WINREVAIR
MSD
sotatercepte
Anti-hipertensivo pulmonar.
Apresentações.
WINREVAIRTM Pó liofilizado para solução injetável de
-45 mg de sotatercepte em frasco-ampola de dose única em embalagem com 1 frasco-ampola, 1 seringa preenchida de diluente, 1 adaptador, 1 seringa, 1 agulha e 4 envelopes com lenços umedecidos com álcool.
-45 mg de sotatercepte em frasco-ampola de dose única em embalagem com 2 frascos-ampolas, 2 seringas preenchidas de diluente, 2 adaptadores, 1 seringa, 1 agulha e 8 envelopes com lenços umedecidos com álcool.
-60 mg de sotatercepte em frasco-ampola de dose única em embalagem com 1 frasco-ampola, 1 seringa preenchida de diluente, 1 adaptador, 1 seringa, 1 agulha e 4 envelopes com lenços umedecidos com álcool.
-60 mg de sotatercepte em frasco-ampola de dose única em embalagem com 2 frascos-ampolas, 2 seringas preenchidas de diluente, 2 adaptadores, 1 seringa, 1 agulha e 8 envelopes com lenços umedecidos com álcool.
USO SUBCUTÂNEO
USO ADULTO
Composição.
WINREVAIRTM 45 mg Frasco-ampola de dose única de 45 mg: Cada frasco contém 45 mg de sotatercepte. Após reconstituição com 1,0 mL de Água para Injetáveis, a concentração resultante é de 50 mg/1,0 mL de sotatercepte e o volume nominal de entrega é de 0,9 mL.
WINREVAIRTM 60 mg Frasco-ampola de dose única de 60 mg: Cada frasco contém 60 mg de sotatercepte. Após reconstituição com 1,3 mL de Água para Injetáveis, a concentração resultante é de 50 mg/1,0 mL de sotatercepte e o volume nominal de entrega é de 1,2 mL.
Excipientes: ácido cítrico monoidratado, polissorbato 80, sacarose e citrato trissódico di-hidratado.
Diluente: água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
WINREVAIRTM, associado a outras terapias, é indicado para o tratamento de adultos com hipertensão arterial pulmonar (HAP, Grupo 1 da Organização Mundial da Saúde [OMS]) para aumentar a capacidade de exercício e retardar a progressão da doença.
2. RESULTADOS DE EFICÁCIA
Hipertensão arterial pulmonar em indivíduos adultos
A eficácia de WINREVAIRTM foi avaliada em pacientes adultos com HAP no estudo STELLAR. O STELLAR foi um estudo clínico global, duplo-cego, controlado por placebo, multicêntrico, de grupos paralelos, no qual 323 pacientes com HAP (Grupo 1 CF II ou III da OMS) foram randomizados 1:1 para receber WINREVAIRTM (dose alvo de 0,7 mg/kg) (n=163) ou placebo (n=160) administrado por via subcutânea uma vez a cada 3 semanas.
As características demográficas e clínicas iniciais foram geralmente comparáveis entre os grupos WINREVAIRTM e placebo. Os participantes deste estudo eram adultos com idade mediana de 48,0 anos (intervalo: de 18 a 82 anos); peso médio 68 kg (intervalo: de 38,0 a 141,3 kg); 89,2% dos participantes eram brancos e 79,3% não eram hispânicos ou latinos; e 79,3% eram do sexo feminino. As etiologias mais comuns de HAP foram HAP idiopática (58,5%), HAP hereditária (18,3%) e HAP associada a doenças do tecido conjuntivo (DTC) (14,9%). O tempo médio entre o diagnóstico de HAP e a seleção foi de 8,76 anos. A maioria dos participantes estava recebendo terapia de base tripla (61,3%) ou dupla (34,7%) para HAP, e mais de um terço (39,9%) estava recebendo infusões de prostaciclina. Todos os pacientes estavam recebendo terapia de base estável para HAP há pelo menos 90 dias e continuaram assim durante todo o estudo. As proporções de participantes em CF II da OMS (48,6%) e em CF III da OMS (51,4%) foram semelhantes em ambos os grupos. O estudo STELLAR excluiu pacientes com diagnóstico de hipertensão pulmonar (HP) dos Grupos 2, 3, 4 ou 5 da OMS; HAP associada ao vírus da imunodeficiência humana (HIV), HAP associada à hipertensão portal, HAP associada à esquistossomose e doença veno-oclusiva pulmonar. Outros critérios de exclusão incluíram, mas não se limitaram a: nível de hemoglobina acima do limite superior de normalidade específico por gênero na triagem, conforme teste laboratorial local; contagem basal de plaquetas < 50.000/mm3 ( < 50,0 × 109/L) na triagem; hipertensão sistêmica não controlada, evidenciada por pressão arterial (PA) sistólica sentada > 160 mmHg ou PA diastólica sentada > 100 mmHg durante a visita após um período de repouso; PA sistólica basal < 90 mmHg na triagem.
O desfecho primário de eficácia foi a mudança em relação ao valor basal na Semana 24 na distância no teste de caminhada de 6 minutos (TC6M). No grupo de tratamento WINREVAIRTM, a mediana da mudança ajustada por placebo no TC6M em relação ao valor basal na Semana 24, foi de 40,8 metros (IC 95%: 27,5; 54,1; p < 0,001). A mediana das mudanças ajustadas por placebo no TC6M na Semana 24, também foi avaliada em subgrupos (veja Figura 1).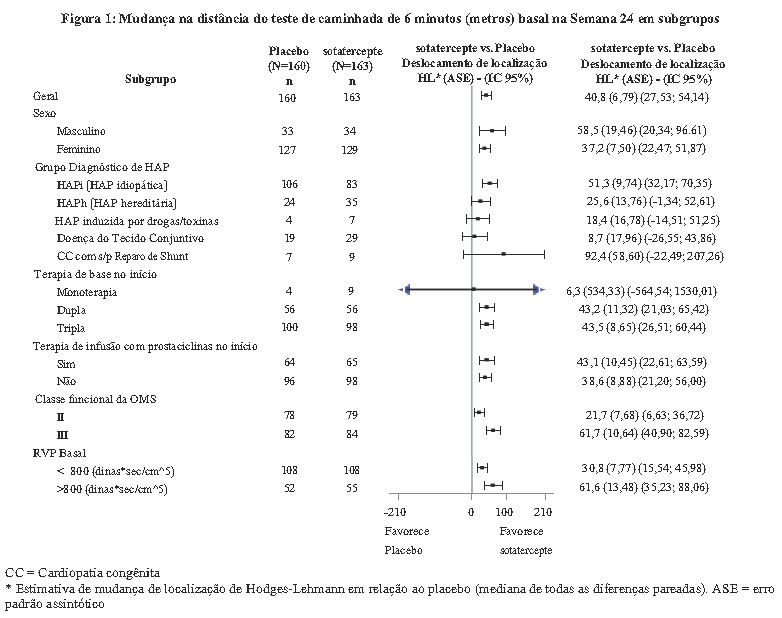
A mudança em relação à linha de base no TC6M na Semana 24 para os indivíduos que morreram recebeu um valor de -2000 metros para receber a pior classificação. A mudança em relação à linha de base no TC6M na Semana 24 para indivíduos que faltaram dados devido a um evento de piora clínica não fatal foi imputada a -1000 metros para receber a próxima pior classificação.
A melhora clínica foi um desfecho pré-definido medido pela proporção de pacientes que atingiram todos os três seguintes critérios na Semana 24 em relação ao valor basal: melhora no TC6M (aumento ≥ 30 m), melhora no peptídeo natriurético pró-tipo B N-terminal (NT-proBNP) (diminuição no NT-proBNP ≥ 30% ou manutenção/obtenção do nível de NT-proBNP < 300 ng/L) e melhora da CF OMS ou manutenção da CF II da OMS. A progressão da doença foi medida pelo tempo até o óbito ou primeira ocorrência de um evento de piora clínica. Os eventos de piora clínica incluíram inclusão na lista para transplante pulmonar e/ou cardíaco, necessidade de iniciar terapia de resgate com uma terapia de HAP de base aprovada ou a necessidade de aumentar a dose de prostaciclina infusional em ≥ 10%, necessidade de septostomia atrial, hospitalização por piora da HAP (≥ 24 horas), ou deterioração da HAP (piora da CF da OMS e redução da distância do TC6M ≥ 15%, com ambos os eventos ocorrendo ao mesmo tempo ou em momentos diferentes). Os eventos de piora clínica e óbito foram registrados até que o último paciente completasse a visita da Semana 24 (dados até a linha de corte dos dados; duração mediana da exposição de 33,6 semanas).
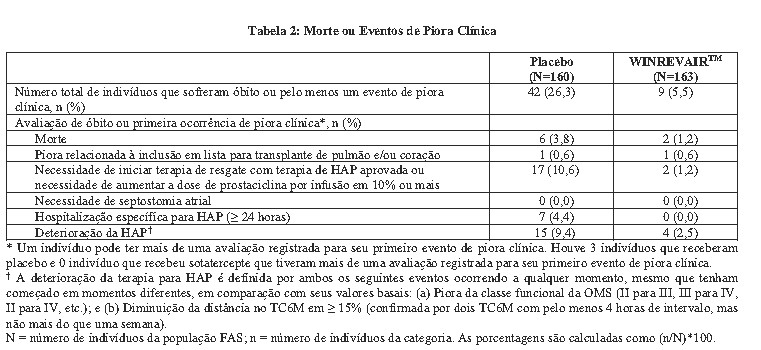
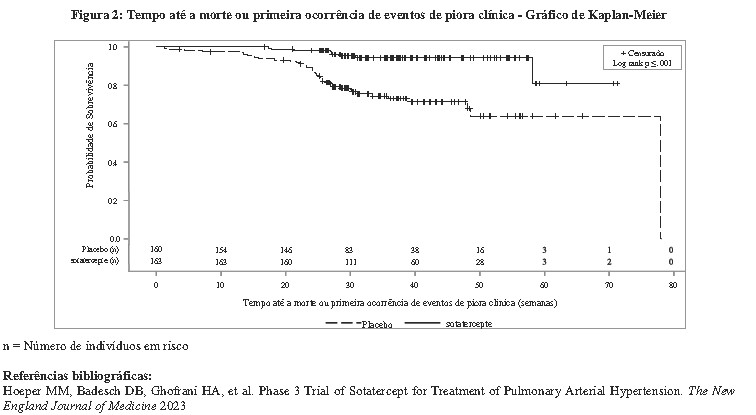
Os pacientes tratados com WINREVAIRTM apresentaram melhora clínica estatisticamente significativa, melhora na CF da OMS e atraso na progressão da doença, incluindo redução do risco de morte e hospitalização em comparação aos pacientes tratados com placebo (veja Tabela 1, Tabela 2 e Figura 2).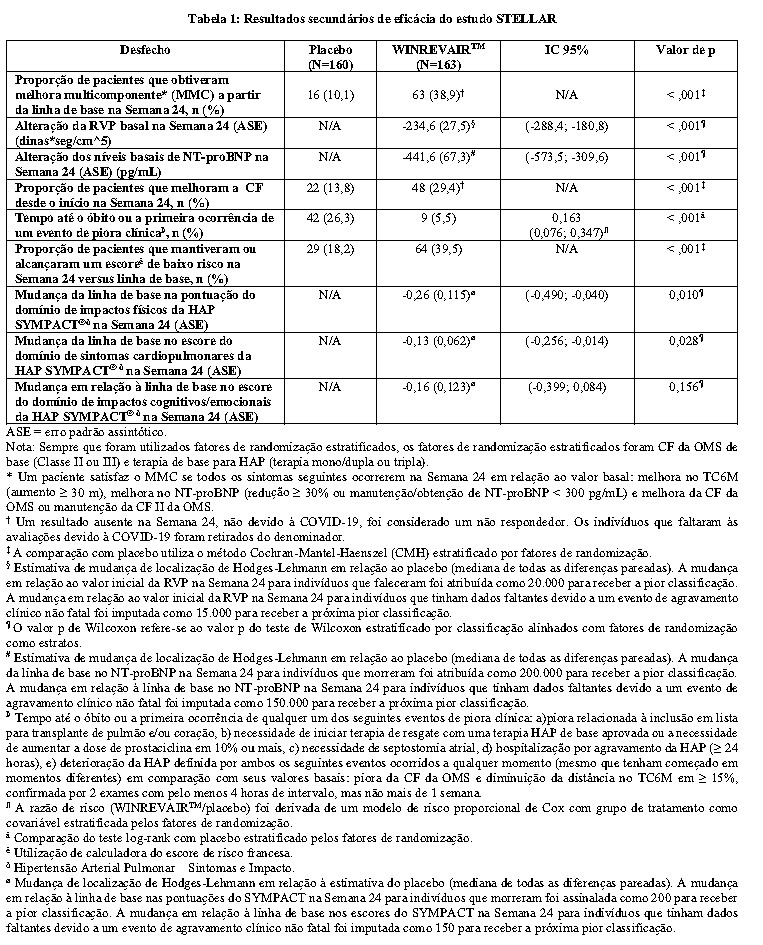
Referências bibliográficas:
Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. The New England Journal of Medicine 2023
3. CARACTERÍSTICAS FARMACOLÓGICAS
Classe terapêutica
WINREVAIRTM (sotatercepte) é um inibidor de sinalização de activina.
Mecanismo de Ação
O sotatercepte é um inibidor da sinalização de activina com alta seletividade para activina A, uma glicoproteína dimérica que pertence à superfamília de ligantes do fator de crescimento transformador b (TGF-b). A activina A se liga ao receptor de activina tipo IIA (ActRIIA) regulando a sinalização chave para inflamação, proliferação celular, apoptose e homeostase tecidual.
Os níveis de activina A estão aumentados em pacientes com HAP. A ligação da activina ao ActRIIA promove sinalização proliferativa enquanto há uma diminuição na sinalização do receptor morfogenético ósseo tipo II (BMPR-II) antiproliferativo. O desequilíbrio da sinalização ActRIIA-BMPRII subjacente à HAP resulta em hiperproliferação de células vasculares, causando remodelamento patológico da parede arterial pulmonar, estreitamento da luz arterial, aumento da resistência vascular pulmonar, aumento da pressão da artéria pulmonar e disfunção ventricular direita.
O sotatercepte consiste em uma proteína de fusão do receptor homodimérico de activina recombinante tipo IIA-Fc (ActRIIA-Fc) que atua como um captador do ligante que elimina o excesso de activina A e outros ligantes para ActRIIA para inibir a sinalização de activina. Como resultado, o sotatercepte reequilibra a sinalização pró-proliferativa (ActRIIA/Smad2/3-mediado) e antiproliferativa (BMPRII/Smad1/5/8mediado) para modular a proliferação vascular. Em modelos de HAP em ratos, um análogo do sotatercepte reduziu a expressão de marcadores pró-inflamatórios na parede arterial pulmonar, reduziu o recrutamento leucocitário, inibiu a proliferação de células endoteliais e musculares lisas e promoveu apoptose na vasculatura doente. Essas alterações celulares foram associadas a paredes mais finas dos vasos, remodelamento arterial e ventricular direito reversos e melhora hemodinâmica. Em estudos clínicos de HAP, WINREVAIRTM diminuiu a resistência vascular pulmonar e reverteu o remodelamento do ventrículo direito.
Farmacodinâmica
Um estudo clínico de fase 2 avaliou a resistência vascular pulmonar (RVP) em pacientes com HAP após 24 semanas de tratamento com sotatercepte. A diminuição em relação ao valor basal na RVP foi significativamente maior nos grupos sotatercepte 0,7 mg/kg e 0,3 mg/kg em comparação com o grupo placebo. A diferença média dos mínimos quadrados (MQ) ajustados por placebo em relação ao valor basal foi de -269,4 dinas*seg/cm5 (IC 95%: -365,8; -173,0) para o grupo sotatercepte 0,7 mg/kg e -151,1 dinas*seg/cm5 (IC 95%: -249,6; -52,6) para o grupo sotatercepte 0,3 mg/kg. No STELLAR, a diminuição em relação ao valor basal da RVP foi também significativamente maior no grupo sotatercepte 0,7 mg/kg em comparação com o grupo placebo (veja item
2. RESULTADOS DE EFICÁCIA).
Farmacocinética
Em pacientes com HAP, a média geométrica (%CV) da área sob a curva (AUC) em estado estável e a concentração de pico em estado estável (Cmáx) na dose de 0,7 mg/kg a cada 3 semanas (Q3W) foram 171,3 mcg×d/mL (34,2%) e 9,7 mcg/mL (30%CV), respectivamente. A AUC e Cmáx do sotatercepte aumentam proporcionalmente com a dose. O estado de equilíbrio é alcançado após aproximadamente 15 semanas com a dosagem múltipla Q3W. A razão de acumulação da AUC do sotatercepte foi de aproximadamente 2,2.
-Absorção
A formulação SC tem uma biodisponibilidade absoluta de aproximadamente 66%. A concentração máxima de sotatercepte é atingida em um tempo mediano até o pico da concentração do medicamento (Tmáx) de aproximadamente 7 dias (intervalo de 2 a 8 dias) após doses SC múltiplas (0,1 mg/kg a cada 4 semanas) em mulheres na pós-menopausa.
-Distribuição
O volume central de distribuição (%CV) do sotatercepte é de aproximadamente 3,6 L (24,7%). O volume periférico de distribuição (%CV) é de aproximadamente 1,7 L (73,3%).
-Eliminação
A depuração do sotatercepte é de aproximadamente 0,18 L/dia. A média geométrica da meia-vida terminal (%CV) é de aproximadamente 21 dias (33,8%).
-Metabolismo
O sotatercepte é catabolizado por processos gerais de degradação de proteínas.
Populações Especiais
Idade, sexo e raça Não foram observadas diferenças clinicamente significativas na farmacocinética (PK) do sotatercepte com base na idade (18 a 81 anos de idade), sexo ou raça.
Peso Corporal
A depuração (CL) e o volume central de distribuição (Vc) do sotatercepte aumentaram com o aumento do peso corporal. O regime posológico baseado no peso recomendado resulta em exposições consistentes ao sotatercepte, independentemente do peso corporal.
Insuficiência Renal A PK do sotatercepte foi comparável em pacientes com HAP com insuficiência renal leve a moderada (TFGe variando entre 30 e 89 mL/min/1,73m2) e naqueles com função renal normal (TFGe ≥ 90 mL/min/1,73 m2). Além disso, a PK do sotatercepte é comparável entre pacientes com doença renal terminal (DRCT) sem HAP e pacientes com função renal normal. WINREVAIRTM não é dialisável durante a hemodiálise. Não é recomendado qualquer ajuste posológico em pacientes com insuficiência renal. O sotatercepte não foi estudado em pacientes com HAP com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2).
Insuficiência hepática
Não é esperado que a insuficiência hepática (determinada pela Classificação de Child-Pugh) influencie o metabolismo do sotatercepte, uma vez que o sotatercepte é metabolizado através do catabolismo celular. O sotatercepte não foi estudado em pacientes com HAP com insuficiência hepática (Classificação de Child-Pugh A a C).
Imunogenicidade
A incidência observada de anticorpos antidrogas é altamente dependente da sensibilidade e especificidade do ensaio. Diferenças nos métodos de ensaio impedem comparações significativas da incidência de anticorpos antidrogas no estudo descrito abaixo com a incidência de anticorpos antidrogas em outros estudos, incluindo os de WINREVAIRTM ou de outros produtos de sotatercepte. Durante o período de tratamento de 24 semanas no estudo pivotal (STELLAR), 44/163 (27%) dos pacientes tratados com sotatercepte desenvolveram anticorpos anti-sotatercepte. Entre esses 44 pacientes, 12 (27%) testaram positivo para anticorpos neutralizantes contra o sotatercepte. Os anticorpos anti-sotatercepte geralmente tinham títulos baixos, com um título mediano de 30 (variação < 20 a 640).
Não foram identificados efeitos clínicos dos anticorpos anti-sotatercepte na farmacocinética, farmacodinâmica, segurança ou efetividade do sotatercepte durante o período de tratamento de 24 semanas.
TOXICOLOGIA ANIMAL
Toxicidade aguda
Nenhuma toxicidade aguda foi observada em estudos de toxicidade SC de dose repetida em dosagens de até 30 mg/kg em ratos e 50 mg/kg em macacos (doses únicas proporcionaram exposições aproximadamente 15 vezes e 38 vezes, respetivamente, à exposição humana na dose humana máxima recomendada (DHMR) (com base na AUC estimada)).
Toxicidade crônica
Em ratos e macacos, os estudos de toxicidade SC de maior duração foram de 3 meses e 9 meses, respectivamente. Em ratos administrados com doses semanais de 0,3, 3 e 30 mg/kg durante 3 meses, os achados adversos incluíram degeneração do ducto eferente/testicular, congestão/necrose da glândula adrenal, glomerulonefrite membranoproliferativa e nefrite tubulointersticial nos rins que ocorreram com uma exposição 18 vezes maior que a DHMR (com base na AUC estimada). Tanto as alterações adrenais quanto as renais demonstraram reversibilidade após um período de recuperação de 1 mês. Em macacos administrados com 1, 2,6 e 10 mg/kg uma vez a cada 4 semanas e 10 mg/kg uma vez a cada 2 semanas, as alterações adversas foram limitadas à glomerulonefrite e nefrite tubulointersticial nos rins que ocorreram em exposições ≥ 6 vezes a DHMR (com base na ASC estimada). As alterações renais em macacos se resolveram parcialmente após um período de recuperação de 3 meses.
Carcinogênese
Não foram realizados estudos de carcinogenicidade com o sotatercepte.
Mutagênese
Não foram realizados estudos de mutagenicidade com o sotatercepte.
Reprodução
Em um estudo de fertilidade e desenvolvimento embrionário inicial em ratas, o sotatercepte foi administrado SC uma vez por semana nas doses de 5, 15 e 50 mg/kg a partir de 2 semanas antes do acasalamento e até ao dia 7 de gestação. Emdoses ≥ 15 mg/kg (≥ 9 vezes o DHMR, com base na AUC estimada), as taxas de gravidez foram reduzidas e houve aumentos na perda pré e pós-implantação e reduções no tamanho da ninhada viva. O aumento da duração do ciclo estral ocorreu apenas com 50 mg/kg (21 vezes o DHMR, com base na AUC estimada).
Em um estudo de fertilidade em ratos machos, o sotatercepte foi administrado SC uma vez por semana nas doses de 0,3, 3 e 30 mg/kg durante 13 semanas (começando 10 semanas antes do acasalamento). Um subgrupo de animais foi examinado após um período de recuperação de 13 semanas. Com≥0,3 mg/kg(0,5 vezeso DHMR, com base na AUC estimada) houve alterações histológicas irreversíveis nos ductos eferentes, testículos e epidídimos. Ocorreram diminuições reversíveis na fertilidade a 30 mg/kg (20 vezes o DHMR, com base na AUC estimada).
Desenvolvimento
Em estudos de toxicidade para o desenvolvimento embriofetal, animais gestantes foram dosados por via subcutânea com sotatercepte durante
o período de organogênese. O sotatercepte foi administrado a ratas nos dias 6 e 13 de gestação nas doses de 5, 15 ou 50 mg/kg e a coelhos nos dias 7e 14 de gestação nas doses de 0,5, 1,5 ou 5 mg/kg. Os efeitos em ambas as espécies incluíram reduções no número de fetos vivos e no peso corporal fetal, atrasos na ossificação e aumentos nas reabsorções e perdas pós-implantação. Em ratos e coelhos, estes efeitos foram observados em exposições (com base na área sob a curva (AUC)) aproximadamente 4 vezes e 0,6 vezes a dose humana máxima recomendada (DHMR), respetivamente. Apenas em ratos, as variações esqueléticas (aumento do número de costelas supranumerárias e mudanças no número de vértebras torácicas ou lombares) ocorreram em uma exposição 15 vezes maior do que a exposição humana no DHMR.
Em um estudo de desenvolvimento pré e pós-natal em ratos, o sotatercepte foi administrado por via subcutânea nas doses de 1,5 e 5 mg/kg nos dias 6 e 13 de gestação, ou nas doses de 1,5, 5 ou 10 mg/kg durante a lactação nos dias 1, 8 e 15. Não houve efeitos adversos em filhotes de primeira geração filial (F1) de mães dosadas durante a gestação em exposições estimadas até 2 vezes a DHMR. Em filhotes F1 de mães dosadas durante a lactação, as diminuições no peso dos filhotes correlacionaram-se com atrasos na maturação sexual em exposições estimadas (com base na AUC) ≥ 2 vezes o DHMR.
4. CONTRAINDICAÇÕES
WINREVAIR™ está contraindicado em pacientes com hipersensibilidade à substância ativa ou a qualquer excipiente do medicamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Eritrocitose
Foram observados aumentos de hemoglobina (Hb) em pacientes durante o tratamento com WINREVAIRTM. A eritrocitose grave pode aumentar o risco de eventos tromboembólicos ou síndrome de hiperviscosidade. Monitorar a Hb antes de cada nas primeiras 5 doses, ou por mais tempo caso os valores estiverem instáveis e, posteriormente, periodicamente, para determinar se são necessários ajustes posológicos (veja os itens 8. POSOLOGIA E MODO DE USAR e 9. REAÇÕES ADVERSAS).
Trombocitopenia grave
Foi observada diminuição da contagem de plaquetas em alguns pacientes usando WINREVAIRTM e foi observada trombocitopenia grave (contagem de plaquetas < 50.000/mm3 ( < 50,0 x 109/L)). A trombocitopenia ocorreu com maior frequência em pacientes que também estavam recebendo infusão de prostaciclina.
Não inicie o tratamento se a contagem de plaquetas for < 50.000/mm3 ( < 50 x 109/L) (veja o item 8. POSOLOGIA E MODO DE USAR).
Monitorar a contagem de plaquetas antes de cada dose nas primeiras 5 doses, ou por mais tempo se os valores estiverem instáveis, e, posteriormente, periodicamente, para determinar se são necessários ajustes posológicos (veja os itens 8. POSOLOGIA E MODO DE USAR e 9. REAÇÕES ADVERSAS).
Sangramento grave
Em estudos clínicos, foram relatados casos graves de hemorragia (por exemplo, hemorragia gastrointestinal, intracraniana) em 4% dos pacientes em terapia com WINREVAIRTM e em 1% daqueles que receberam placebo. Os pacientes com eventos hemorrágicos graves tinham maior probabilidade de estar em terapia com prostaciclina e/ou agentes antitrombóticos, ou apresentar contagens baixas de plaquetas. Aconselhe os pacientes sobre sinais e sintomas de perda de sangue. Avalie e trate o sangramento adequadamente. Não administre WINREVAIRTM se o paciente estiver apresentando um evento hemorrágico grave (veja os itens 5. ADVERTÊNCIAS E PRECAUÇÕES -Trombocitopenia grave e 9. REAÇÕES ADVERSAS -Experiência em Ensaios Clínicos).
Toxicidade embriofetal
Com base em achados em estudos de reprodução animal, WINREVAIRTM pode causar danos fetais quando administrado a uma gestante. Aconselhar as gestantes sobre o risco potencial para o feto. Aconselhar mulheres com potencial reprodutivo a utilizar um método contraceptivo eficaz durante o tratamento com WINREVAIRTM e por, pelo menos, 4 meses após a dose final (veja os itens 5. ADVERTÊNCIAS E PRECAUÇÕES -USO EM POPULAÇÕES ESPECIAIS -Mulheres e homens com potencial reprodutivo e 3. CARACTERÍSTICAS FARMACOLÓGICAS -TOXICOLOGIA ANIMAL).
Fertilidade prejudicada
Com base em achados em animais, WINREVAIRTM pode prejudicar a fertilidade feminina e masculina. Aconselhar os pacientes sobre os potenciais efeitos na fertilidade (veja os itens 5. ADVERTÊNCIAS E PRECAUÇÕES -USO EM POPULAÇÕES ESPECIAIS -Mulheres e homens com potencial reprodutivo e 3. CARACTERÍSTICAS FARMACOLÓGICAS -TOXICOLOGIA ANIMAL).
USO EM POPULAÇÕES ESPECIAIS
Gravidez
Resumo do Risco
Não há dados disponíveis sobre o uso de WINREVAIRTM em gestantes para informar um risco associado ao medicamento de defeitos congênitos graves e aborto espontâneo. No entanto, com base em estudos de toxicidade embriofetal animal, WINREVAIRTM pode causar perda pós-implantação ou danos fetais quando administrado a uma gestante. Aconselhar gestantes o risco potencial para o feto.
O risco de antecedentes de defeitos congênitos graves e aborto espontâneo para a população indicada não é conhecido. Os desfechos adversos na gravidez ocorrem independentemente da saúde da mãe ou do uso de medicamentos. Na população geral dos EUA, o risco basal estimado de defeitos congênitos graves e aborto espontâneo em gestações clinicamente reconhecidas é de 2% a 4% e 15% a 20%, respectivamente (veja o subitem Considerações Clínicas abaixo).
Considerações clínicas
Gestantes com HAP apresentam risco de insuficiência cardíaca, parto prematuro e morte materna e fetal.
Dados
Dados em animais Em ratas e coelhas prenhes, as exposições ao sotatercepte ≥ 4 vezes e ≥ 0,6 vezes a DHMR, respectivamente, resultaram em diminuições no peso fetal, atrasos na ossificação e aumentos nas reabsorções e perda pós-implantação. Com 15 vezes a DHMR, os fetos de ratas tiveram uma incidência aumentada de variações esqueléticas (aumento do número de costelas supranumerárias e alterações no número de vértebras torácicas ou lombares). Em um estudo de desenvolvimento pré e pós-natal, filhotes de ratos de mães lactantes expostos a níveis de sotatercepte ≥ 2 vezes a DHMR tiveram pesos corporais reduzidos que se correlacionaram com atrasos na maturação sexual (veja item 3. CARACTERÍSTICAS FARMACOLÓGICAS -TOXICOLOGIA ANIMAL).
Mães lactantes
Resumo do Risco Não existem dados sobre a presença de sotatercepte no leite humano, os efeitos no lactente ou os efeitos na produção de leite. Uma vez que não se sabe se o sotatercepte é excretado no leite materno, evite o uso de sotatercepte em lactantes.
Uso criterioso no aleitamento ou na doação de leite humano: O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico ou cirurgião-dentista.
Mulheres e homens com potencial reprodutivo
Teste de gravidez
O teste de gravidez é recomendado para mulheres com potencial reprodutivo antes de iniciar o tratamento.
Contracepção
Mulheres
As mulheres com potencial reprodutivo devem utilizar contracepção eficaz durante o tratamento com WINREVAIRTM e durante pelo menos 4 meses após a última dose, se o tratamento for descontinuado (veja os itens 5. ADVERTÊNCIAS E PRECAUÇÕES -USO EM POPULAÇÕES ESPECIAIS -Gravidez e 3. CARACTERÍSTICAS FARMACOLÓGICAS -TOXICOLOGIA ANIMAL).
Categoria: C. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Infertilidade
Com base nos achados em animais, o sotatercepte pode prejudicar a fertilidade feminina e masculina (veja item 3. CARACTERÍSTICAS FARMACOLÓGICAS -Toxicologia Animal). Os efeitos adversos sobre a fertilidade em ratos machos foram reversíveis após um período de 13 semanas.
Uso Pediátrico
A segurança e eficácia de WINREVAIRTM não foram estabelecidas em pacientes com menos de 18 anos de idade.
Uso geriátrico
Nenhum ajuste de dose de WINREVAIRTM é necessário com base na idade. Um total de 81 pacientes com idade ≥ 65 anos participaram de estudos clínicos para HAP, dos quais 52 (16%) foram tratados com WINREVAIRTM.
Não foram observadas diferenças globais na eficácia de WINREVAIRTM entre os subgrupos com idade de < 65 anos e ≥ 65 anos.
Com exceção dos eventos hemorrágicos (um grupo coletivo de eventos adversos de interesse clínico), não houve diferenças na segurança entre os subgrupos com idade de < 65 anos e ≥ 65 anos. Os eventos hemorrágicos ocorreram mais comumente no subgrupo WINREVAIRTM mais velho; no entanto, não houve desequilíbrio notável entre os subgrupos etários para qualquer evento hemorrágico específico.
No estudo STELLAR, 10 de um total de 323 pacientes (3%) tinham 75 anos ou mais.
Insuficiência renal
Não é necessário ajuste posológico de WINREVAIRTM com base na insuficiência renal leve ou moderada. O sotatercepte não foi estudado em pacientes com HAP com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2) (veja os itens 3. CARACTERÍSTICAS FARMACOLÓGICAS e 8. POSOLOGIA E MODO DE USAR).
Insuficiência hepática
A utilização de WINREVAIRTM não foi estudada em pacientes com insuficiência hepática (Classificação de Child-Pugh A a C). Não é esperado que a insuficiência hepática influencie o metabolismo do sotatercepte, uma vez que o sotatercepte é metabolizado através do catabolismo celular (veja os itens 3. CARACTERÍSTICAS FARMACOLÓGICAS e 8. POSOLOGIA E MODO DE USAR).
Este medicamento pode causar doping.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhuma interação medicamentosa foi identificada com base nos dados disponíveis.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenar WINREVAIRTM em geladeira (2°C a 8°C). Não congelar. Manter nesta embalagem até o final do uso. Manter na embalagem original para proteger da luz.
O prazo de validade do medicamento a partir da data de fabricação é de 36 meses.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, utilize a solução reconstituída o mais rapidamente possível, em até 4 horas após a reconstituição, em temperatura de até 30°C. A estabilidade por até 4 horas foi comprovada para os aspectos físico-químicos do medicamento. Do ponto de vista microbiológico, recomenda-se que o medicamento seja utilizado imediatamente após a reconstituição.
WINREVAIRTM é um pó liofilizado estéril, sem conservantes, branco a esbranquiçado, para administração subcutânea após reconstituição. Após a reconstituição, WINREVAIRTM é límpido a opalescente, incolor a ligeiramente amarelado acastanhado e livre de grumos ou pó.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Dosagem inicial recomendada em adultos
WINREVAIRTM é administrado uma vez a cada 3 semanas por injeção subcutânea (SC) de acordo com o peso do paciente. A dose inicial de WINREVAIRTM é de 0,3 mg/kg (veja Tabela 3). Obtenha a hemoglobina (Hb) e a contagem de plaquetas antes da primeira dose de WINREVAIRTM. Foram observados aumentos rápidos de Hb superiores a 2 g/dL após o início do tratamento. Não é recomendado iniciar o tratamento se a contagem de plaquetas for < 50.000/mm3 ( < 50,0 x 109/L) (veja o item 8. POSOLOGIA E MODO DE USAR - Modificações na dosagem em adultos devido ao aumento da hemoglobina ou diminuição da contagem de plaquetas).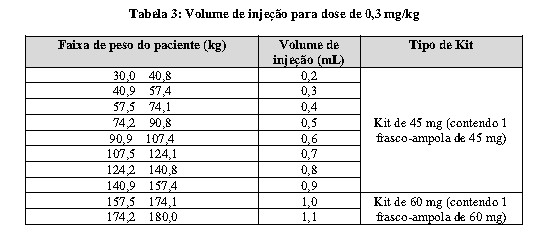
Dose alvo recomendada em adultos
A dose alvo de WINREVAIRTM é de 0,7 mg/kg (veja Tabela 4) administrada a cada 3 semanas. Obtenha e analise a hemoglobina (Hb) e a contagem de plaquetas antes de aumentar para a dose alvo de 0.7 mg/kg. Continuar o tratamento a 0,7 mg/kg a cada 3 semanas, a menos que sejam necessários ajustes posológicos (veja o item 8. POSOLOGIA E MODO DE USAR -Modificações na dosagem em adultos devido ao aumento da hemoglobina ou diminuição da contagem de plaquetas).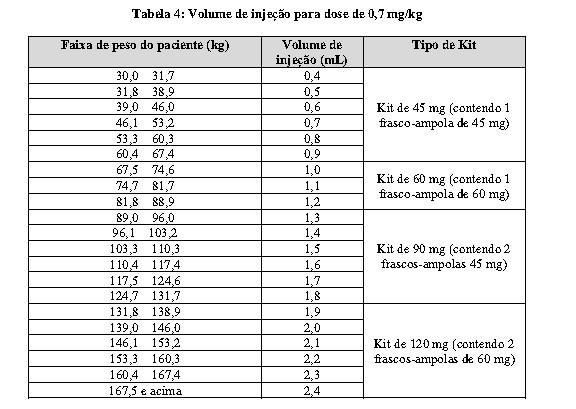
Dose esquecida, superdose ou subdose
Se uma dose de WINREVAIRTM for esquecida, administrar o mais rápido possível. Se a dose esquecida de WINREVAIRTM não for aplicada dentro de 3 dias da data programada, ajuste o cronograma para manter intervalos de dosagem de 3 semanas. Em caso de superdose ou subdose, considere retreinar o paciente ou o cuidador sobre a administração adequada, conforme apropriado. Em caso de superdose, monitorar a eritrocitose (veja o item 10. SUPERDOSE).
Modificações na dosagem em adultos devido ao aumento da hemoglobina ou diminuição da contagem de plaquetas
Foram observados aumentos na Hb para níveis superiores a 2 g/dL acima do limite superior da normalidade (LSN) e reduções na contagem de plaquetas < 50.000/mm3 ( < 50,0 x 109/L). Verifique a contagem de Hb e plaquetas antes de cada dose para as primeiras 5 doses, ou por mais tempo se os valores estiverem instáveis. Depois disso, monitore a Hb e a contagem de plaquetas periodicamente. Considere a avaliação do risco-benefício individualizada para o paciente para determinar se a modificação da dose é adequada (veja o item 5. ADVERTÊNCIAS E PRECAUÇÕES).
Atrasar o tratamento por 3 semanas se ocorrer alguma das seguintes situações:
•Aumento de Hb > 2,0 g/dL em relação à dose anterior e está acima do LSN.
•Aumento de Hb > 4,0 g/dL em relação ao valor basal.
• Aumento de Hb > 2,0 g/dL acima do LSN.
• Diminuição da contagem de plaquetas para < 50.000/mm3 ( < 50,0 x 109/L).
Para atrasos de tratamento com duração > 9 semanas, reiniciar o tratamento a 0,3 mg/kg e aumentar para 0,7 mg/kg após verificar níveis aceitáveis de Hb e contagem de plaquetas.
Pacientes pediátricos
A segurança e eficácia do WINREVAIRTM não foram estabelecidas em pacientes com menos de 18 anos de idade.
Pacientes geriátricos
Não é necessário ajuste de dose de WINREVAIRTM com base na idade (veja os itens 5. ADVERTÊNCIAS E PRECAUÇÕES - USO EM POPULAÇÕES ESPECIAIS e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência renal
Não é necessário ajuste posológico de WINREVAIRTM com base na insuficiência renal leve ou moderada. O sotatercepte não foi estudado em pacientes com HAP com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2) (veja itens 3. CARACTERÍSTICAS FARMACOLÓGICAS e 5. ADVERTÊNCIAS E PRECAUÇÕES - USO EM POPULAÇÕES ESPECIAIS).
Insuficiência hepática
A utilização de WINREVAIRTM não foi estudada em pacientes com insuficiência hepática (Classificação de Child-Pugh A a C). Não se espera que a insuficiência hepática influencie o metabolismo do sotatercepte, uma vez que o sotatercepte é metabolizado através do catabolismo celular (veja os itens 3. CARACTERÍSTICAS FARMACOLÓGICAS e 5. ADVERTÊNCIAS E PRECAUÇÕES - UTILIZAÇÃO EM POPULAÇÕES ESPECIAIS).
Preparação e administração
WINREVAIRTM destina-se ao uso sob a orientação de um profissional de saúde. Pacientes e cuidadores podem administrar WINREVAIRTM quando considerarem apropriado e quando receberem treinamento e acompanhamento do profissional de saúde sobre como reconstituir, preparar, medir e injetar WINREVAIRTM.
Considere confirmar em consultas subsequentes que o paciente ou cuidador pode preparar e administrar WINREVAIRTM corretamente:
• se a dose mudar ou se o paciente necessitar de um kit diferente
• se o paciente desenvolver eritrocitose (veja o item 5. ADVERTÊNCIAS E PRECAUÇÕES).
Consulte as Instruções de Uso (IU) para obter instruções detalhadas sobre a preparação e administração adequadas de WINREVAIRTM.
Selecionando o kit de produto apropriado
Se o peso de um paciente exigir a utilização de dois frascos-ampolas de 45 mg ou dois frascos-ampolas de 60 mg do produto liofilizado, um kit de 2 frascos-ampolas deve ser utilizado em vez de dois kits individuais de 1 frasco-ampola. Um kit de 2 frascos-ampolas inclui instruções para combinar o conteúdo de dois frascos, o que ajuda a medir a dosagem adequada e elimina a necessidade de injeções múltiplas (veja APRESENTAÇÕES).
Instruções de reconstituição
• Retire o kit de injeção da geladeira e aguarde 15 minutos para permitir que a(s) seringa(s) preenchidas e o medicamento cheguem à temperatura ambiente antes do preparo.
• Verifique o frasco-ampola para garantir que o produto não está vencido. O pó deve ser branco a esbranquiçado e pode parecer um pó compactado inteiro ou fragmentado.
• Retire a tampa do frasco contendo o pó liofilizado de WINREVAIRTM e limpe a tampa de borracha com um lenço umedecido com álcool.
• Conecte o adaptador do frasco ao frasco-ampola.
• Inspecione visualmente a seringa preenchida em busca de danos ou vazamentos e a água para injetáveis em seu interior para garantir que não haja partículas visíveis.
• Retire a tampa da seringa preenchida e coloque-a ao adaptador do frasco.
• Injete toda a água para injetáveis da seringa acoplada no frasco-ampola contendo o pó liofilizado. Isso proporcionará uma concentração final de 50 mg/mL.
• Agite suavemente o frasco para reconstituir o medicamento. NÃO agite ou misture vigorosamente.
• Deixe o frasco-ampola em repouso por até 3 minutos para permitir que as bolhas desapareçam.
• Inspecione visualmente a solução reconstituída. Quando devidamente misturada, WINREVAIRTM deve ser límpido a opalescente e incolor a ligeiramente amarelado acastanhado e não apresentar grumos ou pó.
• Desenrosque a seringa do adaptador de frasco e elimine a seringa vazia em um recipiente para materiais perfurocortantes.
• Se for prescrita uma apresentação com 2 frascos-ampolas, repita os passos descritos nesta seção para preparar o segundo frasco.
• Utilizar a solução reconstituída o mais rapidamente possível, mas não mais do que 4 horas após a reconstituição, em temperatura de até 30°C.
Preparação da seringa
• Limpe o adaptador de frasco com um lenço umedecido com álcool.
• Retire a seringa dosadora da embalagem e coloque a seringa no adaptador de frasco.
• Vire a seringa e o frasco-ampola de cabeça para baixo e retire o volume apropriado para injeção, com base no peso do paciente.
o Se a quantidade de dose exigir a utilização de dois frascos-ampolas, retire todo o conteúdo do primeiro frasco-ampola e transfira lentamente o conteúdo completo para o segundo frasco-ampola.
o Vire a seringa e o frasco-ampola de cabeça para baixo e retire a quantidade necessária do medicamento.
• Se necessário, empurre o êmbolo para remover o excesso de medicamento ou ar da seringa.
• Retire a seringa do frasco-ampola e coloque a agulha.
Instruções de administração
WINREVAIRTM é para ser administrado por injeção subcutânea.
• Selecione o local de injeção no abdômen (pelo menos 5 centímetros de distância do umbigo), na parte superior da coxa ou do braço e limpe com um lenço umedecido com álcool. Para cada injeção, selecione um novo local que não esteja com cicatrizes, sensível ou machucado.
o Para administração pelo paciente ou cuidador, use apenas o abdômen e a parte superior da coxa (veja IU).
• Realizar injeção subcutânea.
• Descarte a seringa vazia em um recipiente para materiais perfurocortantes. Não reutilize a seringa.
9. REAÇÕES ADVERSAS
Experiência em Ensaios Clínicos
Como os ensaios clínicos são conduzidos em condições muito variadas, as taxas de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser diretamente comparadas com as taxas nos ensaios clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
Os dados a seguir refletem a exposição a WINREVAIRTM no estudo clínico pivotal STELLAR. Os pacientes (n=323) foram randomizados em uma proporção de 1:1 para receber WINREVAIRTM ou placebo em combinação com terapias padrão de tratamento (SOC). Os pacientes receberam uma dose inicial de 0,3 mg/kg por injeção SC e a dose foi aumentada para a dose alvo de 0,7 mg/kg uma vez a cada 3 semanas durante 24 semanas. Após completar a fase de tratamento primário de 24 semanas, os pacientes continuaram em um período de tratamento duplo-cego de longo prazo (LTDB), mantendo sua terapia atual, até que todos os pacientes completassem o período de tratamento primário. As medianas das durações do tratamento foram semelhantes entre os grupos placebo e WINREVAIRTM (229,5 dias vs 252,0 dias, respetivamente) (veja item 2. RESULTADOS DE EFICÁCIA).
As reações adversas que ocorreram no estudo STELLAR quando todos os pacientes completaram o período primário de 24 semanas do estudo estão resumidas na Tabela 5.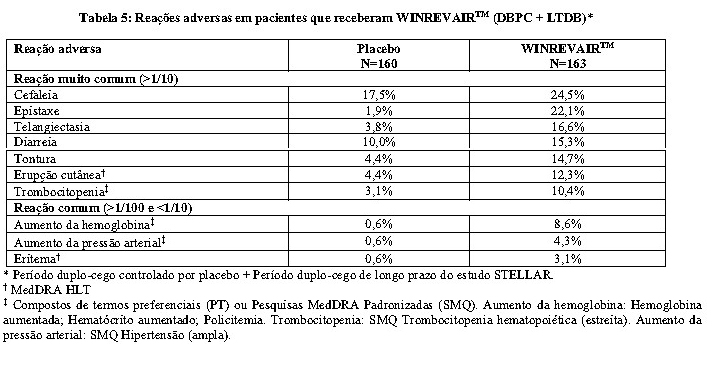
Aumento da hemoglobina
A maioria dos eventos de aumento de Hb (aumento de Hb, policitemia) foram não graves, leves e reversíveis, e não foram associados à descontinuação da terapia. Elevações moderadas na Hb ( > 2 g/dL acima do LSN) ocorreram em 12,3% dos pacientes em uso de WINREVAIRTM. Não foram observadas elevações graves (≥ 4 g/dL acima do LSN). Os aumentos na Hb foram controláveis por atrasos de dose, reduções de dose ou ambos.
Trombocitopenia
A maioria dos eventos de trombocitopenia (trombocitopenia e diminuição de contagem de plaquetas) foram não graves, leves, reversíveis e não foram associados à descontinuação da terapia. Redução acentuada na contagem de plaquetas < 50.000/mm3 ( < 50,0 x 109/L) ocorreu em 1,8% dos pacientes em uso de WINREVAIRTM.
Telangiectasia
Os eventos de telangiectasias não foram graves e não progrediram em gravidade ao longo do tempo. Em todos os pacientes expostos à WINREVAIRTM, a mediana do tempo até o início foi de 47,1 semanas. As descontinuações da terapia devido a telangiectasias foram de 1% no grupo WINREVAIRTM vs 0% no grupo placebo. Nenhum episódio de sangramento grave


