VOTRIENT
NOVARTIS
pazopanibe
Inibidor da proteína quinase.
Apresentações.
Comprimidos revestidos de 200 mg para uso oral, em cartuchos com 30 comprimidos.
Comprimidos revestidos de 400 mg para uso oral, em cartuchos com 30 e 60 comprimidos.
VIA ORAL
USO ADULTO
Composição.
Votrient® 200 mg: Cada comprimido revestido contém 200 mg de pazopanibe equivalente a 216,7 mg de cloridrato de pazopanibe.
Excipientes: estearato de magnésio, celulose microcristalina, povidona (K30), amidoglicolato de sódio, Opadry® Rosa YS-1-14762-A* [hipromelose, óxido de ferro vermelho (E172), macrogol/PEG 400, polissorbato 80, dióxido de titânio (E171)], água purificada.
Votrient® 400 mg: Cada comprimido revestido contém 400 mg de pazopanibe equivalente a 433,4 mg de cloridrato de pazopanibe.
Excipientes: estearato de magnésio, celulose microcristalina, povidona (K30), amidoglicolato de sódio, Opadry® Branco YS-1-7706-G* [hipromelose, macrogol/PEG 400, polissorbato 80, dióxido de titânio (E171)], água purificada.
* Composição do revestimento.
Informações técnicas.
1. INDICAÇÕES
Carcinoma de Células Renais (RCC)
Votrient® é indicado para o tratamento de carcinoma de células renais (RCC) avançado e/ou metastático.
Sarcoma de partes moles (STS)
Votrient® é indicado para o tratamento de pacientes adultos com subtipos específicos de sarcoma de partes moles (STS) avançado que receberam quimioterapia prévia para doença metastática ou que tenham progredido dentro de 12 meses após a terapia neoadjuvante ou adjuvante. A eficácia e a segurança foram estabelecidas apenas para certos subtipos histológicos de STS (ver Resultados de Eficácia).
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
Carcinoma de Células Renais (RCC)
A segurança e a eficácia de Votrient® no carcinoma de células renais (RCC) foram avaliadas em um estudo multicêntrico, randômico, duplocego e controlado com placebo. Pacientes (N=435) com RCC localmente avançado e/ou metastático foram randomizados para receber Votrient® 800 mg ou placebo uma vez ao dia. O endpoint primário do estudo foi avaliar e comparar os dois grupos de tratamento em relação à sobrevida livre de progressão da doença (PFS). O endpoint secundário foi a sobrevida global (OS). Os demais endpoints foram a avaliação da taxa de resposta global e da duração da resposta.
Do total de 435 pacientes desse estudo, 233 eram virgens de tratamento e 202 eram pacientes (de segunda linha) que receberam uma terapia prévia baseada em IL-2 ou INF-a. Observou-se semelhança no performance status (ECOG) entre o grupo de Votrient® e o de placebo (ECOG 0: 42% vs 41%; ECOG 1: 58% vs 59%). Todos os pacientes apresentavam histologia compatível com células clara ou predominantemente clara. Aproximadamente a metade dos pacientes tinha três ou mais órgãos envolvidos em sua doença e a maioria dos pacientes tinha o pulmão (74%) e/ou nódulos linfáticos (54%) como localização metastática da doença na randomização do estudo.
Uma proporção similar de pacientes em cada grupo era virgem de tratamento ou tinha sido pré-tratada a base de citoquinas (53% e 47% no grupo de Votrient® e 54% e 46% no de placebo). No subgrupo pré-tratado com citoquinas, a maioria (75%) tinha sido submetida a tratamento à base de interferon.
Proporções semelhantes de pacientes em cada grupo haviam sido submetidas à nefrectomia (89% no grupo de Votrient® e 88% no de placebo) e/ou a radioterapia prévia (22% e 15%, respectivamente).
A análise principal do endpoint primário (PFS) se baseia na avaliação da doença por análise radiológica independente, na população total do estudo (de primeira linha e de segunda linha). 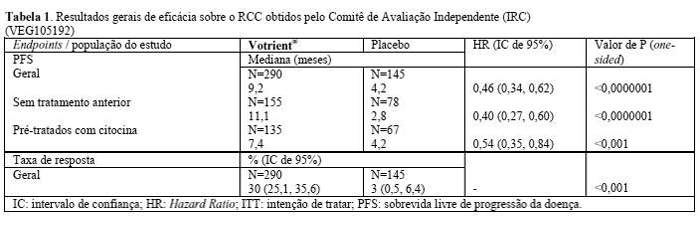
No caso dos pacientes que responderam ao tratamento, a duração média de resposta foi de 58,7 semanas, de acordo com a avaliação do comitê independente.
Os dados da mediana de sobrevida global (OS) na análise de sobrevida final especificada no protocolo foram de 22,9 meses e 20,5 meses [HR=0,91 (IC de 95%: 0,71-1,16; p=0,224)] para os pacientes randomizados nos grupos Votrient® e placebo, respectivamente. Os resultados da OS são sujeitos a possíveis vieses porque 54% dos pacientes do grupo placebo também receberam Votrient® na parte de extensão desse estudo depois da progressão da doença. Sessenta e seis por cento (66%) dos pacientes de placebo receberam terapia posterior ao estudo em comparação com 30% dos pacientes de Votrient® .
No estudo pivotal, as avaliações de qualidade de vida (QoL) foram baseadas nas pontuações globais obtidas de maneira cega e por autorrelato por meio de dois questionários especificados segundo protocolo, EORTC QLQ-C30 e EuroQoL EQ-5D. A análise foi baseada em pacientes de ambos os grupos que continuaram em terapia antes da progressão. As avaliações mostraram que não há diferença entre o tratamento com Votrient® e o tratamento com placebo (p > 0.05), indicando que Votrient® não exerce impacto negativo sobre a qualidade de vida global.
Em um estudo de Fase II com 225 pacientes com carcinoma renal de células claras localmente recorrente ou metastático, a taxa de resposta objetiva foi de 35% e a duração média da resposta foi de 68 semanas, de acordo com a avaliação do comitê independente.
Em um estudo de fase III aberto, randomizado, paralelo e de não inferioridade (VEG108844), foram avaliadas segurança, eficácia e qualidade de vida de Votrient® comparado ao sunitinibe. Pacientes (N=1110) com RCC localmente avançado e/ou metastático que não tinham recebido terapia sistêmica prévia foram randomizados para receber Votrient® 800 mg uma vez ao dia, continuamente, ou sunitinibe 50 mg uma vez ao dia em ciclos de 6 semanas sendo 4 semanas de tratamento e 2 sem tratamento.
O endpoint primário deste estudo foi avaliar e comparar a PFS em pacientes tratados com Votrient® àqueles tratados com sunitinibe. As características demográficas foram similares entre os grupos de tratamento. As características da doença no diagnóstico inicial e no recrutamento foram balanceadas entre os grupos de tratamento, com a maioria dos pacientes apresentando histologia celular clara e doença no estágio IV.
O estudo VEG108844 alcançou o endpoint primário de PFS e demonstrou que Votrient® não foi inferior ao sunitinibe, uma vez que o limite superior do IC 95 % para Hazard Ratio (HR) foi menor que a margem de não inferioridade de 1,25 especificada no protocolo. Os resultados gerais de eficácia estão descritos na Tabela 2.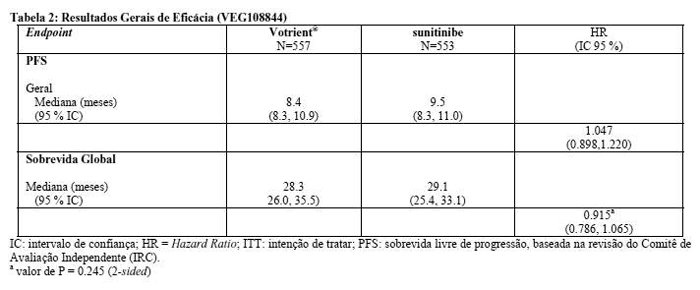
Sarcoma de partes moles (STS):
A segurança e eficácia de pazopanibe em STS foram avaliadas em um estudo pivotal de fase III randomizado, duplo-cego, controlado com placebo, multicêntrico (VEG110727). Um total de 369 pacientes com STS avançado foram randomizados para receber pazopanibe 800 mg uma vez ao dia ou placebo. É importante destacar que foi permitida a participação no estudo apenas de pacientes com subtipos histológicos selecionados de STS, portanto a eficácia e a segurança do pazopanibe podem ser consideradas estabelecidas apenas para estes subgrupos de STS e o tratamento com pazopanibe deve ser restrito a tais subtipos de STS.
Os seguintes tipos de tumor foram elegíveis:
Fibroblástico (fibrossarcoma adulto, mixofibrossarcoma, fribrossarcoma epitelioide esclerosante, tumores fibrosos solitários malignos), os chamados fibrohistiocítica (histiocitoma fibroso maligno pleomórfico [MFH], células gigantes MFH, MFH inflamatório), leiomiossarcoma, tumores gliômicos malignos, músculos esqueléticos (pleomórfico e rabdomiossarcoma alveolar), vascular (hemangioendotelioma epitelioide, angiosarcoma), diferenciação incerta (sinovial, epitelioide, parte mole alveolar, de células claras, desmoplásicas de células pequenas e rodadas, rabdóide extrarrenal, mesenquimoma maligno, PEComa, sarcoma intimal), tumores da bainha dos nervos periféricos malignos, sarcomas de tecidos moles não especificadas (NOS) e outros tipos de sarcoma (não listados como não elegíveis).
Os seguintes tipos de tumor não foram elegíveis:
Sarcoma adipocítico (todos os subtipos), todos os rabdomiossarcoma que não eram alveolares ou pleomórficos, condrossarcoma, osteossarcoma, tumores de Ewing / tumores neuroectodérmicos primitivos (PNET), GIST, sarcoma protuberante, sarcoma miofibroblástico inflamatório, mesotelioma maligno e tumores mesodérmicos mistos do útero.
Os pacientes com sarcoma adipocítico foram excluídos do estudo pivotal de fase III uma vez que em um estudo preliminar de fase II (VEG20002), a atividade (PFS na semana 12) observada com pazopanibe no adipocítico não satisfazia a taxa de pré-requisito para permitir testes clínicos adicionais.
Outros critérios de elegibilidade chave do estudo VEG110727 foram: evidência histológica de STS maligna de grau alto ou intermediário e progressão da doença dentro de 6 meses após a terapia para doença metastática, ou recorrência dentro de 12 meses de terapia (neo) / adjuvante.
Noventa e oito por cento (98%) de pacientes receberam doxorrubicina anteriormente, 70% ifosfamida prévia, e 65% dos pacientes receberam pelo menos três ou mais agentes quimioterápicos antes do recrutamento do estudo.
Pacientes foram estratificados pelos fatores de status de desempenho da OMS (OMS OS) (0 ou 1) na linha basal e o número de linhas antes da terapia sistêmica para doença avançada (0 ou 1 vs. 2+). Em cada grupo de tratamento, houve um pequeno aumento da porcentagem de pacientes nas linhas 2+ da terapia sistêmica prévia para doença sistêmica (58% e 55% respectivamente para os braços de tratamento de placebo e pazopanibe). A duração média do acompanhamento de pacientes (definido como data de recrutamento a data do último contato ou morte) foi similar para ambos os braços de tratamento (9,36 meses para placebo [faixa 0,69 a 23,0 meses] e 10,4 meses para pazopanibe [faixa 0,2 a 24,3 meses]).
O objetivo primário do estudo foi sobrevida livre de progressão (PFS avaliada por revisão radiológica independente); os endpoints secundários incluíram sobrevida global (OS), taxa de resposta global e duração da resposta.
Observou-se uma melhora semelhante na PFS com base na avaliação do investigador no braço pazopanibe em comparação com o braço placebo (na população total ITT HR: 0,39, IC de 95%, 0,30 a 0,52, p < 0,001).
Figura 1: Curva Kaplan-Meier para Sobrevida livre de progressão em STS por avaliação independente para a população geral (VEG110727)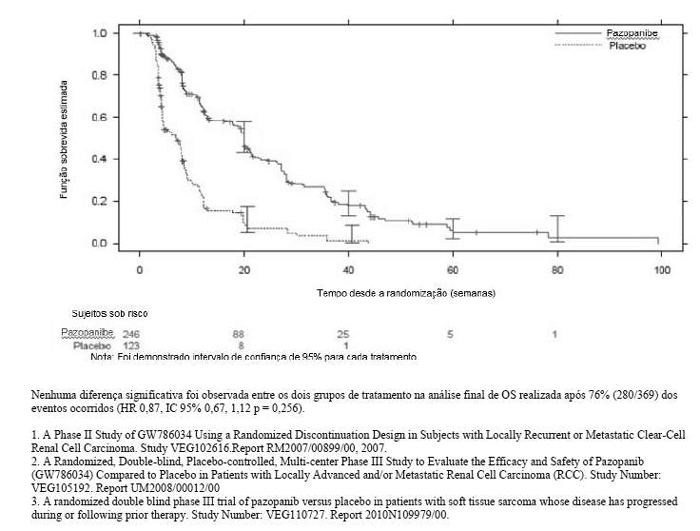
Nenhuma diferença significativa foi observada entre os dois grupos de tratamento na análise final de OS realizada após 76% (280/369) dos eventos ocorridos (HR 0,87, IC 95% 0,67, 1,12 p = 0,256).
1. A Phase II Study of GW786034 Using a Randomized Discontinuation Design in Subjects with Locally Recurrent or Metastatic Clear-Cell Renal Cell Carcinoma. Study VEG102616.Report RM2007/00899/00, 2007.
2. A Randomized, Double-blind, Placebo-controlled, Multi-center Phase III Study to Evaluate the Efficacy and Safety of Pazopanib (GW786034) Compared to Placebo in Patients with Locally Advanced and/or Metastatic Renal Cell Carcinoma (RCC). Study Number: VEG105192. Report UM2008/00012/00
3. A randomized double blind phase III trial of pazopanib versus placebo in patients with soft tissue sarcoma whose disease has progressed during or following prior therapy. Study Number: VEG110727. Report 2010N109979/00.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação:
Votrient®, administrado por via oral, é um potente inibidor multialvo da tirosinoquinase (TKI) de receptores dos fatores de crescimento endotelial vascular 1, 2 e 3 (VEGFR-1, VEGFR-2, VEGFR-3), dos fatores de crescimento derivados de plaquetas a e b (PDGFR-a e PDGFR-b) e do receptor do fator de célula-tronco (c-KIT), cujos valores IC50 são, respectivamente, de 10, 30, 47, 71, 84 e 74 nM.
Em experimentos pré-clínicos, o pazopanibe inibiu de modo dependente de dose a autofosforilação induzida por ligante dos receptores VEGFR-2, c-KIT e PDGFR-b em células. In vivo, o pazopanibe inibiu a fosforilação de VEGFR-2 induzido por VEGF em pulmões de camundongos, a angiogênese em vários modelos animais e o crescimento de múltiplos xenoenxertos de tumor humano em camundongos.
Farmacocinética
Em uma meta-análise farmacogenética de dados de 31 estudos clínicos de Votrient® administrado em monoterapia ou em combinação com outros agentes, valores de ALT 5 X LSN (NCI CTC Grau 3) ocorreram em 19% dos portadores do alelo HLA-B*57:01 e em 10% dos nãoportadores. Nestes dados, 133/2235 (6%) dos pacientes eram portadores do alelo HLA-B*57:01 (ver Advertências e Precauções).
Propriedades Farmacocinéticas
Absorção
O pazopanibe é absorvido oralmente e alcança a sua concentração máxima no prazo mediano de duas a quatro horas após a administração. A dosagem diária resulta em aumento de AUC de 1,23 vez a 4 vezes. Não houve aumento consistente de AUC e de Cmáx quando se usou uma dose de Votrient® acima de 800 mg.
A exposição sistêmica ao pazopanibe é aumentada quando administrado com alimentos. A administração de Votrient® com uma refeição rica ou pobre em gorduras resulta em aumento praticamente duplicado de AUC e Cmáx. Por isso, Votrient® deve ser administrado no mínimo uma hora antes da refeição ou duas horas depois (ver Posologia e Modo de Usar).
A administração de um único comprimido de 400 mg de Votrient® esmagado aumentou a AUC(0-72) em 46% e a Cmáx em aproximadamente duas vezes e diminuiu o tmáx em aproximadamente 90 minutos, em comparação com a administração do comprimido inteiro. Esses resultados indicam que a biodisponibilidade e a taxa de absorção oral de pazopanibe aumentaram depois da administração do comprimido esmagado em comparação com o comprimido inteiro. Portanto, devido a esse potencial de aumento da exposição, os comprimidos não devem esmagados (ver Posologia e Modo de Usar).
Distribuição
A ligação do pazopanibe à proteína de plasma humano in vivo foi superior a 99% sem dependência de concentração acima da faixa de 10-100 mg/ml. Estudos in vitro sugerem que o pazopanibe é um substrato da glicoproteína P (Pgp) e da proteína resistente ao câncer de mama (BCRP).
Metabolismo
Resultados de estudos in vitro demonstraram que o metabolismo do pazopanibe é mediado basicamente pelo CYP3A4, com contribuições menores do CYP1A2 e do CYP2C8.
Eliminação
O pazopanibe é eliminado de forma lenta e possui meia-vida média de 30,9 horas após a administração da dose recomendada de 800 mg. A eliminação ocorre principalmente pelas fezes, e a eliminação renal representa menos que 4% da dose administrada.
Populações especiais
· Insuficiência Renal:
Em uma análise farmacocinética da população, com 408 pacientes diagnosticados com variados tipo de câncer, o clearance de creatinina (30-150 mL/min) não influenciou o clearance de pazopanibe. Não é esperado que a insuficiência renal influencie na farmacocinética do pazopanibe, não havendo necessidade de alterações na dose em pacientes com clearance de creatina ≥ 30 mL/min.
· Insuficiência Hepática:
A mediana no estado de equilíbrio da Cmáx e AUC(0-24) de pazopanibe, em pacientes com insuficiência hepática leve (definida tanto como bilirrubina normal e qualquer grau de elevação de ALT - alaninaminotransferase, quanto como elevação de bilirrubina até 1,5 x LSN - Limite Superior Normal, independente do valor de ALT), após uma dose diária de 800 mg/dia (30,9 mg/mL, no intervalo de 12,5-47,3 e 841,8 mg.h/mL, no intervalo de 600,4-1078), são similares a mediana dos pacientes sem insuficiência hepática (49,4 mg/mL, no intervalo de 17,1-85,7 e 888,2 mg.h/mL, no intervalo de 345.5-1482) (ver Posologia e Modo de Usar).
A dose máxima tolerada (DMT) de Votrient® por pacientes com insuficiência hepática moderada (definida como uma elevação de bilirrubina > 1,5 x a 3 x LSN, independente dos valores de ALT) foi de 200 mg, uma vez ao dia. Os valores da mediana no estado de equilíbrio da Cmáx (22,4 mg/mL, no intervalo de 6,4-32,9) e AUC(0-24) (350,0 mg.h/mL, no intervalo de 131,8-487,7), após a administração de 200 mg de Votrient®, uma vez ao dia, em indivíduos com insuficiência hepática moderada, foram de aproximadamente 45% e 39%, respectivamente, o que corresponde aos valores da mediana após administração de 800 mg, uma vez ao dia, em indivíduos com função hepática normal (ver Posologia e Modo de Usar).
Não existem dados suficientes sobre o uso de Votrient® em pacientes com insuficiência hepática grave (total de bilirrubina > 3 x LSN, independente de qualquer nível de ALT), portanto o uso de Votrient® não é recomendado para esses pacientes.
4. CONTRAINDICAÇÕES
Votrient® é contraindicado a pacientes com hipersensibilidade a qualquer um dos componentes da formulação.
Não existem contraindicações relativas à faixa etária. Entretanto, Votrient® não foi suficientemente estudado em pacientes com menos de 18 anos de idade.
Categoria D de risco na gravidez:
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião-dentista. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Efeitos hepáticos
Foram relatados casos de insuficiência hepática (inclusive com mortes) durante o uso de Votrient® .
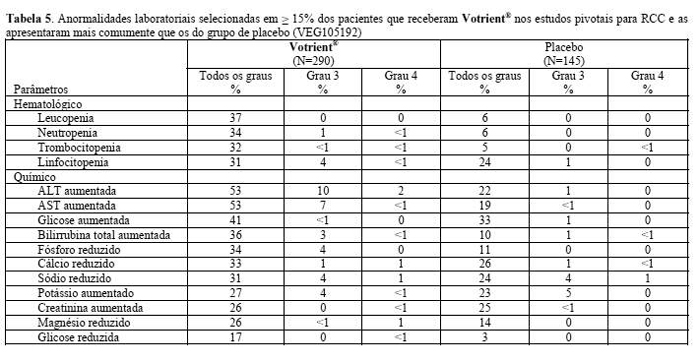
Em ensaios clínicos com Votrient® observaram-se aumentos das transaminases séricas (ALT, AST) e da bilirrubina (ver Reações Adversas). Na maioria dos casos foram relatados aumentos isolados dos valores de ALT (alaninaminotransferase) e AST (aspartatotransaminase), sem elevações concomitantes da fosfatase alcalina ou da bilirrubina. Pacientes com mais de 60 anos de idade podem estar em um risco maior para ALT > 3x LSN. Pacientes portadores do alelo HLA-B*57:01 também têm um risco aumentado de elevações na ALT associadas ao Votrient®. A função hepática deve ser monitorada em todos os pacientes recebendo Votrient®, independente do genótipo ou idade (ver Características Farmacológicas). A grande maioria (mais de 90%) de todas as elevações das transaminases, de qualquer grau, ocorreu nas primeiras 18 semanas. As classificações são baseadas nos Critérios de Terminologia Comum do Instituto Nacional de Câncer.
Recomenda-se a monitorização da função hepática antes de iniciar o tratamento com Votrient® e nas semanas três, cinco, sete e nove do tratamento. Posteriormente, fazer a monitorização no terceiro e quarto mês, bem como quando clinicamente indicado. Depois do quarto mês, realizar monitorização periódica.
Ver Tabela 4 para orientação de modificação de dose para pacientes com valores basais de bilirrubina total ≤ 1,5 x LSN e AST e ALT ≤ 2 x LSN:
O uso concomitante de pazopanibe e sinvastatina aumenta o risco de elevações de ALT (ver item Interações Medicamentosas) e deve ser realizado com precaução e monitoramento rigoroso.
O tratamento com Votrient® não é recomendado em pacientes com insuficiência hepática grave.
Hipertensão
Em estudos clínicos com Votrient® ocorreram eventos de hipertensão, incluindo crises hipertensivas. Aferir cuidadosamente a pressão arterial antes de iniciar o tratamento com Votrient®. Os pacientes devem ser monitorizados quanto à hipertensão logo após o início do tratamento com Votrient® (não mais que uma semana após o início) e com frequência, depois disso, para garantir o controle da pressão arterial. E caso apresentem hipertensão devem ser tratados por meio de terapia anti-hipertensiva padrão e redução ou interrupção de dose de Votrient®, como justificado clinicamente (ver Posologia e Modo de Usar e Reações Adversas). A hipertensão (pressão sanguínea sistólica > 150 ou diastólica > 100 mmHg) surge logo no início do tratamento (aproximadamente 40% dos casos ocorrem no nono dia e em aproximadamente 90% dos casos, nas primeiras 18 semanas).. O tratamento com Votrient® deve ser interrompido caso o paciente apresente crise hipertensiva, hipertensão grave ou persistente mesmo com a terapia anti-hipertensiva e a redução da dose de Votrient®.
Síndrome de encefalopatia posterior reversível (PRES)/ Síndrome de leucoencefalopatia posterior reversível (RPLS)
Casos de PRES e RPLS foram reportados e associados ao uso de Votrient®. Estas síndromes podem se apresentar com cefaleia, hipertensão, convulsão, letargia, confusão, cegueira e outros distúrbios visuais e neurológicos e podem ser fatais. O tratamento com Votrient® deve ser permanentemente descontinuado em pacientes que desenvolvam PRES/RPLS.
Doença pulmonar intersticial (ILD)/pneumonite: ILD, que pode ser fatal, foi reportada e associada a Votrient® (ver item Reações Adversas). Os pacientes devem ser monitorados quanto a sintomas pulmonares indicativos de ILD/pneumonite e Votrient® deve ser descontinuado em pacientes que desenvolvam essas doenças.
Disfunção cardíaca/Insuficiência cardíaca
Os riscos e benefícios do pazopanibe devem ser considerados antes de iniciar a terapia em pacientes com disfunção cardíaca pré-existente. A segurança e a farmacocinética do pazopanibe não foram estudadas em pacientes com insuficiência cardíaca moderada a grave ou aqueles com fração de ejeção ventricular esquerda (LVEF) abaixo do normal.
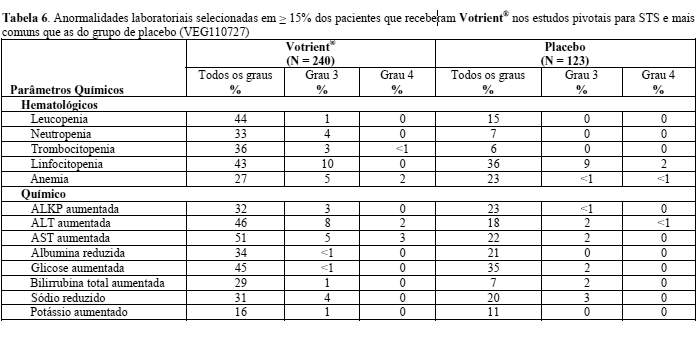
Nos estudos clínicos com Votrient®, ocorreram eventos de disfunção cardíaca, como insuficiência cardíaca congestiva e fração de ejeção ventricular esquerda (LVEF) diminuída. Em um ensaio clínico randomizado que comparou pazopanibe e sunitinibe em RCC (VEG108844), os indivíduos tinham medições de LVEF basais e de acompanhamento. A disfunção miocárdica ocorreu em 13% (47/362) dos sujeitos no braço pazopanibe em comparação com 11% (42/369) dos sujeitos no braço de sunitinibe. Insuficiência cardíaca congestiva foi observada em 0,5% dos indivíduos em cada braço de tratamento. A insuficiência cardíaca congestiva foi relatada em três de 240 indivíduos (1,0%) nos estudos de fase III de STS. As reduções da LVEF em indivíduos submetidos a medições pós-basais e LVEF de acompanhamento foram detectadas nesse estudo em 11% (15/140) no braço tratado com Votrient®, em comparação com 3% (1/39) no braço placebo.
Fatores de risco: Treze dos quinze indivíduos no braço de pazopanibe do estudo de STS de fase III tiveram hipertensão recorrente o que pode ter exacerbado a disfunção cardíaca em pacientes de risco pelo aumento da pós-carga cardíaca. 99% dos pacientes (243/216) recrutados no estudo de STS de fase III, incluindo os 15 indivíduos, receberam antraciclina. Terapia prévia com antraciclina pode ser um fato de risco para disfunção cardíaca.
Resultado: Quatro dos 15 indivíduos tiveram recuperação total (dentro de 5% na linha de base) e 5 tiveram recuperação parcial (dentro da faixa normal, mas > 5% abaixo da linha de base). Um indivíduo não recuperou e dados de acompanhamento não estão disponíveis para os outros 5 indivíduos.
Gerenciamento: Interrupção do pazopanibe e/ou redução de dose deve ser combinada com tratamento da hipertensão (se presente, ver Hipertensão, acima) em pacientes com reduções significativas em LVEF, conforme clinicamente indicado.
Os pacientes devem ser meticulosamente monitorados quanto a sinais ou sintomas clínicos de insuficiência cardíaca congestiva. A avaliação basal e periódica da LVEF é recomendada nos pacientes com risco de disfunção cardíaca.
Prolongamento do intervalo QT e Torsades de Pointes
Em estudos clínicos com Votrient® ocorreram eventos de aumento do QT e Torsades de Pointes (ver Reações Adversas). Votrient® deve ser usado com cautela em pacientes com histórico de prolongamento do QT, em pacientes sob tratamento com antiarrítmicos ou outros medicamentos com potencial de aumentar o QT e em pacientes com doença cardíaca preexistente relevante. Durante a administração de Votrient® recomenda-se realizar a monitorização periódica de eletrocardiogramas e manter os eletrólitos (cálcio, magnésio, potássio) dentro dos limites normais.
Eventos trombóticos arteriais
Em estudos clínicos com Votrient® observou-se a ocorrência de infarto do miocárdio, angina, acidente vascular cerebral isquêmico e ataque isquêmico transitório (ver Reações Adversas). Houve eventos fatais. O medicamento deve ser usado com cautela em pacientes com alto risco de eventos tromboembólicos ou com histórico desses eventos nos últimos 6 meses. A decisão sobre o tratamento deve ser tomada com base na avaliação dos benefícios/riscos individuais para o paciente.
Eventos tromboembólicos venosos:
Nos estudos clínicos com Votrient®, ocorreram eventos tromboembólicos venosos incluindo trombose venosa e embolia pulmonar fatal. A incidência foi superior na população com STS (5%) do que na população com RCC (2%).
Microangiopatia trombótica
Microangiopatia trombótica (MAT) foi relatada em estudos clínicos com Votrient® em monoterapia, em combinação com bevacizumabe e em combinação com topotecana (ver Reações Adversas). O tratamento com Votrient® deve ser permanentemente descontinuado em pacientes que desenvolvam MAT. A reversão dos efeitos da MAT foi observada após a descontinuação do tratamento. Votrient® não é indicado para uso em combinação com outros agentes.
Eventos hemorrágicos
Em estudos clínicos com Votrient® foram relatados eventos hemorrágicos (ver Reações Adversas). Houve eventos hemorrágicos fatais. Votrient® não foi estudado em pacientes com histórico de hemoptise ou de hemorragia cerebral ou gastrointestinal clinicamente significativa nos últimos seis meses.
Votrient® deve ser usado com cautela em pacientes com risco significativo de hemorragia.
Perfurações e fístulas gastrintestinais
Em estudos clínicos com Votrient® ocorreram eventos de perfuração ou fístula gastrintestinal (GI) (ver Reações Adversas). Houve eventos de perfuração fatais.
Votrient® deve ser usado com cautela em pacientes com risco de perfuração ou fístula gastrintestinal.
Cicatrização de feridas
Não foram conduzidos estudos formais sobre o efeito de Votrient® na cicatrização de feridas. Uma vez que os inibidores do fator de crescimento vascular endotelial (VEGF) podem dificultar a cicatrização de feridas, o uso de Votrient® deve ser interrompido pelo menos sete dias antes da data da cirurgia. A decisão de reiniciar o tratamento com Votrient® após a cirurgia deve se basear no julgamento clínico da capacidade de cicatrização. O uso de Votrient® deve ser interrompido em pacientes com deiscência de ferida cirúrgica.
Hipotireoidismo
Em estudos clínicos com Votrient®, ocorreram eventos de hipotireoidismo (ver Reações Adversas). Recomenda-se realizar o monitorização pró ativa da função tireoidiana.
Proteinúria
Existem relatos de proteinúria em estudos clínicos com Votrient® (ver Reações Adversas). Recomendam-se exames de urina no início do tratamento e periodicamente, com monitoramento dos pacientes quanto ao agravamento da proteinúria. Votrient® deve ser suspenso se o paciente desenvolver síndrome nefrótica.
Pneumotórax
Em estudos clínicos com Votrient® em sarcoma avançado de tecido mole, eventos de pneumotórax ocorreram em 3% (8/240) dos pacientes tratados com Votrient® e em nenhum paciente no braço placebo (ver Reações Adversas). Pacientes em tratamento com Votrient® devem ser observados de perto quanto a sinais e sintomas de pneumotórax.
Infecções
Foram relatados casos de infecções sérias (com ou sem neutropenia), em alguns casos com desfecho fatal.
Combinação com outras terapias sistêmicas antineoplásicas
Os estudos clínicos com Votrient® combinado com pemetrexede (câncer de pulmão de células não pequenas [NSCLC]) e lapatinibe (câncer de colo do útero) foram interrompidos antes devido a preocupações com o aumento da toxicidade e/ou mortalidade, e não ficou estabelecida uma dose segura e eficaz, com esses esquemas. Votrient® não é indicado para uso em combinação com outros agentes.
Toxicidade em animais juvenis
Como o mecanismo de ação de Votrient® pode afetar gravemente o crescimento e amadurecimento de órgãos durante o desenvolvimento pós-natal imediato, Votrient® não deve ser administrado a pacientes pediátricos com menos de 2 anos de idade.
Fertilidade
Votrient® pode prejudicar a fertilidade de homens e mulheres. Em estudos sobre a toxicidade reprodutiva feminina realizados com ratos, observou-se a diminuição da fertilidade feminina.
Mutagênese, Carcinogênese, Diminuição da fertilidade
Em estudos de carcinogenicidade de dois anos com pazopanibe, houve um aumento no número de adenomas hepáticos observados em camundongos e adenocarcinomas duodenais observados em ratos. Baseado na patogênese específica aos roedores e no mecanismo desses achados, não se considera que estes representem um aumento no risco carcinogênico para pacientes fazendo uso de Votrient®.
O pazopanibe não causou danos genéticos quando testado em ensaios de genotoxicidade (teste de Ames, ensaio de aberração cromossômica humana de linfócitos periféricos e ensaios in vivo de micronúcleo em ratos).
Em ratas, foi observada uma redução da fertilidade, incluindo um aumento da perda de pré e pós-implantação e reabsorções precoces, em doses ≥ 10 mg / kg / dia (cerca de 0,2 vezes a exposição clínica humana, com base na AUC). Foi observado também diminuição no corpo lúteo de macacas, quando receberam 500 mg /kg/ dia durante até 34 semanas, em camundongos que receberam ≥ 100 mg / kg / dia durante 13 semanas e atrofia ovariana foi observado em ratas que receberam 300 mg / kg / dia durante 26 semanas (aproximadamente igual a, 0,6, 1,4 e 0,9 vezes a exposição clínica humana, com base na AUC, respectivamente).
O pazopanibe não pareceu afetar o acasalamento ou a fertilidade em ratos machos. No entanto, foram observadas reduções nas taxas de produção de espermatozoides, motilidade espermática e concentrações de espermatozoides do epidídimo e testículo em doses ≥ 100 mg / kg / dia (aproximadamente 0,5 vezes a exposição clínica humana, com base na AUC), após 15 semanas de administração. Após 26 semanas de administração, foram observados diminuição do peso dos testículos e epidídimo, atrofia e degeneração dos testículos com aspermia, hipospermia e alteração cribiforme no epidídimo de ratos machos que receberam doses ≥ 30 mg / kg / dia (aproximadamente 0,4 vezes a exposição clínica humana, com base na AUC).
O pazopanibe produziu efeitos fetais teratogênicos (incluindo malformação cardiovascular e atraso na ossificação), redução do peso corporal fetal, e letalidade embrionária em ratos com uma dose de ≥ 3 mg / kg / dia (aproximadamente 0,1 vezes a exposição clínica humana com base na AUC). Em coelhos, toxicidade materna (a perda de peso, redução no consumo de alimentos e aborto) foi observada em doses ≥ 30 mg / kg / dia (cerca de 0,007 vezes a exposição clínica humana com base na AUC), enquanto o peso fetal foi reduzido em doses ≥ 3 mg / kg / dia. (ver Gravidez e Lactação).
Toxicologia animal e/ou Farmacologia:
Em estudos toxicológicos em ratos, houve efeitos em uma variedade de tecidos (ossos, dentes, medula óssea, leito ungueal, órgãos reprodutores, tecidos hematológicos, renais, glândulas adrenais, linfonodos, pituitária e pâncreas) consistentes com a inibição de VEGFR e / ou perturbação das vias de sinalização de VEGF com alguns efeitos que ocorrem com doses de 3 mg / kg / dia (cerca de 0,1 vezes a exposição clínica humana com base na AUC).
Efeitos hepáticos, incluindo discretas elevações das transaminases hepáticas de roedores e elevações de bilirrubina em macacos sem histopatologia associada foram observadas em doses que produziram exposições sistêmicas aproximadamente 0,1 e 0,6 vezes a exposição clínica humana, respectivamente.
Em estudos de toxicidade juvenil, quando ratos em pré-desmame receberam pazopanibe a partir do dia 9 até 14 dias pós-parto, este causou mortalidade e crescimento/maturação anormal de alguns órgãos como rins, pulmão, fígado e coração, em uma dose de aproximadamente 0,1 vezes a exposição clínica baseada na AUC em adultos. Quando ratos em pós desmame foram tratados do dia 21 ao dia 62 dias pós-parto, os resultados toxicológicos foram semelhantes e com exposições comparáveis aos ratos adultos, com alterações em ossos, traqueia, dentes, suprarrenais, pâncreas, estômago, duodeno, linfonodo, glândula mamária masculina e órgãos reprodutivos. Em ratos, o desmame ocorre no dia 21 pós-parto, que equivale a cerca de uma idade pediátrica humana de 2 anos. Pacientes pediátricos humanos apresentam um maior risco de efeitos em ossos e dentes, quando comparados a adultos, incluindo encurtamento em membros, que foi observado em ratos juvenis em doses ≥ 10 mg / kg / dia (igual à cerca de 0,1-0,2 vezes a exposição clínica com base na AUC em adultos).
Gravidez e lactação
Gravidez
Não há dados adequados sobre o uso de Votrient® em mulheres grávidas. Estudos realizados com animais demonstraram toxicidade reprodutiva. O risco potencial em seres humanos é desconhecido. Votrient® não deve ser usado durante a gravidez, a não ser que a condição clínica da mulher requeira o tratamento com Votrient®. Se Votrient® for usado durante a gravidez ou se a paciente ficar grávida no decorrer do tratamento com Votrient®, deve-se informar à paciente os riscos potenciais ao feto.
As mulheres com potencial de ter filhos devem ser avisadas da necessidade de usar a contracepção adequada durante o tratamento com Votrient® e por duas semanas após a descontinuação do tratamento e de evitar a gravidez durante o tratamento com Votrient®.
Os homens (incluindo aqueles que fizeram vasectomia) com parceira sexual que esteja grávida, possivelmente grávida ou com possibilidade de engravidar devem usar preservativos na relação sexual durante o período de tratamento com Votrient® e por pelo menos duas semanas após a última dose do produto.
Lactação
O uso seguro de Votrient® durante a lactação não foi estabelecido. Não se sabe se o pazopanibe é excretado pelo leite humano. A amamentação deve ser interrompida durante o tratamento com Votrient®.
Categoria D de risco na gravidez:
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião-dentista. Informe imediatamente seu médico em caso de suspeita de gravidez.
Interações
O tratamento simultâneo com inibidores fortes da CYP3A4, glicoproteína P ou proteínas resistentes ao câncer de mama (BCRP) deve ser evitado devido ao risco de exposição aumentada ao pazopanibe (ver Interações Medicamentosas). Recomenda-se a seleção de um medicamento alternativo para uso concomitante que tenha pouco ou nenhum potencial de inibição da CYP3A4, glicoproteína P ou BCRP.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não há estudos sobre os efeitos de Votrient® sobre o desempenho ao dirigir veículos ou sobre a capacidade de operar máquinas. A farmacologia do pazopanibe não leva a crer que haja um efeito prejudicial sobre essas atividades. O estado clínico do paciente e o perfil do evento adverso de Votrient® devem ser levados em consideração ao se avaliar a capacidade do paciente de desempenhar tarefas que requeiram discernimento e habilidades motoras e cognitivas.
6. INTERAÇÕES MEDICAMENTOSAS
Drogas inibidoras ou indutoras das enzimas do citocromo P450 3A4:
Estudos in vitro sugeriram que o metabolismo oxidativo do pazopanibe em microssomos do fígado humano é mediado basicamente pelo CYP3A4, com contribuições menores do CYP1A2 e do CYP2C8. Por isso, os inibidores e indutores do CYP3A4 podem modificar o metabolismo do pazopanibe.
Inibidores de CYP3A4, glicoproteína P e BCRP: pazopanibe é o substrato da CYP3A4, glicoproteína P ou BCRP.
A administração simultânea de Votrient® (400 mg uma vez ao dia) com um forte inibidor da CYP3A4 e da glicoproteína P, como cetoconazol (400 mg uma vez ao dia) por 5 dias consecutivos, resulta em um aumento médio de 66% da AUC(0-24) e 45% da Cmáx de pazopanibe, em relação à administração de Votrient® em monoterapia (400 mg uma vez ao dia durante 7 dias). A Cmáx e AUC de pazopanibe aumentam menos que o modelo dose-proporcional quando há um aumento da dose de 50 mg a 2.000 mg. Assim, uma redução de dose para 400 mg de Votrient® uma vez ao dia, na presença de um inibidor forte de CYP3A4 resulta, na maioria dos pacientes, numa exposição sistêmica similar àquela observada após a administração de 800 mg de Votrient® em monoterapia uma vez ao dia. Alguns pacientes, no entanto, podem apresentar uma exposição sistêmica de pazopanibe maior que a observada após a administração de 800 mg Votrient® em monoterapia.
A coadministração de Votrient® com outros inibidores fortes da família CYP3A4 (p. ex. itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, voriconazol) pode aumentar as concentrações de pazopanibe. O suco de toranja também pode aumentar as concentrações plasmáticas do pazopanibe.
A administração de 1.500 mg de lapatinibe, um substrato e inibidor fraco de CYP3A4, Pgp e BCRP, com 800 mg de Votrient® resultou em um aumento de 50% a 60% nos valores médios de AUC(0-24) e Cmáx, em comparação com a admini