realizados.
Fertilidade
Estudos não específicos com crizotinibe foram realizados em animais para avaliar o efeito na fertilidade; contudo, o crizotinibe é considerado como tendo o potencial de prejudicar a função reprodutora e a fertilidade em humanos baseado em descobertas nos estudos de toxidade de dose repetida no rato. Descobertas observadas no trato reprodutivo masculino incluiu a degeneração de espermatócitos no paquíteno testicular em ratos que receberam ≥ 50 mg/kg/dia durante 28 dias (aproximadamente equivalente à exposição clínica humana baseada em AUC). Descobertas observadas no trato reprodutivo feminino incluiu a necrose unicelular dos folículos ovarianos de uma rata que recebeu 500 mg/kg/dia durante 3 dias.
Contraindicações.
O uso de Xalkori® é contraindicado em pacientes com hipersensibilidade ao crizotinibe ou a qualquer um dos excipientes.
O uso de Xalkori® é contraindicado em pacientes com insuficiência hepática grave.
Advertências e precauções.
Hepatotoxicidade
A hepatotoxicidade induzida por fármacos com desfecho fatal ocorreu em menos de 0,5% dos 1669 pacientes tratados com crizotinibe em ensaios clínicos. Elevações simultâneas de ALT e/ou AST ≥ 3 x LSN e bilirrubina total ≥ 2 x LSN sem elevações significativas na fosfatase alcalina ( < 2 x LSN) foram observadas em menos de 1% dos pacientes tratados com crizotinibe. Foram observadas elevações significativas até o Grau 3 ou 4 de ALT ou AST em 184 (11%) e 93 (6%) dos paciente