VYNAXA
SIGMA PHARMA
10 mg
rivaroxabana
Antitrombótico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
Comprimidos revestidos de 10 mg. Embalagem contendo 5, 10, 30, 100* ou 200** unidades.
*Embalagem hospitalar
**Embalagem fracionável.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém: rivaroxabana 10 mg, excipientes* q.s.p. 1 com rev
*celulose microcristalina, lactose monoidratada, hipromelose, laurilsulfato de sódio, croscamelose sódica, estearato de magnésio, hipromelose + macrogol, dióxido de titânio e óxido de ferro vermelho.
Informações técnicas.
1. INDICAÇÕES
VYNAXA® é indicado para a prevenção de tromboembolismo venoso (TEV) em pacientes adultos submetidos à cirurgia eletiva de artroplastia de joelho ou quadril.
VYNAXA® é indicado para o tratamento de trombose venosa profunda (TVP) e prevenção de trombose venosa profunda (TVP) e embolia pulmonar (EP) recorrentes, em adultos.
VYNAXA® é indicado para o tratamento de embolia pulmonar (EP) e prevenção de embolia pulmonar (EP) e trombose venosa profunda (TVP) recorrentes, em adultos.
2. RESULTADOS DE EFICÁCIA
Prevenção de eventos tromboembólicos venosos (TEV)
Prevenção de eventos tromboembólicos venosos (TEV) em pacientes submetidos à cirurgia ortopédica de grande porte dos membros inferiores.
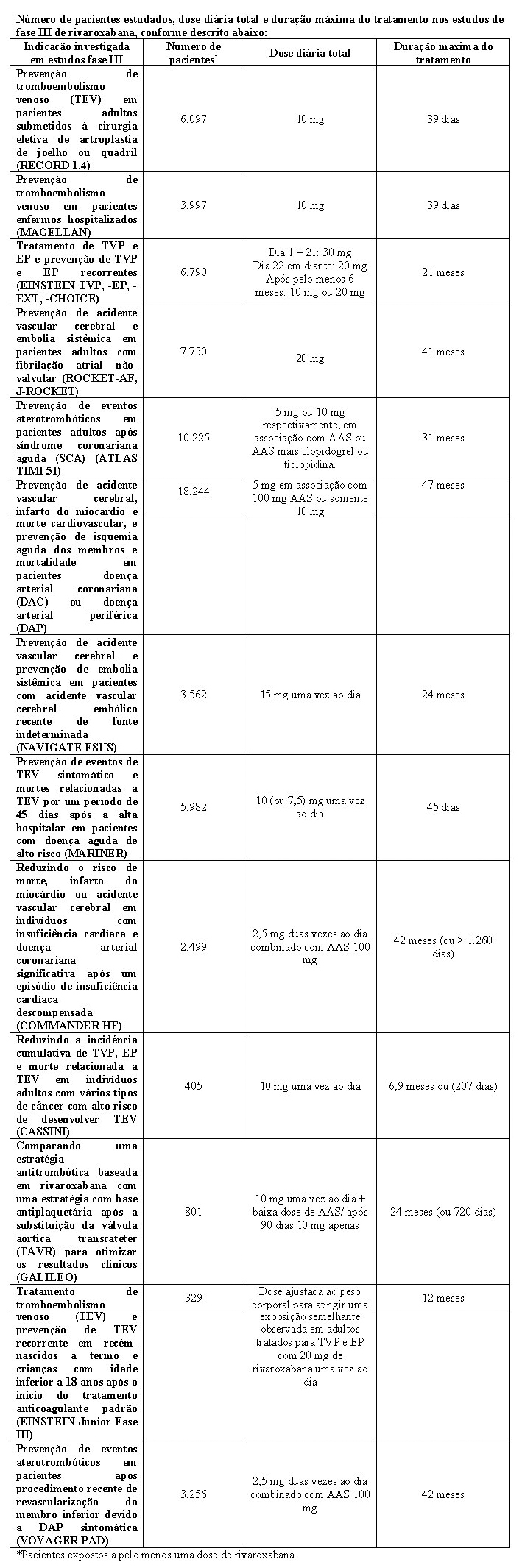
O programa clínico da rivaroxabana foi elaborado para demonstrar a eficácia de rivaroxabana para a prevenção de eventos tromboembólicos venosos (TEV), por exemplo, trombose venosa profunda (TVP) proximal e distal e embolia pulmonar (EP) em pacientes submetidos à cirurgia ortopédica de grande porte dos membros inferiores. Mais de 9.500 pacientes (7.050 em cirurgia de artroplastia total do quadril e 2.531 em cirurgia de artroplastia total do joelho) foram estudados em estudos clínicos de fase III controlados, duplo-cegos, randomizados, o programa RECORD.
Rivaroxabana, em dose de 10 mg uma vez ao dia, iniciada no mínimo 6 horas após a cirurgia, foi comparada a 40 mg de enoxaparina uma vez ao dia, iniciada em 12 horas antes da cirurgia.
Em três estudos de fase III (ver Tabela 1), a rivaroxabana reduziu significativamente a taxa de TEV total (qualquer TVP venograficamente detectada ou sintomática, EP não-fatal ou morte) e de TEV maior (TVP proximal, EP não-fatal e morte relacionada ao TEV), os desfechos finais ("endpoints") de eficácia primária e secundária maior pré-especificados.
Além disso, em todos os três estudos, a taxa de TEV sintomático (TVP sintomática, EP não-fatal, morte relacionada a um TEV) foi menor nos pacientes tratados com rivaroxabana, em comparação aos pacientes tratados com enoxaparina.
O objetivo final principal de segurança, sangramento importante, mostrou taxas comparáveis para pacientes tratados com 10 mg de rivaroxabana, em comparação a 40 mg de enoxaparina.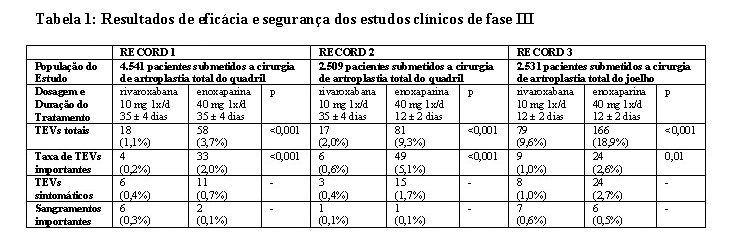
A análise dos resultados agrupados dos ensaios clínicos de fase III corroborou os dados obtidos nos estudos individuais referentes à redução de TEVs totais, de TEVs importantes e de TEVs sintomáticos com 10 mg de rivaroxabana uma vez ao dia, em comparação a 40 mg de enoxaparina uma vez ao dia.
Além do programa RECORD de fase III, foi conduzido um estudo pós-comercialização, coorte, aberto não-intervencional, (XAMOS) em 17.413 pacientes submetidos à cirurgia ortopédica de grande porte de joelho ou quadril, para comparar rivaroxabana com outra tromboprofilaxia farmacológica padrão de tratamento no contexto da vida real. TEV sintomático ocorreu em 57 (0,6 %) pacientes no grupo rivaroxabana (n=8.778) e 88 (1,0 %) pacientes no grupo padrão de tratamento ((n=8.635; HR 0,63; IC 95 % = 0,43 - 0,91); população de segurança). Ocorreram sangramentos importantes em 35 (0,4 %) e 29 (0,3 %) dos pacientes do grupo rivaroxabana e do grupo padrão de tratamento, respectivamente (HR 1,10; IC 95 % = 0,67 - 1,80). Este estudo não-intervencional confirmou os resultados de eficácia e segurança observados no programa RECORD.
Tratamento de trombose venosa profunda (TVP) e embolia pulmonar (EP) e prevenção de TVP e EP recorrentes
O programa clínico de rivaroxabana foi desenhado para demonstrar a eficácia do medicamento no tratamento inicial e continuado de trombose venosa profunda (TVP) aguda e embolia pulmonar (EP) e na prevenção de TVP e de EP recorrentes.
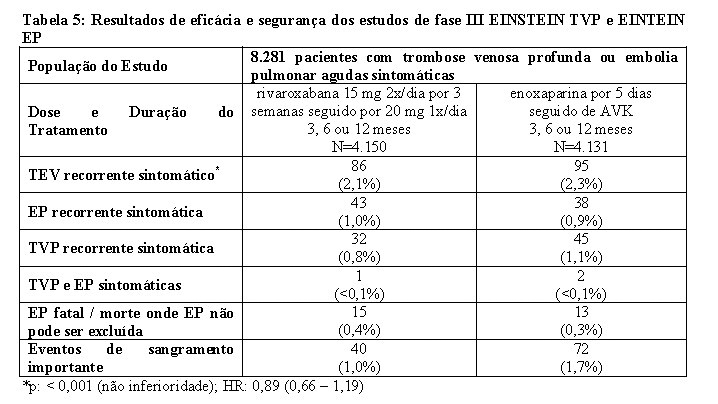
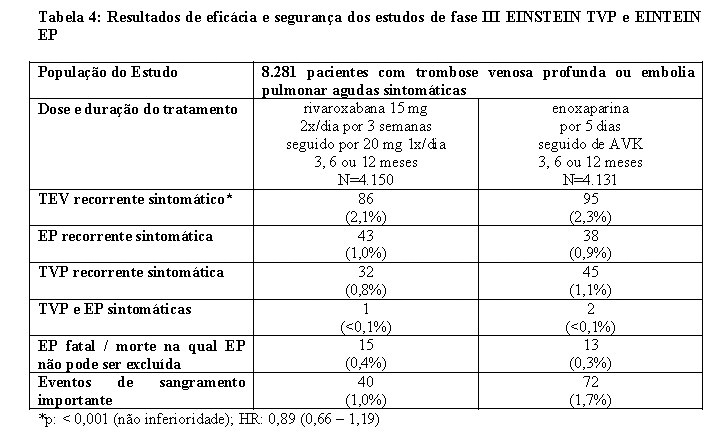
Mais de 12.800 pacientes foram estudados em quatro estudos clínicos de fase III, randomizados, controlados (EINSTEIN TVP, EINSTEIN EP, EINSTEIN Extension e EINSTEIN CHOICE) e adicionalmente foi realizada uma análise combinada predefinida dos estudos Einstein TVP e Einstein EP (veja Tabela 4). A duração total do tratamento combinado em todos os estudos foi de até 21 meses.
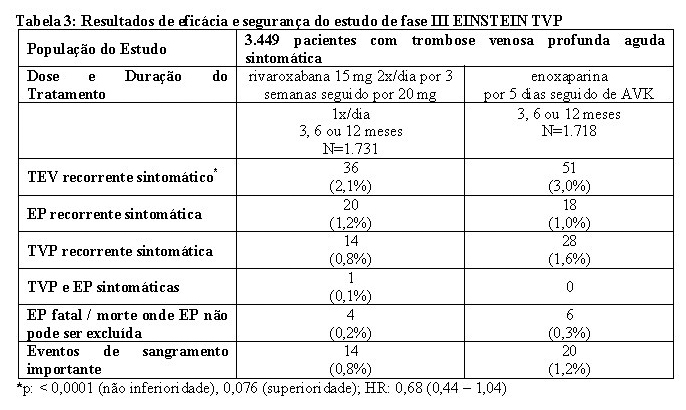
No estudo EINSTEIN TVP, 3.449 pacientes com TVP aguda foram estudados para o tratamento de TVP e prevenção de TVP e de EP recorrentes. A duração do tratamento foi de até 12 meses dependendo do julgamento clínico do investigador.
Para as três semanas iniciais de tratamento da TVP aguda, uma dose de 15 mg de rivaroxabana foi administrada duas vezes ao dia. Isto foi seguido por uma dose de 20 mg de rivaroxabana uma vez ao dia.
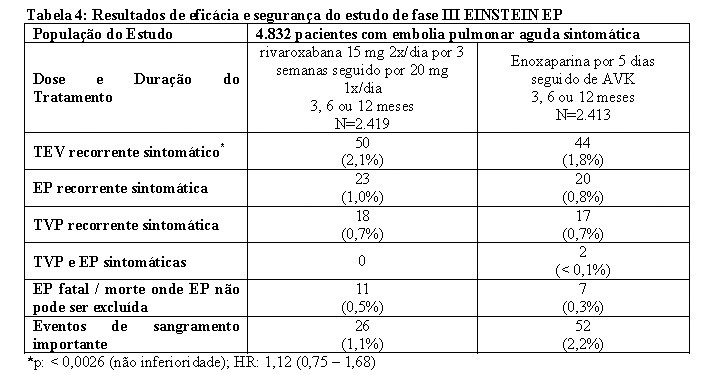
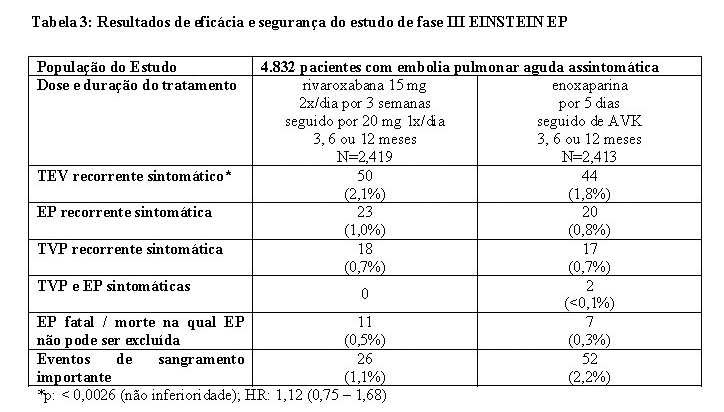
No estudo EINSTEIN EP, 4.832 pacientes com EP aguda foram estudados para o tratamento de EP e prevenção de TVP e EP recorrentes. A duração do tratamento foi de até 12 meses dependendo do julgamento clínico do investigador.
Para o tratamento inicial de EP aguda, uma dose de 15 mg de rivaroxabana foi administrada duas vezes ao dia por três semanas. Isso foi seguido por uma dose de 20 mg de rivaroxabana uma vez ao dia.
Em ambos os estudos EINSTEIN TVP e EINSTEIN EP, o regime de tratamento comparador consistiu em administrar enoxaparina por pelo menos cinco dias em combinação com antagonista da vitamina K até que o valor de TP/RNI atingisse a faixa terapêutica (≥ 2,0). O tratamento foi continuado com o antagonista da vitamina K com dose ajustada para manter os valores de TP/RNI dentro da faixa terapêutica de 2,0 a 3,0.
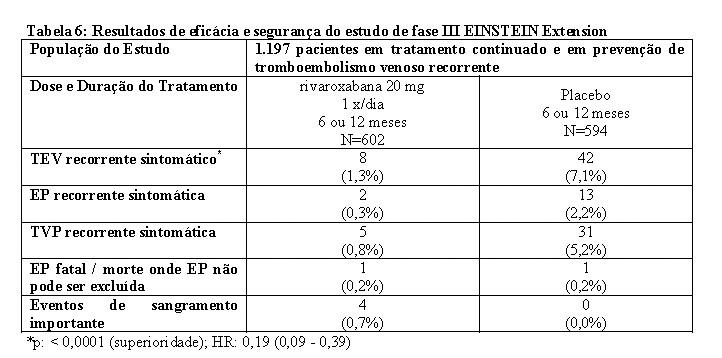
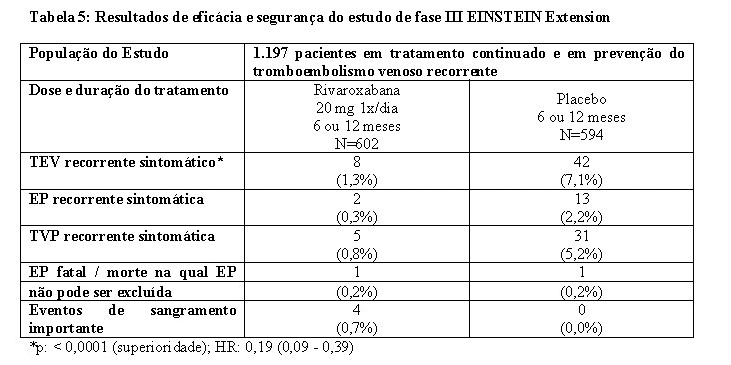
No estudo EINSTEIN Extension, 1.197 pacientes com TVP ou EP foram estudados para a prevenção de TVP e de EP recorrentes. A duração do tratamento foi de até 12 meses, dependendo do julgamento clínico do investigador. Rivaroxabana 20 mg uma vez ao dia foi comparado com placebo.
Os estudos EINSTEIN TVP, EP e Extension usaram os mesmos desfechos primário e secundário de eficácia predefinidos. O desfecho primário de eficácia foi TEV recorrente sintomático, definido como o composto de TVP recorrente ou EP fatal ou não fatal. O desfecho secundário de eficácia foi definido como o composto de TVP recorrente, EP não fatal e mortalidade por todas as causas.
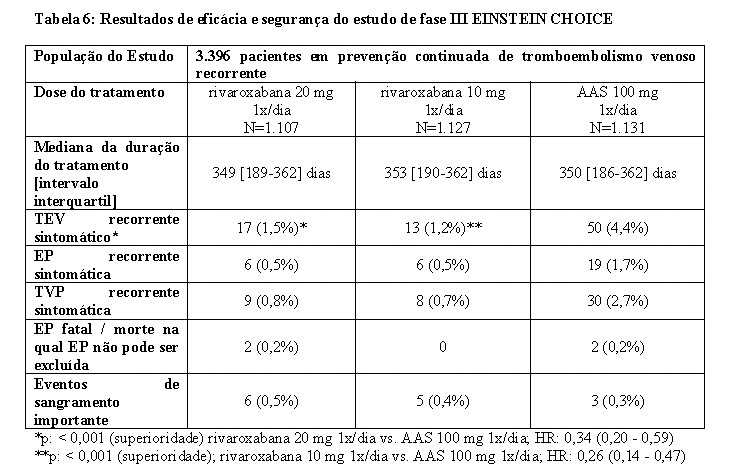
No estudo EINSTEIN CHOICE, 3.396 pacientes com TVP e/ou EP sintomática confirmada que completaram 6-12 meses de tratamento anticoagulante foram estudados para a prevenção de EP fatal ou TVP ou EP recorrente sintomática não fatal. Os pacientes com indicação de anticoagulação com dose terapêutica continuada foram excluídos do estudo. A duração do tratamento foi de até 12 meses dependendo da data de randomização individual (mediana: 351 dias). Rivaroxabana 20 mg uma vez ao dia e rivaroxabana 10 mg uma vez ao dia foram comparados com 100 mg de ácido acetilsalicílico uma vez ao dia.
O desfecho primário de eficácia foi a TEV recorrente sintomático, definido como o composto de TVP recorrente ou EP fatal ou não fatal. O desfecho secundário de eficácia foi o composto do desfecho primário de eficácia, infarto do miocárdio, acidente vascular cerebral isquêmico ou embolia sistêmica não sistema nervoso central.
No estudo EINSTEIN TVP (veja Tabela 2), rivaroxabana demonstrou ser não inferior à enoxaparina/AVK para o desfecho primário.
O benefício clínico líquido (NCB - Net Clinical Benefit) pré-especificado (desfecho primário de eficácia e eventos de sangramento importante) foi reportado com HR de 0,67 ((IC 95% = 0,47 - 0,95), valor nominal p = 0,027) a favor da rivaroxabana.
As taxas de incidência para o desfecho principal de segurança (eventos de sangramento importante ou não importante clinicamente relevante) assim como o desfecho secundário de segurança (eventos de sangramento importante), foram semelhantes para ambos os grupos de tratamento.
No estudo EINSTEIN EP (veja Tabela 3) rivaroxabana demonstrou ser não inferior à enoxaparina/AVK para o desfecho primário (p= 0,0026 (teste para não-inferioridade); hazard ratio: 1,12 (0,75 - 1,68)).
O benefício clínico global pré-especificado (desfecho primário de eficácia e eventos de sangramento importante) foi reportado com um HR de 0,85 ((IC 95% = 0,63 - 1,14), valor nominal p= 0,275)).
Foi conduzida uma análise agrupada pré-especificada do resultado dos estudos EINSTEIN TVP e EINSTEIN EP (veja Tabela 4).
No estudo EINSTEIN Extension (veja Tabela 5), rivaroxabana foi superior ao placebo para os desfechos primário e secundário de eficácia. Para o desfecho principal de segurança (eventos de sangramento importante) houve uma taxa de incidência mais alta, numericamente não significativa, para pacientes tratados com rivaroxabana 20 mg uma vez ao dia comparado com placebo. O desfecho de segurança secundário (eventos de sangramento importante ou não importante clinicamente relevante) demonstrou taxas mais altas para pacientes tratados com rivaroxabana 20 mg uma vez ao dia comparado com placebo.
No estudo EINSTEIN CHOICE, rivaroxabana 20 mg e 10 mg foram ambos superiores a 100 mg de ácido acetilsalicílico para o desfecho primário de eficácia. O desfecho secundário de eficácia foi significativamente reduzido quando comparado com rivaroxabana 20 mg ou 10 mg vs. 100 mg ácido acetilsalicílico. O desfecho principal de segurança (eventos de sangramento importantes) foi semelhante nos pacientes tratados com rivaroxabana 20 mg e 10 mg 1x/dia, em comparação com 100 mg de ácido acetilsalicílico. O desfecho secundário de segurança (sangramento não importante associado à interupção do tratamento por mais de 14 dias) foi semelhante quando comparado rivaroxabana 20 mg ou 10 mg vs. 100 mg de ácido acetilsalicílico.
Os resultados foram consistentes entre os pacientes com TEV provocado e não provocado (ver Tabela 6). Em uma análise pré-especificada do benefício clínico líquido (NCB - Net Clinical Benefit) (desfecho primário de eficácia mais eventos de sangramento importantes) do EINSTEIN CHOICE, foram relatados uma HR de 0,44 (IC 95% 0,27 - 0,71; p = 0,0009) para rivaroxabana 20 mg 1x/dia vs. 100 mg de ácido acetilsalicílico 1x/dia e uma HR de 0,32 (IC 95% 0,18 - 0,55; p < 0,0001) para rivaroxabana 10 mg 1x/dia vs. 100 mg de ácido acetilsalicílico 1x/dia.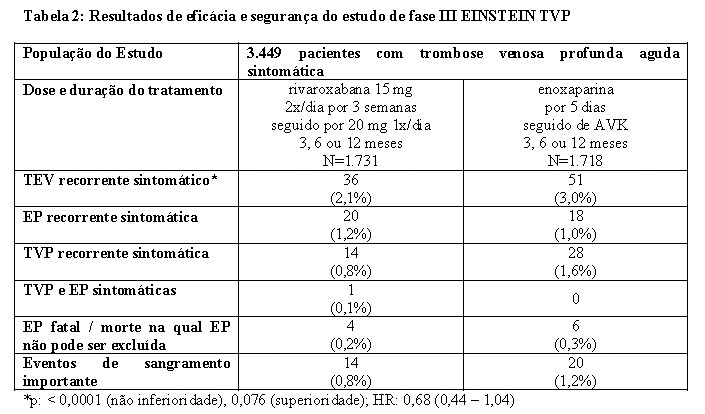
Além do programa EINSTEIN de fase III, foi realizado um estudo de coorte aberto, prospectivo, não intervencionista (XALIA) com adjudicação central de desfecho, incluindo TEV recorrente, sangramento importante e morte. Foram incluídos 5.142 pacientes com TVP aguda para investigar a segurança a longo prazo de rivaroxabana em comparação com a terapia de anticoagulação padrão em condições de mundo real. As taxas de sangramento importante, TEV recorrente e mortalidade por todas as causas de rivaroxabana foram de 0,7%, 1,4% e 0,5%, respectivamente. Foram ajustados os "hazard ratios" comparando rivaroxabana e padrão de cuidados para levar em conta as diferenças nas características basais do paciente. Os "hazard ratios" ajustados para sangramento importante, TEV recorrente e mortalidade por todas as causas foram 0,77 (IC 95% 0,40-1,50), 0,91 (IC 95% 0,54-1,54) e 0,51 (IC 95% 0,24-1,07), respectivamente.
A rivaroxabana mostrou segurança e eficácia semelhantes em comparação com a anticoagulação padrão. Estes resultados em pacientes que foram observados na prática clínica de rotina são consistentes com aqueles observados no estudo EINSTEIN TVP.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
- Mecanismo de ação
A rivaroxabana é um inibidor direto altamente seletivo do fator Xa com biodisponibilidade oral.
A ativação do fator X a fator Xa (FXa) por meio das vias intrínseca e extrínseca desempenha um papel central na cascata da coagulação sanguínea. O FXa converte diretamente a protrombina em trombina por meio do complexo de protrombinase e, finalmente, esta reação leva à formação do coágulo de fibrina e à ativação das plaquetas pela trombina. Uma molécula de FXa é capaz de gerar mais de 1.000 moléculas de trombina devido à natureza amplificadora da cascata da coagulação. Além disso, a taxa de reação do FXa ligado à protrombinase aumenta 300.000 vezes, em comparação à do FXa livre, e causa uma descarga explosiva de geração de trombina. Os inibidores seletivos de FXa podem encerrar a descarga amplificada de geração de trombina. Consequentemente, diversos testes de coagulação específicos e globais são afetados pela rivaroxabana.
- Efeitos farmacodinâmicos
Foi observada inibição dose-dependente da atividade do fator Xa em humanos.
O tempo de protrombina (TP) é influenciado pela rivaroxabana de um modo dose dependente com uma correlação estreita com as concentrações plasmáticas (o valor de r é igual a 0,98) se for usado o reagente Neoplastin® (tromboplastina liofilizada obtida a partir de cérebro de coelho) para a realização deste ensaio.
Outros reagentes proporcionariam resultados diferentes. A leitura do TP deve ser feita em segundos porque a RNI (Relação Normatizada Internacional) é calibrada e validada somente para cumarínicos e não pode ser usada para qualquer outro anticoagulante. Em pacientes submetidos a cirurgia ortopédica de grande porte, os percentis 5/95 para TP (Neoplastin®) 2-4 horas depois da ingestão do comprimido (por exemplo, no momento de efeito máximo) variaram de 13 a 25 segundos.
Em um estudo de farmacologia clínica na reversão farmacodinâmica de rivaroxabana em voluntários adultos sadios (n=22), os efeitos de doses únicas (50 UI/Kg) de dois tipos diferentes de CCPs (concentrados de complexo protrombínico), um CCP 3-fatores (fatores II, IX e X) e um CPCP 4-fatores (fatores II, VII, IX e X) foram avaliados. O CCPP 3-fatores reduziu o valor do TP (Neoplastin®) em aproximadamente 1,0 segundo em 30 minutos, comparado a reduções de aproximadamente 3,5 segundos observadas com o CCP 4-fatores. Em contrapartida, o CPP 3-fatores teve um efeito global maior e mais rápido em reverter alterações na geração de trombina endógena que o CCP 4-fatores (ver "Superdose").
O tempo de tromboplastina parcial ativada (TTPa) e o HepTest® também se prolongam dependendo da dose; entretanto, não são recomendados para avaliar o efeito farmacodinâmico da rivaroxabana. A atividade anti-fator Xa também é influenciada pela rivaroxabana; todavia, não existe padrão para calibração.
Não há necessidade de monitorar os parâmetros de coagulação durante o tratamento clínico de rotina com VYNAXA®.
- Populações especiais de pacientes
- Pacientes com próteses valvulares cardíacas submetidos recentemente a TAVR
No estudo randomizado, aberto, controlado por ativo, orientado por evento, multicêntrico de Fase III GALILEO, 1644 pacientes foram randomizados, tanto para uma estratégia baseada em rivaroxabana quanto para uma estratégia baseada em antiagregante plaquetário, de 1 a 7 dias após uma sucedida substituição da válvula aórtica transcateter. Pacientes com fibrilação atrial prévia ou com indicação para anticoagulantes orais em curso foram excluídos.
O principal objetivo foi avaliar a eficácia e a segurança da estratégia de tratamento baseada em rivaroxabana (10 mg de rivaroxabana, uma vez ao dia, mais 75-100 mg de ácido acetilsalicílico (AAS), uma vez ao dia, por 90 dias seguido por rivaroxabana 10 mg uma vez ao dia) comparado ao tratamento padrão (75 mg de clopidogrel uma vez ao dia mais 75-100 mg de ácido acetilsalicílico, uma vez ao dia, por 90 dias seguido por ácido acetilsalicílico, uma vez ao dia). O estudo foi encerrado precocemente devido a um desequilíbrio em eventos tromboembólicos e óbito.
Na análise de intenção de tratar (ITT), o desfecho primário de eficácia, por exemplo, eventos tromboembólicos e óbito, ocorreu em 105 pacientes (9,8 por 100 pacientes-ano) no braço rivaroxabana e em 78 pacientes (7,21 por 100 pacientes-ano) no braço de antiagregante plaquetário. A razão de risco (HR) foi de 1,35 (IC 95%: 1,01; 1,81). Na análise durante o tratamento, o desfecho primário de eficácia ocorreu em 68 pacientes (8,11 por 100 pacientes-ano) no braço rivaroxabana comparado com 63 pacientes (6,6 por 100 pacientes-ano) no braço de antiagregante plaquetário; a razão de risco (HR) foi 1,21 (IC 95%: 0,86; 1,70).
Na análise de intenção de tratar (ITT), o desfecho primário de segurança, por exemplo, composto de risco de vida, incapacitante ou sangramento maior, ocorreu em 46 pacientes (4,29 por 100 pacientes-ano) no braço de rivaroxabana em comparação com 31 pacientes (2,83 por 100 pacientes-ano) no braço de antiagregante plaquetário; a razão de risco (HR) foi 1,50 (IC 95% 0,95; 2,37).
- Pacientes com síndrome antifosfolípide triplo positivo de alto risco
Em um estudo patrocinado pelo investigador multicêntrico randomizado, aberto, com o desfecho de adjudicação cego, a rivaroxabana foi comparada à varfarina em pacientes com histórico de trombose, com diagnóstico de síndrome antifosfolípide e com alto risco de eventos tromboembólicos (positivo para todos os três testes antifosfolípides: anticoagulante lúpico, anticorpos anticardiolipina e anticorpos anti-beta 2-glicoproteína I). O estudo foi encerrado prematuramente após a inclusão de 120 pacientes devido a um excesso de eventos dentre os pacientes no braço da rivaroxabana. O seguimento médio foi de 569 dias. Cinquenta e nove pacientes foram randomizados para 20 mg de rivaroxabana (15 mg para pacientes com clearance de creatinina < 50 mL / min) e 61 para varfarina (INR 2,0-3,0). Eventos tromboembólicos ocorreram em 12% dos pacientes randomizados para rivaroxabana (4 AVCs isquêmico e 3 infartos do miocárdio). Nenhum evento foi relatado em pacientes randomizados para varfarina. Sangramento importante ocorreu em 4 pacientes (7%) do grupo rivaroxabana e 2 pacientes (3%) do grupo varfarina.
Propriedades farmacocinéticas
- Absorção e biodisponibilidade
A rivaroxabana é rapidamente absorvida, atingindo concentrações máximas (Cmáx) 2 a 4 horas após a ingestão do comprimido.
A absorção oral da rivaroxabana é quase completa e a biodisponibilidade oral é alta (80- 100%) para dose de 2,5 mg e 10 mg, independente das condições de jejum/alimentação.
A ingestão com alimentos não afeta a AUC ou a Cmáx da rivaroxabana na dose de 10 mg. O comprimido de 10 mg de VYNAXA® pode ser tomado com ou sem alimento (ver "Posologia e Modo de Usar").
A variabilidade da farmacocinética da rivaroxabana é moderada, com variabilidade interindividual (CV%) de 30% a 40%.
A absorção da rivaroxabana é dependente do local de liberação do princípio ativo no trato gastrintestinal. Foi relatada uma diminuição de 29% e 56% na AUC e Cmáx quando o granulado de rivaroxabana é liberado no intestino delgado proximal em comparação com o comprimido. A exposição é ainda mais reduzida quando o princípio ativo é liberado no intestino delgado distal, ou no cólon ascendente. Deve-se evitar a administração da rivaroxabana distante ao estômago que pode resultar em redução da absorção e da exposição ao princípio ativo.
A biodisponibilidade (AUC e Cmáx) foi comparável entre 20 mg de rivaroxabana administrados por via oral como comprimido triturado misturado a purê de maçã, ou suspensão em água e administrada por sonda gástrica seguido por uma refeição líquida, comparada a um comprimido inteiro. Como o perfil farmacocinético da rivaroxabana é previsível e dose-proporcional, os resultados de biodisponibilidade desse estudo são provavelmente aplicáveis para as doses mais baixas de rivaroxabana.
- Distribuição
A ligação às proteínas plasmáticas em humanos é alta, aproximadamente de 92% a 95%, sendo a albumina sérica o principal componente de ligação. O volume de distribuição é moderado, sendo Vss de aproximadamente 50 L.
- Metabolismo e eliminação
Aproximadamente 2/3 da dose administrada de rivaroxabana, sofrem degradação metabólica, com metade sendo eliminada via renal e a outra metade, via fecal. Os demais 1/3 da dose administrada são diretamente excretados pelos rins como fármaco inalterado na urina, principalmente por secreção renal ativa.
A rivaroxabana é metabolizada por meio de CYP3A4, CYP2J2 e de mecanismos independentes do CYP. A degradação oxidativa da fração morfolinona e a hidrólise das ligações amida são os principais locais de biotransformação.
Com base em investigações in vitro, a rivaroxabana é um substrato das proteínas transportadoras gp-P (glicoproteína-P) e Bcrp (proteína de resistência ao câncer de mama).
A rivaroxabana inalterada é o composto mais importante no plasma humano, não estando presentes metabólitos maiores ou ativos circulantes. Com uma depuração sistêmica de cerca de 10 L/h, a rivaroxabana pode ser classificada como um fármaco de baixa depuração. A eliminação da rivaroxabana do plasma ocorreu com meias-vidas terminais de 5 a 9 horas em indivíduos jovens e com meias-vidas terminais de 11 a 13 horas em idosos.
- Pacientes geriátricos
Pacientes idosos apresentaram concentrações plasmáticas mais altas que pacientes mais jovens, com valores médios de AUC aproximadamente 1,5 vezes maiores, devido principalmente à redução (aparente) da depuração total e renal (ver "Posologia e Modo de Usar").
- Gênero
Não há diferenças clinicamente relevantes da farmacocinética entre pacientes homens e mulheres (ver "Posologia e Modo de Usar").
- Peso corporal
Pesos corpóreos extremos ( < 50 kg vs > 120 kg) tiveram apenas pequena influência nas concentrações plasmáticas de rivaroxabana (menos de 25%) (ver "Posologia e Modo de Usar").
Dados agrupados obtidos dos estudos clínicos RECORD 1, RECORD 2 e RECORD 3 demonstraram que existe uma tendência a aumento do risco de sangramento em pacientes com peso corpóreo acima de 110 kg.
- Crianças e adolescentes
A segurança e a eficácia não foram estabelecidas para crianças e adolescentes com idade inferior a 18 anos (ver "Posologia e Modo de Usar").
- Diferenças étnicas
Não foram observadas diferenças clinicamente relevantes entre pacientes caucasianos, afro americanos, hispânicos, japoneses ou chineses em relação à farmacocinética e farmacodinâmica (ver "Posologia e Modo de Usar").
- Insuficiência hepática
O efeito da insuficiência hepática na farmacocinética da rivaroxabana foi estudado em indivíduos categorizados de acordo com a classificação Child Pugh, um procedimento padrão no desenvolvimento clínico. O propósito original da classificação Child Pugh é avaliar o prognóstico da doença hepática crônica, principalmente cirrose. Em pacientes nos quais o uso de anticoagulantes é pretendido, o aspecto crítico da insuficiência hepática é a redução da síntese de fatores de coagulação normais no fígado. Uma vez que este aspecto é considerado em apenas uma das cinco medições clínicas/bioquímicas que compõem o sistema de classificação Child Pugh, o risco de sangramento em pacientes pode não ser claramente correlacionado com esta classificação. A decisão de tratar os pacientes com anticoagulantes deve ser, portanto, tomada independentemente da classificação Child Pugh.
Rivaroxabana é contraindicada em pacientes com doença hepática associada à coagulopatia, levando a um risco de sangramento clinicamente relevante.
Pacientes cirróticos com insuficiência hepática leve (classificados como Child Pugh A) apresentaram apenas pequenas alterações na farmacocinética da rivaroxabana (aumento de 1,2 vezes da AUC, em média), próximas das de seu respectivo grupo controle saudável.
Nenhuma diferença relevante nas propriedades farmacodinâmicas foi observada entre estes grupos.
Em pacientes cirróticos com insuficiência hepática moderada (classificados como Child Pugh B), a média da AUC da rivaroxabana foi significativamente aumentada em 2,3 vezes comparada com voluntários sadios, devido à importante insuficiência na depuração do fármaco, o que indica uma significante doença hepática. A AUC da fração não-ligada foi aumentada em 2,6 vezes. Não há dados em pacientes com insuficiência hepática grave.
A inibição da atividade do fator Xa foi aumentada por um fator de 2,6 quando comparada a voluntários sadios; o prolongamento do TP foi similarmente aumentado por um fator de 2,1. O teste global de coagulação TP avalia a via extrínseca que compreende os fatores de coagulação VII, X, V, II e I que são sintetizados no fígado. Pacientes com insuficiência hepática moderada foram mais sensíveis à rivaroxabana, resultando em uma relação mais acentuada de Farmacocinética / Farmacodinâmica entre concentração e TP.
Não há dados disponíveis para pacientes Child Pugh C (ver "Posologia e Modo de Usar" e "Contraindicações").
- Insuficiência renal
Houve um aumento na exposição à rivaroxabana inversamente correlacionada com a diminuição da função renal, como avaliado pela medida da depuração de creatinina.
Em indivíduos com insuficiência renal leve (ClCr ≤80-50 mL/min), moderada (ClCr < 50- 30 mL/min) ou grave (ClCr < 30-15 mL/min), as concentrações plasmáticas de rivaroxabana (AUC) foram 1,4; 1,5 e 1,6 vezes maiores, respectivamente, comparadas com voluntários sadios (ver "Posologia e Modo de Usar" e "Advertências e Precauções").
Aumentos correspondentes nos efeitos farmacodinâmicos foram mais pronunciados (ver "Posologia e Modo de Usar" e "Advertências e Precauções").
Em indivíduos com insuficiência renal leve, moderada ou grave, a inibição total da atividade do fator Xa foi aumentada por um fator de 1,5; 1,9 e 2,0, respectivamente, quando comparada com voluntários sadios; o prolongamento do TP foi similarmente aumentado por um fator de 1,3; 2,2 e 2,4, respectivamente. Não há dados em pacientes com ClCr < 15 mL/min.
O uso não é recomendado em pacientes com depuração de creatinina < 15 mL/min.A rivaroxabana deve ser utilizado com cautela em pacientes com insuficiência renal grave (depuração de creatinina 15-30 mL/min) (ver "Posologia e Modo de Usar" e "Advertências e Precauções").
Devido à doença de base, pacientes com insuficiência renal grave apresentam risco aumentado de sangramento e trombose.
- Administração Concomitante de Potentes Indutores da CYP3A4
Em um estudo de fase I, a coadministração de rivaroxabana com a rifampicina, um potente indutor da isoenzima CYP3A4 e gp-P (glicoproteína-P), levou a uma redução de aproximadamente 50% na AUC média da rivaroxabana, com reduções paralelas em seus efeitos farmacodinâmicos (ver "Interações Medicamentosas").
Em um estudo fase IIa, a relação PK/PD de um regime de dose adaptado de rivaroxabana adaptada (30 mg duas vezes ao dia nas primeiras três semanas de tratamento, seguidos por 20 mg duas vezes ao dia) foi estudada em 19 pacientes tratados para TVP ou EP e que, concomitantemente foram medicados com um potente indutor da isoenzima CYP3A4 e gp-P (rifampicina ou fenitoína). O regime de dose adaptado nesses pacientes levou a uma exposição e farmacodinâmica similares, quando comparado a pacientes tratados para TVP (15 mg duas vezes ao dia nas primeiras três semanas de tratamento, seguido por 20 mg uma vez ao dia) sem a administração concomitante de um potente indutor da isoenzima CYP3A4.
- Dados de segurança pré-clínicos
A avaliação de segurança pré-clínica em dados de estudos convencionais e apropriados de segurança farmacológica, toxicidade de dose única e de doses repetidas, genotoxicidade, fototoxicidade, carcinogenicidade e toxicidade para a reprodução não revelaram riscos especiais para humanos. Não foi observada toxicidade órgão-específica da rivaroxabana até a mais alta dose testada.
- Segurança farmacológica
As funções cardiovascular, respiratória e do SNC não foram afetadas. Não se observou potencial pró-arritmogênico.
Não foram observados efeitos clinicamente relevantes na motilidade gastrintestinal, função hepática, função renal e níveis de glicose sanguínea.
- Toxicidade aguda e de doses repetidas
A rivaroxabana mostrou baixa toxicidade aguda em ratos e camundongos.
A rivaroxabana foi testada em estudos de doses repetidas por até 6 meses em ratos e por até 12 meses em cães. Com base no modo de ação farmacológico, não se pôde estabelecer NOEL (Nível de efeito não observado) em razão dos efeitos sobre o tempo de coagulação.
Todos os achados adversos, exceto uma discreta redução do ganho de peso corporal em ratos e cães, puderam ser relacionados a um efeito farmacológico exagerado do composto.
Em cães com exposições muito altas, foram observados sangramentos importantes espontâneos. Os NOAELs (Níveis de efeitos adversos não observados) após exposição crônica são 12,5 mg/kg em ratos e 5 mg/kg em cães.
- Carcinogenicidade
A rivaroxabana foi testada até 60 mg/kg/dia, atingindo níveis de exposição semelhantes aos seres humanos (camundongo) ou até 3,6 vezes maiores (ratos) do que nos seres humanos.
A rivaroxabana não apresentou potencial carcinogênico em ratos e camundongos.
- Toxicologia para a reprodução
A rivaroxabana foi testada em estudos de toxicidade para o desenvolvimento em níveis de exposição de até 14 vezes (rato) e de até 33 vezes (coelho) acima da exposição terapêutica em humanos. O perfil toxicológico se caracteriza principalmente por toxicidade materna causada por efeitos farmacodinâmicos exagerados. Até a dose mais alta testada, não se identificou potencial teratogênico primário (ver "Gravidez e lactação").
A radioatividade relacionada à [C14] rivaroxabana penetrou a barreira placentária em ratos. Em nenhum dos órgãos e tecidos fetais, a exposição, em termos de concentrações máximas ou AUC, excedeu a exposição sanguínea materna. A exposição média nos fetos, baseada na AUC (0-24), alcançou cerca de 20% da exposição no sangue materno. As glândulas mamárias tinham uma AUC aproximadamente equivalente à do sangue, o que indica secreção de radioatividade no leite (ver "Gravidez e lactação").
A rivaroxabana não mostrou efeito sobre a fertilidade masculina ou feminina até 200 mg/kg (ver "Gravidez e lactação").
- Lactação
Administrou-se [C14] rivaroxabana por via oral a ratas Wistar lactantes (dias 8 a 10 do pós-parto) em dose oral única de 3 mg/kg de peso corporal.
A radioatividade relacionada à [C14] rivaroxabana foi secretada no leite das ratas lactantes apenas em uma pequena extensão em relação à dose administrada: a quantidade estimada de radioatividade excretada com o leite foi de 2,12% da dose materna no prazo de 32 horas após a administração (ver "Gravidez e lactação").
- Genotoxicidade
Não se observou genotoxicidade num teste para mutação genética em bactérias (Teste de Ames), um teste in vitro para aberrações cromossômicas ou no teste in vivo do micronúcleo.
4. CONTRAINDICAÇÕES
VYNAXA® é contraindicado em pacientes com hipersensibilidade à rivaroxabana ou a qualquer outro componente do produto (ver "Composição"); em pacientes com sangramento ativo clinicamente significativo (por exemplo, sangramento intracraniano, sangramento gastrintestinal); e ainda em pacientes com doença hepática associada à coagulopatia, levando a um risco de sangramento clinicamente relevante (ver "Propriedades Farmacocinéticas").
Não foram estabelecidas segurança e eficácia de rivaroxabana em mulheres grávidas. Dados em animais mostram que a rivaroxabana atravessa a barreira placentária. Portanto, o uso de VYNAXA® é contraindicado durante toda a gravidez (ver "Gravidez e lactação" e "Dados de segurança pré-clínicos").
Não foram estabelecidas segurança e eficácia de rivaroxabana em mulheres lactantes. Dados em animais indicam que a rivaroxabana é secretada no leite materno. Portanto, VYNAXA® só pode ser administrado depois que for descontinuada a amamentação (ver "Gravidez e lactação" e "Dados de segurança pré-clínicos").
5. ADVERTÊNCIAS E PRECAUÇÕES
-Risco de sangramento
VYNAXA® como outros antitrombóticos, deve ser utilizado com cautela em pacientes com risco aumentado de sangramento, tais como:
- distúrbios hemorrágicos adquiridos ou congênitos;
- hipertensão arterial grave não controlada;
- doença gastrintestinal ulcerativa ativa;
- ulcerações gastrintestinais recentes;
- retinopatia vascular;
- hemorragia intracraniana ou intracerebral recente;
- anormalidades vasculares intraespinais ou intracerebrais;
- cirurgia cerebral, espinhal ou oftalmológica recente;
- bronquiectasia ou história de sangramento pulmonar.
O sangramento durante o tratamento antitrombótico pode desmascarar malignidades subjacentes ainda desconhecidas, em particular no trato gastrointestinal ou geniturinário. Pacientes com doença maligna podem, simultaneamente, apresentar maior risco de sangramento e trombose. O benefício individual do tratamento antitrombótico deve ser avaliado em relação ao risco de sangramento em pacientes com câncer ativo, dependendo da localização do tumor, terapia antineoplásica e estágio da doença.
Deve-se ter cuidado se os pacientes forem tratados concomitantemente com fármacos que interferem na hemostasia, como os anti-inflamatórios não-esteroidais (AINEs), ácido acetilsalicílico, os inibidores da agregação plaquetária (ou seja, agentes antiplaquetários), outros antitrombóticos ou inibidores seletivos da recaptação de serotonina (ISRSs) e inibidores da receptação de serotonina e noradrenalina (IRSNs) (ver "Interações Medicamentosas").
Pode-se considerar tratamento profilático adequado para pacientes com risco de doença ulcerativa gastrintestinal (ver "Interações Medicamentosas").
Qualquer queda de hemoglobina ou da pressão arterial sem explicação deve levar à investigação de um local com sangramento.
-Anestesia neuraxial (epidural/espinhal)
Quando anestesia neuraxial (epidural/espinhal) ou uma punção espinhal é realizada, os pacientes tratados com antitrombóticos para prevenção de complicações tromboembólicas correm o risco de desenvolver hematoma epidural ou espinhal que pode resultar em paralisia prolongada.
O risco destes eventos é ainda maior pelo uso de cateteres epidurais de demora ou pelo uso concomitante de medicamentos que afetem a hemostasia. O risco também pode aumentar por punção epidural ou espinhal traumática ou repetida.
Pacientes devem ser frequentemente monitorados para sinais e sintomas de alteração neurológica (por exemplo, torpor ou fraqueza das pernas, disfunção intestinal ou da bexiga). Se forem observados déficits neurológicos, serão necessários diagnóstico e tratamento urgentes.
O médico deve considerar o benefício em potencial em relação ao risco antes da intervenção neuraxial em pacientes anticoagulados ou que vão ser anticoagulados para tromboprofilaxia.
Para reduzir o risco potencial de sangramento associado ao uso concomitante de rivaroxabana e anestesia neuraxial (epidural/espinhal) ou punção espinhal, considerar o perfil farmacocinético de rivaroxabana. A inserção ou remoção de um cateter epidural ou punção lombar é melhor realizada quando o efeito anticoagulante de rivaroxabana é estimado ser baixo (ver "Propriedades Farmacocinéticas").
Um cateter epidural não deve ser removido antes de 18 horas após a última administração de VYNAXA®.
VYNAXA® deve ser administrado, pelo menos, 6 horas após a remoção do cateter.
Se ocorrer punção traumática, a administração de VYNAXA® deverá ser adiada por 24 horas.
- Cirurgia e intervenções
Se um procedimento invasivo ou uma intervenção cirúrgica forem necessários, VYNAXA® 10 mg deve ser interrompido pelo menos 24 horas antes da intervenção, se possível, e com base no julgamento clínico do médico.
Se o procedimento não puder ser adiado, o aumento do risco de sangramento deve ser avaliado em relação à urgência de tal intervenção.
A administração de VYNAXA® deve ser reiniciada o mais rapidamente possível após o procedimento invasivo ou a intervenção cirúrgica, desde que a situação clínica do paciente permita e a hemostasia adequada tenha sido estabelecida (ver "Propriedades Farmacocinéticas").
- Pacientes com próteses valvulares cardíacas
VYNAXA® não é recomendado para tromboprofilaxia em pacientes que foram recentemente submetidos a substituição da válvula aórtica transcateter (TAVR), baseado nos dados de um estudo clínico randomizado, controlado comparando um regime de rivaroxabana a um regime de antiagregante plaquetário (ver "Propriedades Farmacodinâmicas").
A segurança e a eficácia de rivaroxabana não foram estudadas em pacientes com outras próteses de válvulas cardíacas ou outros procedimentos valvulares; portanto, não há dados para suportar que rivaroxabana forneça anticoagulação adequada nestas populações de pacientes.
- Pacientes com síndrome antifosfolípide triplo positivo de alto risco
VYNAXA® não é recomendada em pacientes com antecedentes de trombose diagnosticados com síndrome antifosfolípide e com persistência tripla positiva (para anticoagulante lúpico, anticorpos anticardiolipina e anticorpos anti-beta 2-glicoproteína I), uma vez que o tratamento com rivaroxabana está associado a um aumento da taxa de eventos trombóticos recorrentes comparados com antagonistas da vitamina K (AVK) (ver "Propriedades Farmacodinâmicas").
-Insuficiência renal
VYNAXA® deve ser utilizado com cautela em pacientes com insuficiência renal moderada (ClCr < 50-30 mL/min) que estejam recebendo comedicações que levam ao aumento da concentração de rivaroxabana no plasma (ver "Interações Medicamentosas").
Em pacientes com insuficiência renal grave (ClCr < 30 mL/min), os níveis plasmáticos de rivaroxabana podem elevar-se significativamente (1,6 vezes em média), o que pode levar a um aumento do risco de sangramento. Em razão da doença de base, estes pacientes têm um aumento do risco de sangramento e de trombose. Em virtude dos dados clínicos limitados, VYNAXA® deve ser usado com cautela nos pacientes com ClCr < 30-15 mL/min (ver "Propriedades Farmacocinéticas").
Não há dados clínicos disponíveis para pacientes com insuficiência renal grave (ClCr < 15 mL/min). Portanto o uso de VYNAXA® não é recomendado nestes pacientes (ver "Posologia e Modo de Usar" e "Propriedades Farmacocinéticas").
Após início do tratamento, os pacientes com insuficiência renal grave ou risco aumentado de sangramentos e aqueles que recebem tratamento sistêmico concomitante com antimicóticos azólicos ou inibidores das proteases do HIV devem ser cuidadosamente monitorados quanto a sinais de complicações hemorrágicas (ver "Interações Medicamentosas"). Isto pode ser feito por exame físico regular dos pacientes, observação atenta da drenagem da incisão cirúrgica e dosagens periódicas da hemoglobina.
- Medicação concomitante
VYNAXA® não é recomendado em pacientes recebe