VIRAMUNE
BOEHRINGER
nevirapina
Antiviral.
Apresentações.
Suspensão: embalagem com 1 frasco plástico contendo 240 ml de suspensão oral acompanhado de seringa dosadora e de 1 adaptador;
Comprimido: embalagem com 60 comprimidos.
USO ORAL.
USO ADULTO E PEDIÁTRICO.
Composição.
Cada ml de suspensão oral contém 10,35 mg de nevirapina hemiidratada correspondentes a 10,0 mg de nevirapina. Excipientes: carbômer, polissorbato 80, sorbitol, sacarose, metilparabeno, propilparabeno, hidróxido de sódio e água purificada. Cada comprimido contém 200 mg de nevirapina. Excipientes: celulose microcristalina, lactose monoidratada, povidona, amidoglicolato de sódio, dióxido de silício, estearato de magnésio e água purificada.
Indicações.
VIRAMUNE é indicado em associação com outros agentes anti-retrovirais no tratamento de pacientes infectados pelo HIV-1. A resistência do vírus aparece rápida e uniformemente quando se administra VIRAMUNE como monoterapia. Portanto, VIRAMUNE deve ser sempre administrado em associação com, no mínimo, dois agentes anti-retrovirais adicionais.
Na prevenção da transmissão materno-infantil do HIV-1 no caso de mulheres grávidas que não estiverem recebendo tratamento anti-retroviral durante o trabalho do parto, VIRAMUNE é indicado e pode ser administrado isoladamente à mãe em dose oral única durante o trabalho de parto e ao recém-nascido.
A monoterapia com VIRAMUNE tem sido associada ao desenvolvimento de resistência aos INNTRs.
Em mulheres e crianças previamente tratadas com dose única de nevirapina para a prevenção da transmissão materno-infantil do HIV-1, a eficácia de VIRAMUNE como parte da terapia combinada recebida para cuidado de sua própria saúde, pode ser reduzida (vide também Advertências).
Quando outros medicamentos anti-retrovirais estiverem acessíveis, o regime de VIRAMUNE em dose única deve ser combinado com medicamentos anti-retrovirais eficazes adicionais (como recomendado nas diretrizes internacionais reconhecidas).
Resultados de eficácia.
Documentou-se num estudo clínico uma resposta durável por no mínimo 1 ano para o tratamento com a associação tripla de nevirapina, zidovudina e didanosina, comparada com zidovudina + didanosina ou nevirapina + zidovudina, em 151 pacientes infectados por HIV-1 virgens de tratamento, com contagem de células CD4 de 200-600 células/mm3 (média 376 células/mm3) e uma concentração basal média de RNA pertencente ao HIV-1 de 4,41 log10 cópias/ml (25.704 cópias/ml).ref
Um outro estudo clínico comparou o tratamento constituído pela associação de VIRAMUNE + AZT + ddI com a associação de AZT + ddI em 398 pacientes infectados por HIV-1 com contagem de células CD4+ 350 células/mm3 (média de 153 células/mm3), com concentração plasmática inicial média de RNA viral de 4,59 log10 cópias/ml (38.905 cópias/ml), que haviam recebido no mínimo 6 meses de tratamento prévio com nucleosídeo (média 115 semanas). Na 48ª semana de tratamento observou-se um benefício significativo do tratamento constituído pela associação tripla contendo VIRAMUNE em relação à associação medicamentosa dupla, evidenciado na contagem de células CD4, em % CD4, em microcultura quantitativa de PBMC e em DNA viral plasmático. Observaram-se respostas favoráveis ao tratamento composto pela associação tripla contendo VIRAMUNE em todos os níveis de contagem de CD4.ref
Um outro estudo clínico controlado com placebo comparou o tratamento com VIRAMUNE + AZT + ddI com AZT + ddI. Esse mesmo estudo avaliou o tratamento com AZT + ddC e AZT alternado mensalmente com ddI. O estudo foi realizado em 1.298 pacientes infectados pelo HIV-1 (idade média 37 anos, 51% caucasianos, 87% do sexo masculino) com contagem de células CD4+ 50 células/mm3 (média de 25 células/mm3). 84% dos pacientes haviam recebido tratamento prévio com nucleosídeos (média de 15 meses).ref
As doses dos tratamentos foram de 200 mg de VIRAMUNE uma vez ao dia por 2 semanas, seguidos por 200 mg duas vezes ao dia, ou placebo; 200 mg de AZT três vezes ao dia; 0,75 mg de ddC três vezes ao dia; 200 mg de ddI duas vezes ao dia (ou 125 mg duas vezes ao dia para pacientes pesando menos de 60 kg).
O tempo médio de progressão do evento ou morte relacionados ao HIV foi significativamente maior no grupo que recebeu o tratamento de VIRAMUNE + AZT + ddI do que no grupo AZT + ddI (82 semanas contra 62 semanas). Durante o estudo, a taxa de mortalidade foi similar nos dois grupos. Os tempos médios de progressão da doença ou morte relacionados ao HIV foram menores para os grupos que receberam AZT + ddC (53 semanas) ou AZT e ddI alternadamente (57 semanas).
Avaliação da eficácia clínica na prevenção de transmissão materno-infantil
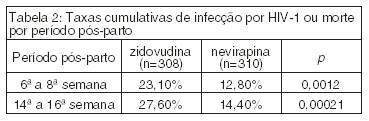
O estudo clínico HIVNET 012 comparou a prevenção da transmissão vertical da infecção do HIV-1 de 618 mulheres grávidas para seus recém-nascidos utilizando VIRAMUNE ou AZT.ref4
As mães receberam 200 mg de nevirapina via oral no início do trabalho de parto e os recém-nascidos receberam 2 mg/kg dentro de 72 horas após o nascimento ou administrou-se zidovudina via oral para a mãe, 600 mg no início do trabalho de parto e 300 mg a cada 3 horas até o parto e administraram-se 4 mg/kg via oral duas vezes ao dia aos recém-nascidos durante 7 dias após o nascimento. Nenhum outro medicamento anti-retroviral foi administrado.
As mães avaliadas nesse estudo possuíam as seguintes características: idade média de 24 anos, contagem média de 448 células CD4/mcl e média dos níveis plasmáticos de RNA de HIV-1 igual a 26.943 cópias/ml. 98,8% das crianças foram amamentadas e 95,6% ainda estavam sendo amamentadas nas 14ª a 16ª semanas.
O parâmetro primário de infecção por HIV baseou-se no teste PCR HIV RNA, confirmado por um segundo teste de RNA de HIV ou por cultura de HIV. A Tabela 1 apresenta as taxas de infecção confirmada por PCR e a Tabela 2 apresenta as taxas de infecção ou morte.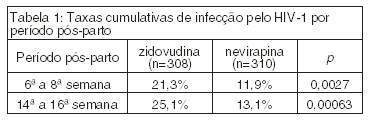
Caract. farmacológicas.
Modo de ação
O princípio ativo de VIRAMUNE é a nevirapina, um inibidor não-nucleosídeo da transcriptase reversa (INNTR) do vírus HIV-1. A nevirapina liga-se diretamente à transcriptase reversa e bloqueia as atividades da DNA-polimerase RNA e DNA-dependente, causando a ruptura do sítio catalítico da enzima. A atividade inibitória da nevirapina não é competitiva em relação ao molde ou aos nucleosídeos trifosfatos. A transcriptase reversa do vírus HIV-2 e as DNA-polimerases eucarióticas (como as DNA-polimerases humanas alfa, beta, gama, ou delta) não são inibidas pela nevirapina.
Em estudos clínicos, associou-se VIRAMUNE® com aumento do colesterol-HDL e melhora geral na proporção de colesterol-HDL, o que na população geral poderia ser associado a um menor risco cardiovascular. Entretanto, na ausência de estudos específicos com VIRAMUNE® a respeito da modificação do risco cardiovascular em pacientes com infecção por HIV, se desconhece o impacto clínico destes achados. A seleção de uma medicação anti-retroviral deve ser primariamente guiada pela sua eficácia antiviral.
Susceptibilidade do HIV in vitro
A atividade antiviral da nevirapina in vitro tem sido determinada em várias linhagens de células incluindo células mononucleares de sangue periférico, macrófagos derivados de monócitos e linhagem de linfoblastos. Em estudos recentes usando linfócitos sanguíneos de cordão umbilical humano e células 293 de rim de embrião humano, os valores de CI50 (concentrações inibitórias de 50%) variaram de 14 a 302 nM contra isolados clínicos e laboratoriais de HIV-1.
A nevirapina demonstrou atividade antiviral in vitro contra isolados do grupo M HIV-1 de subtipos A, B, C, D, F, G e H e formas recombinantes circulantes (CRF), CRF01_AE, CRF02_AG e CRF12_BF (valor mediano de CI50 de 63 nM). A nevirapina não teve atividade antiviral in vitro contra isolados do grupo O HIV-1 e HIV-2.
Em combinação com efavirenz a nevirapina demonstrou uma forte atividade antagonista anti-HIV-1 in vitro e foi aditiva à atividade antagonista com o inibidor de protease ritonavir ou o inibidor de fusão enfuvirtide. A nevirapina demonstrou aditiva atividade sinérgica anti-HIV-1 em combinação com os inibidores de protease amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, saquinavir e tipranavir; e com os inibidores nucleosídeos da transcriptase reversa (INTRs) abacavir, didanosina, emtricitabina, lamivudina, stavudina, tenofovir e zidovudina. A atividade anti-HIV da nevirapina foi antagonizada pela droga anti-HBV adefovir e pela droga anti-HCV ribavirina in vitro.
Resistência
Testes in vitro demonstraram o crescimento de cepas de HIV com susceptibilidade reduzida (100 a 250 vezes) à nevirapina. Análises genotípicas demonstraram mutações no gene HIV-1 RT Y181C e/ou V106A dependendo da cepa de vírus e da linhagem celular utilizadas. Em testes in vitro, o tempo de aparecimento de resistência à nevirapina não se alterou quando se administrou a nevirapina em associação com vários outros INNTRs.
Alterações genotípicas e fenotípicas em cepas isoladas de HIV-1 de pacientes tratados com VIRAMUNE (n=24) ou VIRAMUNE + ZDV (n=14) foram avaliadas em estudos de fase I/II por períodos de 1 a 12 semanas. Após uma semana de monoterapia de VIRAMUNE, as cepas isoladas de 3/3 dos pacientes apresentaram diminuição da susceptibilidade à nevirapina in vitro. Detectou-se em alguns pacientes uma ou mais mutações na transcriptase reversa nas posições dos aminoácidos 103, 106, 108, 181, 188 e 190 em menos de 2 semanas após o início do tratamento. Na 8ª semana de monoterapia com VIRAMUNE, 100% dos pacientes testados (n=24) apresentaram cepas com a susceptibilidade in vitro à nevirapina diminuída em mais de 100 vezes, em comparação com as cepas iniciais, e apresentaram uma ou mais mutações na transcriptase reversa associadas à resistência à nevirapina. 19 de 24 pacientes (80%) apresentaram isolados com mutação na posição 181 independentemente da dose.
A análise genotípica de isolados de pacientes virgens em terapia anti-retroviral com rebote virológico (n=71) recebendo nevirapina uma vez ao dia (n=25) ou duas vezes ao dia (n=46), em combinação com lamivudina e estavudina (estudo 2NN) por 48 semanas, demonstrou que os isolados de 8/25 e 23/46 pacientes, respectivamente, continham uma ou mais das seguintes mutações associadas à resistência INNTR: Y18C, K101E, G190A/S, K103N, V106A/M, V108I, Y188C/L, A98G, F227L e M230L.
Resistência em Prevenção de Transmissão Materno-Infantil de HIV
Detectou-se mutações relacionadas à resistência à nevirapina em 6-8 semanas administrando-se doses únicas em 21 das 111 (19%) mulheres testadas. K103N foi a mutação de resistência à nevirapina mais freqüentemente observada (57%) nessas mulheres, seguida pela combinação de K103N e Y181C (19%).
Não se detectou mutações de resistência à nevirapina em nenhuma das mulheres (n=11) nas quais se detectou mutações na 6ª a 8ª semanas, e que foram submetidas a novos testes no período de 12 a 24 meses após o parto.
Observou-se resistência à nevirapina em 11 dos 24 (46%) recém-nascidos infectados, sendo que a mutação Y181C foi a mais comumente detectada. Não se detectou mutações de resistência à nevirapina em nenhum dos recém-nascidos (n=7) em que se detectou mutações no período de 6 a 8 semanas, e que foram submetidos a novos testes com 12 meses de idade.
Em um estudo, mulheres que haviam recebido uma dose única de nevirapina para prevenção da transmissão materno-infantil, e que foram tratadas com VIRAMUNE combinado com outros fármacos anti-retrovirais para cuidado de sua própria saúde, 29 das 123, ou 24%, apresentaram falha virológica, e cinco (38%) de 13 mulheres com HIV 1 e resistência de base detectada ao VIRAMUNE apresentaram falha virológica. A partir de um estudo em que crianças de mães infectadas receberam tanto placebo quanto dose única de nevirapina, 30 crianças infectadas pelo HIV, das quais 15 receberam placebo e 15 receberam nevirapina, foram posteriormente tratadas com nevirapina combinada com outros medicamentos anti-retrovirais. Após 6 meses de tratamento de nevirapina combinada com outros medicamentos antiretrovirais, ocorreu falha virológica mais significativa em crianças que tinham recebido anteriormente uma dose única de nevirapina (10 de 15) do que em crianças que receberam anteriormente placebo (1 de 15). A combinação de outros anti-retrovirais com uma dose única de nevirapina atenua o aparecimento de resistência à nevirapina.
Resistência cruzada
Observou-se in vitro um rápido aparecimento de cepas de HIV com resistência cruzada aos INNTRs. Os dados sobre a resistência cruzada entre a nevirapina (INNTR) e os inibidores nucleosídeos análogos da transcriptase reversa são muito limitados. Em quatro pacientes, os isolados resistentes à ZDV testados in vitro mantiveram a susceptibilidade à nevirapina e em seis pacientes os isolados resistentes à nevirapina foram susceptíveis ao ZDV e ao ddI. A resistência cruzada entre a nevirapina e os inibidores da protease do vírus HIV é improvável porque as enzimas-alvo envolvidas são diferentes.
A resistência cruzada para delavirdina e efavirenz é esperada após falha virológica com nevirapina. Dependendo dos resultados dos testes de resistência, um regime contendo etravirina pode ser utilizado posteriormente.
A nevirapina não deve ser usada como um agente único para tratar o HIV ou adicionada como um agente único a um regime ineficaz. Como ocorre com todos os outros inibidores não-nucleosídeos de transcriptase reversa, vírus resistentes desenvolvem-se rapidamente quando a nevirapina é administrada como monoterapia. A escolha dos novos agentes anti-retrovirais a serem usados concomitantemente à nevirapina deve levar em consideração o potencial para resistência cruzada.
Na descontinuação de um regime anti-retroviral contendo nevirapina, deve-se considerar a longa meia-vida da nevirapina; se anti-retrovirais com meias-vidas menores que a da nevirapina forem descontinuados simultaneamente, as baixas concentrações plasmáticas da nevirapina podem persistir por uma semana ou mais e posteriormente pode ocorrer resistência do vírus.
Farmacocinética em pacientes adultos
A nevirapina é rapidamente absorvida ( > 90%) após administração oral em voluntários sadios e em adultos infectados com HIV-1. A biodisponibilidade absoluta em 12 adultos saudáveis após administração de uma dose única de um comprimido de 50 mg foi de 93 ± 9% (média ± desvio padrão) e 91 ± 8% após administração de uma solução oral. Os picos de concentração plasmática de nevirapina, de 2 ± 0,4 mcg/ml (7,5 mcM) foram atingidos 4 horas após a administração de uma dose única de 200 mg.
Após múltiplas doses, o pico de concentração de nevirapina parece aumentar de forma linear na faixa de dose situada entre 200 e 400 mg/dia. O estado de equilíbrio com concentrações de nevirapina de 4,5 ± 1,9 mcg/ml (17 ± 7 mcM), (n=242) foi atingido com 400 mg/dia.
A absorção de nevirapina não é afetada por alimentos, antiácidos ou medicamentos com tampão alcalino em suas formulações (ex.: didanosina).
A nevirapina é altamente lipofílica e é essencialmente não-ionizável em pH fisiológico. Após administração intravenosa em adultos saudáveis, o volume de distribuição aparente (Vdss) de nevirapina foi de 1,21 ± 0,09 l/kg, sugerindo que a nevirapina se distribui amplamente em humanos.
A nevirapina atravessa a placenta rapidamente e é encontrada no leite materno. Na faixa de concentração plasmática de 1 a 10 mcg/ml de nevirapina, cerca de 60% liga-se às proteínas plasmáticas. As concentrações de nevirapina no líquido cérebro-espinhal humano (n=6) foram de 45% (± 5%) das concentrações plasmáticas. Esta proporção é aproximadamente igual à fração não-ligada às proteínas plasmáticas.
Estudos in vivo em humanos e in vitro com microssomos de fígado humano demonstraram que a nevirapina é extensamente biotransformada em vários metabólitos hidroxilados por meio de metabolismo oxidativo via citocromo P450. Estudos in vitro com microssomos de fígado humano sugerem que o metabolismo oxidativo da nevirapina seja mediado primariamente pelas isoenzimas da família CYP3A do citocromo P450, embora outras isoenzimas também possam ter uma função secundária. Num estudo de equilíbrio de massa/excreção, oito homens saudáveis tiveram suas concentrações séricas dosadas no estado de equilíbrio após administração de duas doses diárias de 200 mg de nevirapina seguidas pela administração de uma dose única de 50 mg de nevirapina marcada com 14C. Aproximadamente 91,4 ± 10,5% da dose marcada foi recuperada, sendo 81,3 ± 11,1% com a urina, representando a via de excreção primária, e 10,1 ± 1,5% com as fezes. Mais de 80% da radioatividade na urina foi detectada em conjugados de metabólitos hidroxilados de glucuronida. Portanto, o metabolismo via citocromo P450, a conjugação com glucuronida e a excreção urinária de metabólitos glucuronidados representam a via primária de biotransformação e eliminação da nevirapina em humanos. Somente uma pequena fração ( < 5%) da radioatividade na urina (representando < 3% da dose total) foi detectada no composto não-metabolizado; portanto, a excreção renal contribui muito pouco na eliminação do composto não-metabolizado.
Demonstrou-se que a nevirapina é um indutor das enzimas metabólicas hepáticas do citocromo P450. A farmacocinética de auto-indução foi caracterizada por um aumento aproximado de 1,5 a 2 vezes no clearence aparente de nevirapina oral, comparando-se a administração de uma dose única com a administração de 200 a 400 mg/dia por duas a quatro semanas. A auto-indução também promove uma diminuição correspondente na fase terminal da meia-vida plasmática da nevirapina, de aproximadamente 45 horas (dose única) para aproximadamente 25 a 30 horas após a administração de doses múltiplas de 200 a 400 mg/dia.
Gênero: no estudo multinacional 2NN, um sub-estudo farmacocinético populacional de 1077 pacientes foi desenvolvido e incluiu 391 mulheres. As mulheres apresentaram um clearance de nevirapina 13,8% menor do que o apresentado pelos homens. Esta diferença não é considerada clinicamente relevante. O efeito do gênero não pode ser explicado pelo tamanho do corpo uma vez que nem o peso corporal nem o índice de Massa Corpórea (IMC) tiveram influência no clearance de nevirapina.
Com a idade (faixa de 18 a 68 anos) ou a raça, a farmacocinética da nevirapina em adultos infectados por HIV-1 aparentemente não se altera. Esta informação é baseada na avaliação dos dados acumulados a partir de vários estudos clínicos.
Disfunção renal: a farmacocinética de uma dose única de VIRAMUNE foi avaliada em 23 indivíduos portadores de disfunção renal leve (50 ml/min ≤ clearance de creatinina < 80 ml/min) ou moderada (30 ml/min ≤ clearance de creatinina < 50 ml/min) ou grave (clearance de creatinina < 30 ml/min) ou deficiência renal ou doença renal terminal que requeiram hemodiálise e em 8 indivíduos portadores de função renal normal (clearence de creatinina > 80 ml/min). A deficiência renal (leve, moderada e grave) não promoveu nenhuma alteração significativa na farmacocinética de VIRAMUNE. Entretanto, indivíduos portadores de doença renal terminal dialítica apresentaram uma redução de 43,5% na AUC de VIRAMUNE durante o período de exposição de uma semana. Houve também acúmulo de hidroximetabólitos de nevirapina no plasma. Esses resultados sugerem que a suplementação com uma dose adicional de 200 mg de VIRAMUNE após cada hemodiálise compensaria os efeitos da hemodiálise no clearence de VIRAMUNE. Por outro lado, pacientes com clearence de creatinina ≥ 20 ml/min não necessitam de ajustes na dose de VIRAMUNE.
Lesão hepática: foi conduzido um estudo no estado de equilíbrio comparando 46 pacientes com fibrose hepática:
Leve (n=17: Escore de Ishak 1-2),
Moderada (n=20: Escore de Ishak 3-4),
ou grave (n=9: Escore de Ishak 5-6, Child-Pugh A em 8 pontos, para escore 1 de Child-Pugh não aplicável)
Os pacientes estudados receberam terapia anti-retroviral contendo VIRAMUNE 200 mg duas vezes ao dia por, no mínimo, 6 semanas antes da amostragem farmacocinética, com uma terapia de duração mediana de 3,4 anos. Neste estudo, a disposição farmacocinética de doses múltiplas de nevirapina e os 5 metabólitos oxidativos não foram alterados. Entretanto, aproximadamente 15% destes pacientes com fibrose hepática tiveram concentrações-vale de nevirapina acima de 9000 ng/ml (2 vezes a média usual de concetração-vale). Pacientes com lesão hepática devem ser cuidadosamente monitorados quanto à evidência da toxicidade induzida pela droga.
Em um estudo farmacocinético de dose única de nevirapina 200 mg em pacientes HIV-negativos com lesão hepática leve a moderada (Child-Pugh A, n=6; Child-Pugh B, n=4), foi observado aumento significativo na AUC de nevirapina em um paciente Child-Pugh B com ascite sugerindo que pacientes com piora da função hepática e ascite podem estar sob risco de acúmulo de nevirapina na circulação.
Devido à nevirapina induzir seu próprio metabolismo com doses múltiplas, este estudo de dose única pode não refletir o impacto da lesão hepática na farmacocinética de doses múltiplas (Vide item Precauções).
Farmacocinética em pacientes pediátricos
Dados referentes à farmacocinética da nevirapina foram obtidos a partir de duas fontes principais: um estudo clínico pediátrico de 48 semanas na África do Sul envolvendo 123 pacientes HIV-1 positivos e virgens de tratamento anti-retroviral com idade de 3 meses a 16 anos; e uma análise consolidada de 5 protocolos do PACTG (Paediatric AIDS Clinical Trials Group), compreendendo 495 pacientes com idade de 14 dias a 19 anos.
Os resultados da análise de 48 semanas do estudo Sul-africano confirmaram que os grupos que receberam doses de 4/7 mg/kg e 150 mg/m2 de nevirapina foram bem tolerados e eficazes nos pacientes pediátricos virgens de tratamento anti-retroviral. Uma acentuada melhora no percentual de células CD4 + foi observada na semana 48 nos dois grupos. Além disto, as duas formas de dosagem foram eficazes na redução da carga viral. Neste estudo de 48 semanas nenhum achado inesperado de segurança foi observado em qualquer um dos dois grupos de dose.
Dados farmacocinéticos de 33 pacientes (idade entre 0,77 a 13,7 anos) no grupo de amostragem intensiva demonstraram que após a administração oral, o clearance de nevirapina aumentou com o aumento da idade de maneira consistente com o aumento da área de superfície corporal. A dose de nevirapina de 150 mg/m2 duas vezes ao dia (após a administração da dose de introdução gradual de 150 mg/m2 uma vez ao dia por 2 semanas) produziu média geométrica ou vales médios de concentrações de nevirapina entre 4-6 mg/ml (conforme focado baseado nos dados de adultos). Além disto, as concentrações-vale de nevirapina observadas foram comparáveis nos dois métodos.
A análise consolidada dos protocolos PACTG 245, 356, 366, 377 e 403 permitiu a avaliação de pacientes pediátricos com menos de 3 meses de idade (n=17) incluídos nestes estudos. As concentrações plasmáticas de nevirapina encontradas estiveram dentro da faixa observada em adultos e no restante da população pediátrica, mas foram mais variáveis particularmente naqueles pacientes do segundo mês de idade.
Contraindicações.
VIRAMUNE é contra-indicado em pacientes com hipersensibilidade clinicamente significativa ao princípio ativo ou a qualquer outro componente da fórmula.
VIRAMUNE não deve ser administrado a pacientes com grave disfunção hepática (Child-Pugh C) ou que apresentarem níveis de ALT ou AST > 5 vezes o limite superior dos valores de referência antes do tratamento, até que os níveis basais de AST/ALT estabilizem abaixo de 5 vezes o limite superior de referência.
VIRAMUNE não deve ser readministrado a pacientes que necessitaram de descontinuação do tratamento devido a graves erupções cutâneas, erupções cutâneas acompanhadas de sintomas constitucionais, reações de hipersensibilidade ou hepatite clínica causadas pela nevirapina.
VIRAMUNE não deve ser readministrado a pacientes que apresentaram inicialmente níveis de alanina aminotransferase ou aspartato aminotransferase maiores que 5 vezes o limite superior dos valores de referência durante o tratamento com nevirapina e apresentaram rápida recorrência de anormalidades nas funções hepáticas após a readministração de nevirapina.
O uso de VIRAMUNE é contra-indicado em casos de condições hereditárias raras que podem ser incompatíveis com os excipientes do produto (Vide Advertências).
Fitoterápicos que contenham erva de São João (hypericum perforatum) não devem ser utilizados durante o tratamento com VIRAMUNE, em razão do risco de diminuição das concentrações plasmáticas e diminuição dos efeitos clínicos da nevirapina (vide também Interações).
Advertências e precauções.
As primeiras 18 semanas de tratamento com nevirapina são um período crítico que requer minuciosa monitoração dos pacientes para detectar o aparecimento latente de reações cutâneas graves e potencialmente letais (incluindo casos de síndrome de Stevens-Johnson e necrólise epidérmica tóxica) ou graves hepatites/deficiências hepáticas.
O maior risco de eventos hepáticos e reações cutâneas ocorre nas primeiras 6 semanas de tratamento. Entretanto, o risco de qualquer evento hepático continua após este período e a monitoração deve continuar a intervalos freqüentes. Pacientes do sexo feminino e pacientes com maiores contagens de células CD4 no início da terapia estão expostos a maior risco de reações adversas hepáticas.
Com base na hepatotoxicidade grave ou potencialmente letal observada em estudos controlados e não controlados, a administração de VIRAMUNE não deve ser iniciada em mulheres adultas com contagem de células CD4+ > 250 células/mm³ ou em homens adultos com contagem de células CD4+ > 400 células/mm³, a menos que o benefício supere o risco.
Em alguns casos, a lesão hepática tem progredido mesmo após a interrupção do tratamento. Pacientes desenvolvendo sinais ou sintomas de hepatite, reações cutâneas graves ou reações de hipersensibilidade devem descontinuar o tratamento com VIRAMUNE e procurar orientação médica imediatamente. O tratamento com VIRAMUNE não deve ser reiniciado após reações hepáticas, cutâneas ou de hipersensibilidade graves.
O regime posológico deve ser rigorosamente cumprido principalmente nos primeiros 14 dias de tratamento.
Reações cutâneas
Nos pacientes tratados com VIRAMUNE ocorreram reações dermatológicas graves ou potencialmente letais, incluindo casos fatais. Essas reações dermatológicas incluíram casos de síndrome de Stevens-Johnson (SSJ), necrólise epidérmica tóxica (NET) e síndrome de hipersensibilidade caracterizada por erupções cutâneas, sintomas constitucionais e comprometimentos viscerais. Os pacientes devem ser cuidadosamente monitorados durante as primeiras 18 semanas de tratamento. Também devem ser minuciosamente monitorados se apresentarem uma ocorrência isolada de erupção cutânea.
VIRAMUNE deve ser permanentemente descontinuado em qualquer paciente que apresente erupções cutâneas graves ou erupções cutâneas acompanhadas de sintomas constitucionais (tais como febre, erupções vesiculares, lesões bucais, conjuntivite, inchaços, dores musculares ou das articulações, mal-estar geral) incluindo síndrome de Stevens-Johnson ou necrólise epidérmica tóxica. VIRAMUNE deve ser permanentemente descontinuado em qualquer paciente que apresente reações de hipersensibilidade, caracterizadas por erupções cutâneas associadas a sintomas constitucionais e alterações viscerais tais como hepatite, eosinofilia, granulocitopenia e disfunção renal ou sinais de outras alterações viscerais.
Os pacientes devem ser avisados que erupções cutâneas constituem a principal reação adversa ao VIRAMUNE. A fase de introdução deve ser observada porque demonstrou diminuir a freqüência de erupções cutâneas. A maioria das erupções cutâneas associadas ao VIRAMUNE ocorre nas primeiras seis semanas de tratamento; portanto, os pacientes devem ser monitorados cuidadosamente em relação ao aparecimento de erupções cutâneas durante esse período. Os pacientes devem ser advertidos de que a dose não deve ser aumentada se alguma erupção cutânea aparecer durante a fase de introdução, até que as erupções cutâneas tenham desaparecido. O tratamento com a dosagem de 200 mg/dia não deve ultrapassar 28 dias, quando um regime alternativo deve ser adotado.
Raros casos de rabdomiólise foram observados em pacientes com reações de pele e/ou de fígado associados ao VIRAMUNE.
O uso concomitante de prednisona (40 mg/dia nos 14 primeiros dias de administração de VIRAMUNE) demonstrou não diminuir a incidência de erupções cutâneas associadas ao VIRAMUNE, podendo ser associada a um aumento de erupções cutâneas durante as 6 primeiras semanas de tratamento com VIRAMUNE.
Os fatores que propiciam o desenvolvimento de reações cutâneas graves incluem o descumprimento do regime posológico inicial de 200 mg/dia durante o período de introdução. A longa demora em consultar um médico após o aparecimento dos sintomas iniciais pode aumentar o risco do desenvolvimento de reações cutâneas mais graves. Aparentemente, mulheres sob terapia anti-retroviral com ou sem VIRAMUNE apresentam maiores riscos de desenvolverem reações cutâneas do que homens.
Qualquer paciente que apresente erupções cutâneas graves ou erupções cutâneas acompanhadas de sintomas constitucionais tais como febre, erupções vesiculares, lesões bucais, conjuntivite, inchaços, dores musculares ou das articulações, ou mal-estar geral, deve descontinuar o tratamento e procurar orientação médica imediatamente. Não se deve readministrar VIRAMUNE nestes pacientes.
Devem-se realizar testes de função hepática nos pacientes com suspeita de erupções cutâneas associadas ao VIRAMUNE. Pacientes com elevações moderadas a graves de AST ou ALT (maiores que 5 vezes o limite superior dos valores de referência) devem descontinuar VIRAMUNE permanentemente.
Na ocorrência de qualquer reação de hipersensibilidade, caracterizada por erupções cutâneas associadas a sintomas constitucionais tais como febre, artralgia, mialgia, linfadenopatia e alterações viscerais, tais como hepatite, eosinofilia, granulocitopenia e disfunção renal ou sinais de outras alterações viscerais, o tratamento com nevirapina deve ser permanentemente descontinuado e não deve ser reiniciado.
Reações hepáticas
Ocorreu hepatotoxicidade grave ou potencialmente letal, incluindo hepatite fulminante fatal, em pacientes tratados com VIRAMUNE. As primeiras 18 semanas de tratamento são um período crítico que requer cuidadosa monitoração. O risco de eventos hepáticos é maior nas primeiras 6 semanas de tratamento.
Contudo, o risco continua após esse período e a monitoração deve continuar em intervalos freqüentes durante o tratamento. Os pacientes devem ser informados que as reações hepáticas consistem na maior toxicidade de VIRAMUNE.
Pacientes com sinais ou sintomas de hepatite devem ser aconselhados a descontinuar o tratamento com VIRAMUNE e buscar avaliação médica imediatamente, que deve incluir testes de função hepática.
Raros casos de rabdomiólise foram observados em pacientes com reações de pele e/ou de fígado associados com VIRAMUNE.
Relatou-se hepatotoxicidade grave, incluindo insuficiência hepática com necessidade de transplante nos pacientes não infectados com HIV e que receberam doses múltiplas de VIRAMUNE na profilaxia pós-exposição, uma indicação não aprovada e, portanto, enfaticamente desaconselhada.
O aumento nos níveis maiores que 2,5 vezes os limites superiores de referência de AST ou ALT e/ou infecção concomitante de hepatite crônica (B e/ou C) no início do tratamento anti-retroviral está associado a maior risco de eventos adversos hepáticos durante os tratamentos anti-retrovirais em geral, incluindo os regimes de tratamento contendo VIRAMUNE.
Pacientes do sexo feminino e pacientes com maiores contagens de células CD4 estão expostos a maior risco de eventos adversos hepáticos.
Mulheres estão sujeitas a um risco três vezes maior que o dos homens de apresentarem eventos adversos hepáticos sintomáticos, freqüentemente associados a erupções cutâneas (5,8% vs. 2,2%), e pacientes com maiores contagens de CD4 no início do tratamento com VIRAMUNE apresentam maior risco de eventos hepáticos sintomáticos com VIRAMUNE. Em uma revisão retrospectiva, mulheres com contagens de CD4 > 250 células/mm³ apresentaram um risco 12 vezes maior de eventos adversos hepáticos sintomáticos em comparação com mulheres com contagens de CD4 < 250 células/mm³ (11,0% vs. 0,9%). Observou-se um maior risco em homens com contagens de CD4 > 400 células/mm³ (6,3% vs. 1,2% para homens com contagens de CD4 < 400 células/mm³).
Monitoração hepática
Relataram-se testes de funções hepáticas anormais durante o uso de VIRAMUNE, alguns tendo ocorrido nas primeiras semanas de tratamento. Elevações assintomáticas de enzimas hepáticas são relatadas freqüentemente e não são necessariamente uma contra-indicação ao uso de VIRAMUNE. Elevações assintomáticas de gama-glutamil-transferase não são uma contra-indicação à continuação do tratamento.
Recomenda-se enfaticamente realizar testes de monitoração da função hepática em intervalos freqüentes, adequados às necessidades clínicas do paciente, especialmente durante as primeiras 18 semanas de tratamento. Os testes de monitoração clínica e laboratorial devem ser mantidos durante o tratamento com VIRAMUNE. Médicos e pacientes devem estar atentos aos primeiros sinais ou sintomas de hepatite, como anorexia, náusea, icterícia, bilirrubinúria, acolia fecal, hepatomegalia ou dor à palpação do fígado. Os pacientes devem ser instruídos a procurar auxílio médico se esses sintomas ocorrerem.
Com valores de AST e ALT maiores que 2,5 vezes o limite superior dos valores de referência antes ou durante o tratamento, os testes hepáticos deverão ser monitorados mais freqüentemente durante as visitas clínicas regulares. VIRAMUNE não deve ser administrado a pacientes previamente tratados que apresentaram níveis de AST ou ALT maiores que 5 vezes o limite superior dos valores de referência até que os níveis basais de AST ou ALT se estabilizem em níveis menores que 5 vezes o limite superior dos valores de referência.
Se os valores de AST e ALT forem maiores que 5 vezes o limite superior dos valores de referência, o tratamento com nevirapina deve ser imediatamente interrompido. Se os valores de AST e ALT retornarem para os valores basais e se o paciente não apresentou sinais clínicos ou sintomas de hepatite ou sintomas constitucionais ou outros sintomas sugestivos de disfunção orgânica, é possível reintroduzir VIRAMUNE, conforme a necessidade clínica e avaliação caso a caso. Neste caso, VIRAMUNE deverá ser reiniciado sob altíssima vigilância clínica e laboratorial durante o regime de introdução de 200 mg/dia por 14 dias seguidos por 400 mg/dia. Se as anormalidades nas funções hepáticas reaparecerem rapidamente, o tratamento com nevirapina deve ser permanentemente interrompido.
Se ocorrerem casos de hepatite clínica, caracterizados por anorexia, náuseas, vômitos, icterícia e testes laboratoriais anormais (tais como testes anormais de função hepática moderada ou grave, excluindo gama-glutamil-transferase), o tratamento com nevirapina deve ser permanentemente interrompido. VIRAMUNE não deve ser readministrado a pacientes que precisarem descontinuar o tratamento permanentemente devido à ocorrência de hepatite clínica causada pela nevirapina.
Outras advertências
Relataram-se também os seguintes eventos quando se utilizou VIRAMUNE em combinação com outros agentes anti-retrovirais: pancreatite, neuropatia periférica e trombocitopenia. Esses eventos são usualmente associados aos outros agentes anti-retrovirais e espera-se que ocorram quando se utiliza VIRAMUNE em combinação com outros agentes; contudo, é improvável que esses eventos sejam devidos ao tratamento com nevirapina.
Pacientes sob tratamento com VIRAMUNE ou com outro agente anti-retroviral podem continuar desenvolvendo infecções oportunistas e outras complicações da infecção pelo HIV e devem, portanto, permanecer sob rigorosa observação clínica de médicos experientes no tratamento de pacientes com doenças associadas ao HIV. O tratamento com VIRAMUNE não demonstrou reduzir o risco de transmissão horizontal de HIV-1.
Embora a utilidade de VIRAMUNE na prevenção da transmissão materno-infantil do HIV-1 tenha sido demonstrada em mulheres que não estão em tratamento com outros anti-retrovirais, recomenda-se, quando possível, estender o tratamento à mãe antes do parto em combinação com agentes anti-retrovirais, para minimizar a transmissão do HIV-1 ao recém-nascido.
Em mulheres e crianças previamente tratadas com dose única de nevirapina para a prevenção da transmissão materno-infantil do HIV-1, a eficácia de VIRAMUNE como parte da terapia combinada recebida para cuidado da sua própria saúde, pode ser reduzida.
Métodos hormonais de prevenção de gravidez exceto DMPA não devem ser utilizados isoladamente como métodos contraceptivos em mulheres tratadas com VIRAMUNE. A nevirapina pode diminuir os níveis plasmáticos desses medicamentos. (vide também Interações). Portanto, ao ser usada terapia hormonal na pós-menopausa durante a administração de VIRAMUNE, seu efeito terapêutico deve ser monitorado.
Síndrome de Reativação Imune
Em pacientes infectados por HIV com imunodeficiência grave no momento da instituição da terapia anti-retroviral combinada, pode ocorrer uma reação inflamatória a patógenos assintomáticos ou oportunistas residuais e provocar problemas clínicos sérios ou agravamento dos sintomas. Normalmente, estas reações têm sido observadas dentro das primeiras semanas ou meses após o início da terapia anti-retroviral combinada. Os principais exemplos são retinite por citomegalovírus, infecções micobacterianas generalizadas e/ou focais e pneumonia por Pneumocystis carinii. Quaisquer sintomas inflamatórios devem ser avaliados e o tratamento instituído quando necessário.
Advertência sobre uso concomitante com outras medicações (para uma descrição detalhada, vide Interações).
VIRAMUNE pode alterar a exposição plasmática a outros fármacos e outros fármacos podem modificar a exposição plasmática de VIRAMUNE.
A combinação dos seguintes compostos com VIRAMUNE não é recomendada: efavirenz, rifampicina, cetoconazol; se não co-administrado com baixa dose de ritonavir: fosamprenavir, saquinavir, atazanavir.
VIRAMUNE comprimidos contém 636 mg de lactose por dose diária máxima recomendada. Os pacientes portadores de condições hereditárias raras de intolerância à galactose, como por e.x. galactosemia, deficiência de Lapp lactase ou má absorção de glicose-galactose, não devem tomar este medicamento.
VIRAMUNE suspensão contém 6 g de sacarose por dose diária máxima recomendada. Os pacientes portadores de condições hereditárias raras de intolerância à frutose, má absorção de glicose-galactose ou insuficiência de sacarase-isomaltase, não devem tomar este medicamento.
VIRAMUNE suspensão contém 6,5