XALATAN®
VIATRIS
latanoprosta
Antiglaucomatoso.
Apresentações.
Xalatan® solução oftálmica estéril 50 mcg/mL (0,005%) em embalagem contendo frasco gotejador de 2,5 mL
VIA DE ADMINISTRAÇÃO: USO OFTÁLMICO
USO ADULTO E PEDIÁTRICO ACIMA DE 1 ANO
Composição.
Cada mL da solução oftálmica estéril de Xalatan® contém 50 mcg de latanoprosta. Excipientes: cloreto de sódio, fosfato de sódio monobásico monoidratado, fosfato de sódio dibásico anidro, cloreto de benzalcônio e água para injetáveis.
Uma gota da solução contém aproximadamente 1,5 mcg de latanoprosta.
Cada 1 mL da solução oftálmica de Xalatan® corresponde a aproximadamente 37 gotas.
Indicações.
Xalatan® (latanoprosta) solução oftálmica é indicado para a redução da pressão intraocular (PIO) elevada em pacientes com glaucoma de ângulo aberto e hipertensão ocular. Xalatan® também está indicado para a redução da pressão intraocular elevada em pacientes pediátricos com pressão intraocular elevada e glaucoma pediátrico.
Resultados de eficácia.
Pacientes com pressão intraocular (PIO) média de 24 - 25 mmHg no momento inicial do estudo que foram tratados por 6 meses em ensaios multicêntricos, randomizados, controlados demonstraram reduções de 6 - 8 mmHg na pressão intraocular. A redução da PIO com Xalatan® 0,005% Solução Oftálmica Estéril (1 gota, uma vez ao dia) foi equivalente ao efeito de 0,5% de timolol administrado duas vezes ao dia.
Em relação à segurança, um estudo (n= 519) prospectivo, aberto, com duração de 3 anos, extendido por mais 2 anos, avaliou a progressão do aumento da pigmentação da íris devido ao uso contínuo de Xalatan® (1 gota, 1 vez ao dia) para o tratamento de glaucoma de ângulo aberto. A análise de segurança usou a população de 380 pacientes avaliados na fase de extensão.
O aumento da pigmentação da íris teve início no primeiro ano de uso da medicação na maioria dos pacientes em que esse evento foi observado. Ao longo dos 5 anos em que os pacientes usaram a medicação, os sinais de hiperpigmentação se mantiveram. A hiperpigmentação não acarretou nenhum outro evento adverso - ou alterou sua incidência, natureza ou gravidade -exceto a própria alteração da coloração da íris. Pacientes com aumento da coloração da íris evoluíram com redução da PIO semelhante à dos pacientes que não apresentaram o quadro.
Caract. farmacológicas.
Xalatan® é um análogo da prostaglandina F2
Estudos clínicos mostraram que a latanoprosta não tem efeito significativo sobre a produção do humor aquoso, sobre a barreira hemato-humoral aquosa. A latanoprosta não induziu extravasamento de fluoresceína no segmento posterior de olhos humanos pseudofácicos durante tratamento em curto prazo.
Não foram observados quaisquer efeitos farmacológicos significativos sobre o sistema cardiovascular e respiratório com doses clínicas de latanoprosta.
População Pediátrica
A eficácia de Xalatan® em pacientes pediátricos ≤ 18 anos, foi demonstrada num estudo clínico duplomascarado com duração de 12 semanas, de latanoprosta em comparação com timolol, em 107 pacientes com diagnóstico de hipertensão ocular e glaucoma pediátrico. Os recém-nascidos deveriam ter, pelo menos, 36 semanas de idade gestacional. Os pacientes receberam latanoprosta 0,005% uma vez por dia ou timolol 0,5% (ou opcionalmente 0,25% para os indivíduos com idade inferior a 3 anos), diariamente. O endopoint de eficácia primário foi a redução média da pressão intraocular (PIO) da linha de base à Semana 12 do estudo. As reduções médias da PIO nos grupos latanoprosta e timolol foram semelhantes. Em todas as faixas etárias estudadas (0 a < 3 anos, 3 a < 12 anos e 12 a 18 anos) a redução média da PIO na Semana 12, no grupo da latanoprosta, foi semelhante ao do grupo timolol. No entanto, os dados de eficácia na faixa etária de 0 a < 3 anos, foram baseados em apenas 13 pacientes para latanoprosta e, não foi demonstrada eficácia relevante a partir dos 4 pacientes representando a faixa etária de 0 a < 1 ano no estudo clínico pediátrico. Não existem dados disponíveis para recém-nascidos prematuros (com idade gestacional inferior a 36 semanas).
As reduções da PIO entre os indivíduos do subgrupo de glaucoma primário congénito/glaucoma infantil (GPC) foram semelhantes entre o grupo latanoprosta e o grupo timolol. O subgrupo não-GPC (por exemplo, o glaucoma de ângulo aberto juvenil, glaucoma afáquico) apresentou resultados semelhantes aos do subgrupo GPC.
O efeito na PIO foi observado após a primeira semana de tratamento e foi mantido durante todo o período de 12 semanas de estudo, assim como nos adultos.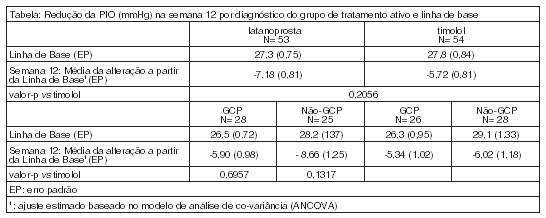
Propriedades Farmacocinéticas
Absorção
A latanoprosta é absorvida pela córnea, onde o pró-fármaco do éster isopropílico é hidrolisado a forma ácida e torna-se biologicamente ativo. Estudos em humanos indicam que a concentração máxima no humor aquoso é alcançada cerca de 2 horas após administração tópica.
Distribuição
O volume de distribuição em humanos é 0,16 ± 0,02 L/kg. O ácido da latanoprosta pode ser medido no humor aquoso durante as primeiras 4 horas após a administração local e no plasma somente durante a primeira hora. Metabolismo
A latanoprosta, um pró-fármaco do éster isopropílico, é hidrolisado por estearases presentes na córnea para o ácido biologicamente ativo. O ácido ativo de latanoprosta alcança a circulação sistêmica e é principalmente metabolizado pelo fígado para os metabólitos 1,2-dinor e 1, 2, 3, 4-tetranor via b-oxidação de ácidos graxos.
Excreção
A eliminação do ácido da latanoprosta do plasma humano é rápida (t1/2 = 17 min) após administração intravenosa e tópica. O clearance sistêmico é de aproximadamente 7 mL/min/kg. Após b-oxidação hepática, os metabólitos são eliminados principalmente por via renal. Aproximadamente 88% e 98% da dose administrada é recuperada na urina após administração tópica e intravenosa, respectivamente.
População pediátrica
Foi realizado um estudo aberto de farmacocinética das concentrações plasmáticas do ácido latanoprosta em 22 pacientes adultos e 25 pacientes pediátricos (do nascimento até < 18 anos de idade) com hipertensão ocular e glaucoma. Todas as faixas etárias foram tratadas com uma gota de latanoprosta 0,005%, por dia, em cada olho, por um período mínimo de 2 semanas. A exposição sistêmica ao ácido latanoprosta foi, aproximadamente, 2 vezes superior no grupo de crianças de 3 a < 12 anos e 6 vezes superior no grupo de crianças < 3 anos, em comparação com os adultos. No entanto, foi mantida uma ampla margem de segurança para efeitos adversos sistêmicos (vide item 10 - Superdose). O tempo médio para atingir a concentração plasmática foi de 5 minutos após a dose, em todas as faixas etárias. O tempo médio de meia-vida de eliminação plasmática foi curto ( < 20 minutos), semelhante em pacientes pediátricos e adultos, e não resultou em acumulação de ácido latanoprosta na circulação sistêmica sob condições de estado de equilíbrio.
Dados de Segurança Pré-Clínicos
Efeitos Sistêmicos / Oculares
A toxicidade ocular, assim como a sistêmica de latanoprosta, foram investigadas em várias espécies animais. Geralmente, a latanoprosta é bem tolerada, com uma margem de segurança entre a dose clínica ocular e a toxicidade sistêmica de, no mínimo, 1.000 vezes. Altas doses de latanoprosta, aproximadamente 100 vezes a dose clínica/kg de peso corporal, administradas intravenosamente a macacos não anestesiados, aumentaram a frequência respiratória, refletindo provavelmente uma broncoconstrição de curta duração. Nos macacos, a latanoprosta foi infundida intravenosamente em doses de até 500 mcg/kg sem maiores efeitos sobre o sistema cardiovascular. Em estudos animais, a latanoprosta não demonstrou propriedades sensibilizantes. Não foram detectados efeitos tóxicos nos olhos com doses de até 100 mcg/olho/dia em coelhos ou macacos (a dose clínica é aproximadamente 1,5 mcg/olho/dia). A latanoprosta não produziu efeitos, ou os produziu de modo desprezível sobre a circulação sanguínea intraocular quando utilizada com doses clínicas e estudada em macacos. Em estudos de toxicidade ocular crônica, a administração de latanoprosta na dose de 6 mcg/olho/dia também mostrou induzir aumento da fissura palpebral. Esse efeito é reversível e ocorre com doses acima do nível da dose clínica. O efeito não foi observado em humanos.
Carcinogenicidade
Estudos de carcinogenicidade em camundongos e ratos foram negativos.
Mutagenicidade
A latanoprosta foi negativa em testes de mutação reversa em bactérias, mutação genética em linfoma de camundongo e testes de micronúcleo de camundongo. Foram observadas aberrações cromossômicas in vitro com linfócitos humanos. Foram observados efeitos similares com prostaglandinas F2a, uma prostaglandina que ocorre naturalmente e indica que este é um efeito de classe. Estudos adicionais de mutagenicidade sobre a síntese de DNA não-esquematizada in vitro/in vivo em ratos foram negativos e indicam que a latanoprosta não tem potencial mutagênico.
Alterações na fertilidade
Não foi observado qualquer efeito sobre a fertilidade de machos e fêmeas em estudos com animais. No estudo de embriotoxicidade em ratos, não foi observada embriotoxicidade em doses intravenosas (5, 50 e 250 mcg/kg/dia) de latanoprosta. Contudo, a latanoprosta induziu efeitos letais em embriões de coelhos em doses iguais ou superiores a 5 mcg/kg/dia.
Foi observado que a latanoprosta pode causar toxicidade embrio-fetal em coelhos, caracterizada pelo aumento de incidências de aborto e reabsorção tardia e peso fetal reduzido quando administrado em doses intravenosas de aproximadamente 100 vezes a dose humana.
Teratogenicidade
Não foi detectado potencial teratogênico.
Contraindicações.
Xalatan® é contraindicado a pacientes que apresentam hipersensibilidade a latanoprosta ou a qualquer componente da fórmula.
Xalatan® é contraindicado para pacientes menores de 1 ano.
Advertências e precauções.
Gerais
Xalatan® contém cloreto de benzalcônio, que pode ser absorvido por lentes de contato (vide Posologia). As lentes de contato devem ser removidas antes da instilação do colírio e podem ser recolocadas após 15 minutos.
Ocular
Xalatan® pode gradualmente aumentar o pigmento castanho da íris. A alteração da cor do olho é devido ao conteúdo aumentado de melanina nos melanócitos estromais da íris, ao invés do aumento no número de melanócitos. Tipicamente, a pigmentação castanha ao redor da pupila se difunde concentricamente em direção à periferia da íris e toda a íris, ou parte dela, pode ficar mais acastanhada. A alteração na cor da íris é leve na maioria dos casos e pode não ser clinicamente detectada. O aumento na pigmentação da íris em um ou ambos os olhos foi documentada predominantemente em pacientes com íris de cores mistas, que contenham a cor castanha como base. Nevos e lentigens da íris não foram afetados pelo tratamento. Não se observou acúmulo de pigmento na malha trabecular ou em outras partes da câmara anterior em estudos clínicos.
Em um estudo clínico destinado a avaliar a pigmentação da íris por mais de 5 anos, não houve evidências de consequências adversas devido ao aumento de pigmentação mesmo quando a administração de Xalatan® continuou. Estes resultados são consistentes com a experiência clínica pós-comercialização desde 1996. Além disso, a redução da PIO foi similar em pacientes independente do aumento da pigmentação da íris. Portanto, o tratamento com Xalatan® pode continuar em pacientes que desenvolveram aumento da pigmentação da íris. Estes pacientes devem ser examinados regularmente e, dependendo da situação clínica, o tratamento pode ser interrompido.
O início do aumento da pigmentação da íris ocorre tipicamente dentro do primeiro ano de tratamento, raramente durante o segundo ou terceiro ano, e não foi observado após o quarto ano de tratamento. A taxa de progressão da pigmentação da íris diminui com o tempo e é estável por 5 anos. Os efeitos do aumento da pigmentação além dos 5 anos não foram avaliados. Durante os estudos clínicos, aumento no pigmento castanho da íris não foi observado após descontinuação do tratamento, mas a alteração da cor resultante pode ser permanente.
O escurecimento da pálpebra, que pode ser reversível, foi relatado com o uso de Xalatan®.
Xalatan® pode gradualmente alterar os cílios e a lanugem da pálpebra no olho tratado, estas alterações incluem aumento do comprimento, espessura, pigmentação e quantidade dos cílios e lanugem e crescimento irregular dos cílios. Alterações dos cílios são reversíveis após descontinuação do tratamento.
Antes de o tratamento ser instituído, os pacientes devem ser informados quanto à possibilidade de alteração na coloração dos olhos. O potencial para heterocromia existe para pacientes recebendo tratamento unilateral. Durante o tratamento com Xalatan®, foram relatados edema macular, incluindo edema macular cistóide. Estes relatos ocorreram principalmente em pacientes afácicos, pseudofácicos com ruptura da cápsula posterior do cristalino ou em pacientes com fatores de risco conhecidos para edema macular. Xalatan® deve ser utilizado com cautela nesses pacientes.
Após a interrupção do tratamento, houve melhora na acuidade visual, utilizando-se em alguns casos tratamento simultâneo com medicamentos antiinflamatórios tópicos esteróides e não-esteróides. Há experiência limitada com Xalatan® no tratamento de glaucoma inflamatório ou neovascular. Portanto, recomenda-se que Xalatan® seja utilizado com cuidado nessas condições até que se disponha de maiores dados nesse aspecto.
Xalatan® deve ser utilizado com cautela em pacientes com história pregressa de ceratite herpética e deve ser evitado em casos de ceratite em atividade causada pelo vírus da herpes simples e em pacientes com história de ceratite herpética recorrente especificamente associado com análogos da prostaglandina.
População pediátrica
Os dados de eficácia e segurança para a faixa etária < 1 ano (4 pacientes) são muito limitados (vide item 3 - Características Farmacológicas). Não existem dados disponíveis para recém-nascidos prematuros (com idade gestacional inferior a 36 semanas).
Em crianças de 0 a < 3 anos de idade, que sofrem principalmente de GCP (Glaucoma Congênito Primário), a cirurgia (por exemplo, trabeculotomia / goniotomia) continua a ser o tratamento de primeira linha. A segurança a longo prazo em crianças ainda não foi estabelecida.
Fertilidade
Não foi observado qualquer efeito sobre a fertilidade de machos e fêmeas em estudos com animais (vide item 3 - Características Farmacológicas - Alterações na fertilidade).
Uso durante a Gravidez
A latanoprosta mostrou causar toxicidade embrio-fetal em coelhos, caracterizada por aumento na incidência de reabsorção tardia, aborto e peso fetal reduzido quando administrada em doses intravenosas de, aproximadamente, 100 vezes a dose humana.
Xalatan® não aumenta a incidência espontânea de defeitos congênitos, mas tem efeitos farmacológicos prejudiciais potenciais em relação ao período da gravidez, para o feto ou neonato.
Não foram realizados estudos adequados e bem controlados em mulheres grávidas. Xalatan® deve ser usado durante a gravidez apenas se o benefício previsto justificar o risco potencial para o feto (vide item 3 - Características Farmacológicas - Dados de Segurança Pré-Clínicos).
Xalatan® é um medicamento classificado na categoria C de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Uso durante a Lactação
A latanoprosta e seus metabólitos podem passar para o leite materno, portanto, Xalatan® deve ser utilizado com cautela em mulheres lactantes.
Efeitos na Habilidade de Dirigir e Operar Máquinas
A instilação de Xalatan® pode embaçar transitoriamente a visão. Até que isto seja resolvido, os pacientes não devem dirigir ou operar máquinas.
Interações medicamentosas.
Estudos in vitro mostraram que ocorre precipitação quando colírios contendo timerosal são misturados com Xalatan®. Se tais produtos forem utilizados, o colírio deve ser administrado com um intervalo de, no mínimo, 5 minutos.
Um estudo clínico de 3 meses mostrou que o efeito redutor da pressão intraocular da latanoprosta é aditivo ao dos antagonistas beta-adrenérgicos (timolol). Outros estudos de curto prazo sugerem que o efeito de latanoprosta é aditivo ao dos agonistas adrenérgicos (dipivalilepinefrina), inibidores da anidrase carbônica (acetazolamida) e, pelo menos parcialmente, ao dos agonistas colinérgicos (pilocarpina). No caso de terapia combinada, os colírios devem ser administrados com um intervalo mínimo de 5 minutos.
Houve relatos de elevações paradoxais da PIO após administrações oftálmicas concomitantes de 2 prostaglandinas análogas. Portanto, o uso de 2 ou mais prostaglandinas, análogas ou derivadas, não é recomendado. Interações com outras medicações não foram investigadas.
População pediátrica
Estudos de interação só foram realizados em adultos.
Cuidados de armazenamento.
Xalatan® solução oftálmica deve ser conservado sob refrigeração (entre 2 e 8°C), protegido da luz e pode ser utilizado por 36 meses a partir da data de fabricação. Após a abertura do frasco, o produto pode ser conservado à temperatura ambiente (até 25°C) por até 10 semanas.
Após aberto, válido por 10 semanas.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido.
Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas do produto: solução límpida, incolor, isenta de partículas visíveis.
Posologia e modo de usar.
Cada 1 mL da solução oftálmica de Xalatan® corresponde a aproximadamente 37 gotas.
Uso em adultos (incluindo idosos)
A dose recomendada é 1 gota de Xalatan® no(s) olho(s) afetado(s), uma vez ao dia. O efeito ótimo é obtido se o produto for administrado à noite.
A dose de Xalatan® não deve exceder 1 dose diária, uma vez que uma administração mais frequente diminui o efeito redutor da pressão intraocular.
Xalatan® pode ser utilizado concomitantemente com outras classes de medicamentos oftálmicos tópicos para redução da PIO. Se outros medicamentos oftálmicos tópicos são utilizados, esses devem ser administrados com um intervalo de, pelo menos, 5 minutos.
Lentes de contato devem ser removidas antes da instilação da solução oftálmica e podem ser recolocadas após 15 minutos (vide item 5 - Advertências e Precauções - Geral).
Uso Pediátrico
Xalatan® pode ser utilizado em pacientes pediátricos na mesma posologia que nos adultos. Não existem dados disponíveis para recém-nascidos prematuros (com idade gestacional inferior a 36 semanas). Dados para a faixa etária < 1 ano (4 pacientes) são muito limitados (vide item 3 - Característica Farmacológicas).
Dose Omitida
Caso o paciente esqueça-se de utilizar Xalatan® no horário estabelecido, deve fazê-lo assim que lembrar. Entretanto, se já estiver perto do horário de administrar a próxima dose, deve desconsiderar a dose esquecida e utilizar a próxima. Neste caso, o paciente não deve utilizar a dose duplicada para compensar doses esquecidas. O esquecimento de dose pode comprometer a eficácia do tratamento.
Reações adversas.
Estudos clínicos
Os seguintes eventos foram considerados relacionados ao fármaco:
Ocular: irritação ocular (sensação de queimação, areia, prurido, picada e corpo estranho), blefarite, hiperemia conjuntival, dor ocular, aumento de pigmentação da íris (vide item 5 - Advertências e Precauções), erosões epiteliais puntiformes transitórias e edema de pálpebra.
Pele e tecido subcutâneo: rash cutâneo.
Experiência Pós-Comercialização
Os seguintes eventos também foram relatados:
Sistema nervoso: tontura e cefaléia.
Ocular: edema e erosões da córnea, conjuntivite, alteração nos cílios e lanugem da pálpebra (aumento do comprimento, espessura, pigmentação e número), irite/uveíte, ceratite, edema macular, incluindo edema macular cistóide, cílios irregulares que podem causar irritação ocular, visão embaçada, fotofobia, alterações periorbitais e na pálpebra que resultam em aprofundamento do sulco da pálpebra (vide item 5 - Advertências e Precauções).
Sistema Respiratório: asma, agravamento da asma, crises agudas de asma e dispnéia.
Pele e tecido subcutâneo: escurecimento da pele da pálpebra, reação cutânea local na pálpebra.
Musculoesquelético e tecido conjuntivo: dor muscular/articulação.
Geral: dor torácica não-específica.
Infecções e infestações: (Frequência desconhecida) ceratites herpéticas.
População pediátrica
Em dois ensaios clínicos de curta duração (≤ 12 semanas), envolvendo 93 pacientes pediátricos (25 e 68) o perfil de segurança foi semelhante ao dos adultos e não foram identificados novos eventos adversos. Os perfis de segurança de curto prazo também foram semelhantes nos diferentes subgrupos pediátricos (vide item 3 - Características Farmacológicas). Os eventos adversos observados mais frequentemente na população pediátrica em comparação aos adultos são: nasofaringite e febre.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Superdose.
Além da irritação ocular e hiperemia conjuntival, não são conhecidos outros efeitos adversos oculares no caso de superdosagem com Xalatan® solução oftálmica.
Se Xalatan® for acidentalmente ingerido, as seguintes informações podem ser úteis: um frasco de 2,5 mL contém 125 mcg de latanoprosta. Mais de 90% é metabolizado durante a primeira passagem pelo fígado. A infusão intravenosa de 3 mcg/kg em voluntários sadios não induziu sintomas, mas uma dose de 5,5-10 mcg/kg causou náuseas, dor abdominal, tontura, fadiga, ondas de calor e sudorese. Contudo, em pacientes com asma brônquica moderada, a latanoprosta não induziu broncoconstrição, quando aplicada topicamente, por via oftálmica, em uma dose equivalente a 7 vezes a dose clínica (vide item 3 - Características Farmacológicas -Dados de Segurança Pré-Clínicos - Efeitos Sistêmicos/Oculares).
Se ocorrer superdosagem com Xalatan®, deve-se instituir tratamento sintomático.
Em caso de intoxicação, ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.0216.0129
VENDA SOB PRESCRIÇÃO MÉDICA.
Esta bula foi aprovada pela Anvisa em (19/09/2011)