VELCADE*
JANSSEN-CILAG
bortezomibe
Antineoplásico.
Apresentações.
Pó liofilizado em embalagem com 1 frasco-ampola de 1,0 mg ou 3,5 mg de bortezomibe.
USO INTRAVENOSO
USO ADULTO
Composição.
VELCADE* 3,5 mg. Cada frasco-ampola de pó liofilizado contém 3,5 mg de bortezomibe como éster boronato de manitol. Excipiente: manitol. Após a reconstituição com 3,5 mL de solução salina (0,9%), cada mL contém 1 mg de bortezomibe. VELCADE* 1,0 mg. Cada frasco-ampola de pó liofilizado contém 1,0 mg de bortezomibe como éster boronato de manitol. Excipiente: manitol. Após a reconstituição com 1,0 mL de solução salina (0,9%), cada mL contém 1 mg de bortezomibe.
Indicações.
VELCADE* (bortezomibe) é indicado para o tratamento de pacientes com mieloma múltiplo:
- que não receberam tratamento prévio e impossibilitados de receberem tratamento com alta dose de quimioterapia e transplante de medula óssea. Nesses pacientes, VELCADE* é utilizado em combinação com melfalano e prednisona.
- que receberam pelo menos um tratamento anterior.
Resultados de eficácia.
Estudo Clínico aberto, randomizado em pacientes com mieloma múltiplo sem tratamento prévio:
Um estudo clínico prospectivo de fase 3, aberto, randomizado (1:1) de 682 pacientes foi conduzido para determinar se VELCADE* (1.3 mg/m2) em combinação com melfalna (9 mg/m2) e prednisona (60 mg/m2) resultou em melhora no tempo de progressão (TTP) quando comparado com melfalano (9 mg/m2) e prednisona (60 mg/m2) em pacientes com mieloma múltiplo sem tratamento prévio. Este estudo incluiu pacientes que não foram candidatos a transplante de células tronco. O tratamento foi administrado para um máximo de 9 ciclos (aproximadamente 54 semanas) e foi descontinuado em caso de progressão da doença ou toxicidade inaceitável. A condição basal demográfica e as características dos pacientes estão resumidas na Tabela 1.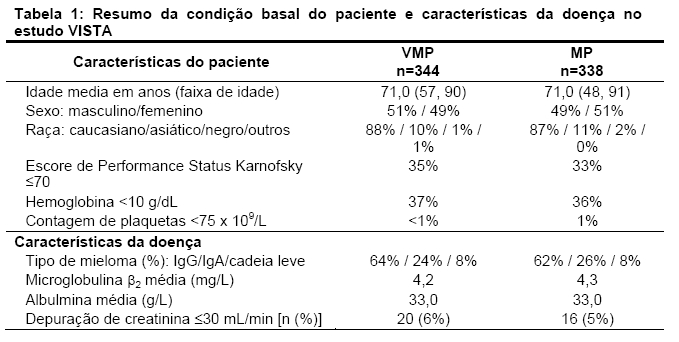
O desfecho primário (tempo para progressão) foi atingido em análise interina pré-determinada do estudo, sendo oferecido aos pacientes do braço MP o tratamento VMP. A mediana de acompanhamento foi 16,3 meses. A atualização final na sobrevida foi feita com uma duração mediana de acompanhamento aos 60,1 meses. Um benefício da sobrevida estatisticamente significante a favor do tratamento VMP foi observado (HR=0,695; p=0,00043) apesar das terapias subsequentes que incluíram regimes baseados em VELCADE*. A sobrevida mediana do grupo de tratamento MP foi estimada como 43,1 meses e a sobrevida mediana no grupo de tratamento VMP foi estimada como 56,4 meses. Resultados de eficácia são apresentados na Tabela 2.
Pacientes que não conseguiram uma resposta ótima no tratamento com VELCADE* isolado (doença progressiva ou estável após 2 ou 4 ciclos respectivamente) foram capazes de receber uma alta dose de dexametasona em conjunto com VELCADE* (exemplo: 40 mg de dexametasona administrada via oral para cada dose de VELCADE* sendo que 20 mg no dia da administração de VELCADE* e 20 mg no dia após a administração (exemplo: dias 1, 2, 4, 5, 8, 9, 11 e 12) mais 160 mg após 3 semanas. Um total de 74 pacientes recebeu dexametasona administrada em combinação com VELCADE* e foram avaliados através da resposta. 18% dos pacientes (13 em 74) obtiveram sucesso ou tiveram um aumento na resposta (CR 11% ou PR 7%) com tratamento combinado.
Estudos Clínicos de Fase 2 em Mieloma Múltiplo Recidivante ou Refratário
A segurança e a eficácia de VELCADE* foram avaliadas em um estudo multicêntrico aberto, com braço único de tratamento, em 202 pacientes que haviam recebido pelo menos 2 tratamentos anteriores e demonstraram progressão durante a terapia mais recente. O número mediano de terapias anteriores foi 6. As características do paciente e da doença na condição basal estão resumidas na Tabela 3.
Uma injeção intravenosa em bolus de VELCADE* 1,3 mg/m2/dose foi administrada 2 vezes por semana por 2 semanas, seguida por um período de descanso de 10 dias (ciclo de tratamento de 21 dias), por no máximo 8 ciclos de tratamento. O estudo usou modificações da dose em função da toxicidade. Pacientes que apresentaram expectativa de resposta ao tratamento com VELCADE* continuaram o tratamento em um estudo de extensão.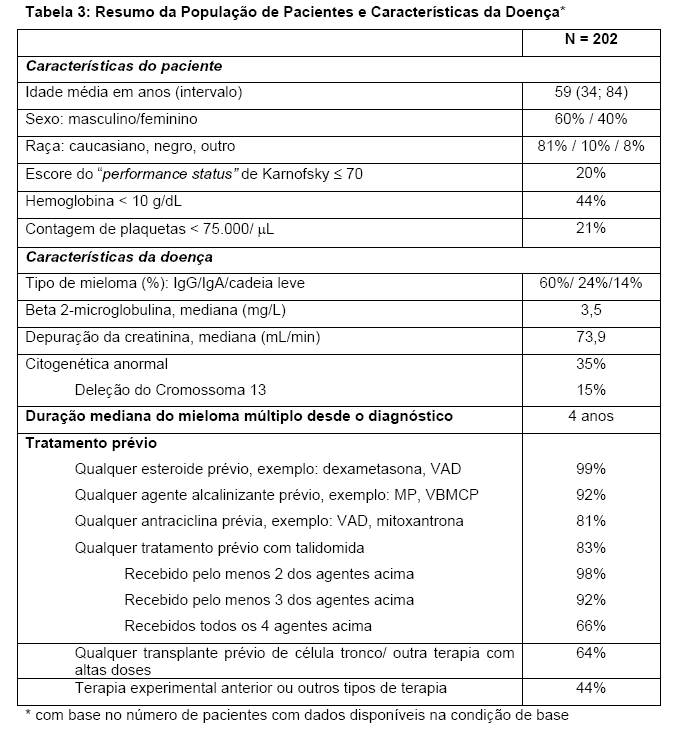
As taxas de resposta ao VELCADE* isolado (Tabela 4) foram determinadas por um Comitê de Revisão Independente (CRI) com base nos critérios publicados por Bladé e outros1. Resposta completa exigiu < 5% de células plasmáticas na medula, 100% de redução na proteína M e teste de imunofixação negativo. As taxas de resposta utilizando os critérios do "SWOG" também são mostradas. A resposta de acordo com o "SWOG" (Southwest Oncology Group) exigiu uma redução ≥ 75% no nível sérico de proteína do mieloma e/ou ≥ 90% da proteína na urina2. Um total de 188 pacientes foram avaliados quanto à resposta; 9 pacientes com doença não mensurável não puderam ser avaliados quanto a resposta pelo CRI. Cinco pacientes foram excluídos das análises de eficácia por terem terapia prévia mínima.
Noventa e oito porcento dos pacientes do estudo receberam uma dose inicial de 1,3 mg/m2. Vinte e oito porcento destes pacientes receberam uma dose de 1,3 mg/m2 ao longo do estudo, enquanto que em 33% dos pacientes que iniciaram com uma dose de 1,3 mg/m2 foi necessário reduzir a dose durante o estudo. Sessenta e três porcento dos pacientes tiveram pelo menos uma dose suspensa durante o estudo. Em geral, pacientes com resposta completa confirmada receberam 2 ciclos adicionais do tratamento com VELCADE* após a confirmação. O número médio de ciclos administrados foi 6.
O tempo mediano para resposta foi 38 dias (variação de 30 a 127 dias). A sobrevida mediana de todos os pacientes recrutados foi 16 meses (variação < 1 a 18+ meses).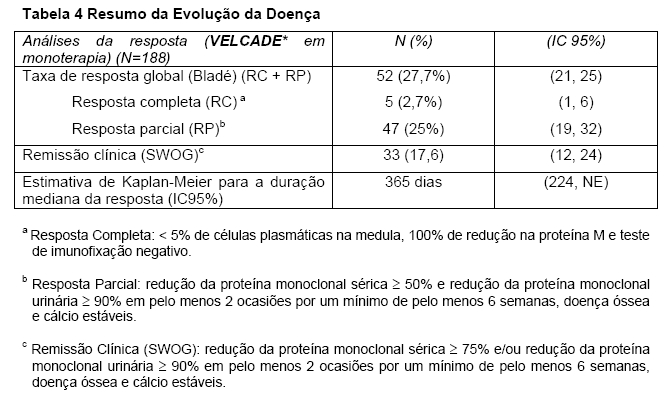
Neste estudo, a taxa de resposta ao VELCADE* foi independente do número e tipos de tratamentos anteriores. Houve uma probabilidade reduzida de resposta em pacientes com células plasmáticas > 50% ou citogenética anormal na medula óssea. A resposta foi observada em pacientes com anomalias do cromossomo 13.
Um estudo de dose-resposta foi realizado em 54 pacientes com mieloma múltiplo que receberam 1,0 mg/m2/dose ou 1,3 mg/m2/dose, duas vezes por semana durante 2 de 3 semanas. Resposta isolada completa foi observada em cada dose e foram observadas taxas de resposta (RC + RP) global de 30% (8/27) na dose de 1,0 mg/m2 e 38% (10/26) na dose de 1,3 mg/m2.
Estudo Clínico Aberto e Randomizado para Mieloma Múltiplo recidivado
Um estudo clínico fase 3 prospectivo, randomizado (1:1), internacional, aberto, estratificado, contou com 669 pacientes que foram designados para determinar se o tratamento com VELCADE* resultou num crescimento em tempo de progressão quando comparado a alta dose de dexametasona em pacientes com mieloma múltiplo progressivo após a utilização de uma a três terapias anteriores. Pacientes considerados tolerantes ao tratamento com altas doses de dexametasona foram excluídos como se fossem aqueles com base de grau ≥ 2 de neuropatia periférica ou contagem plaquetária < 50.000 mL. Um total de 627 pacientes foi avaliado.
Os fatores de estratificação foram baseados no número de linhas de terapias anteriores que os pacientes receberam previamente (uma linha prévia versus mais que uma linha de terapia), tempo de progressão relativo aos tratamentos anteriores (progressão durante ou dentro do período de 6 meses de parada da terapia mais recente versus recaída > 6 meses após receber terapia mais recente e níveis identificados de Beta-2-microglobina (≤2.5 mg/L versus > 2.5 mg/L).
Dados dos pacientes e características da doença estão descritos na Tabela 5.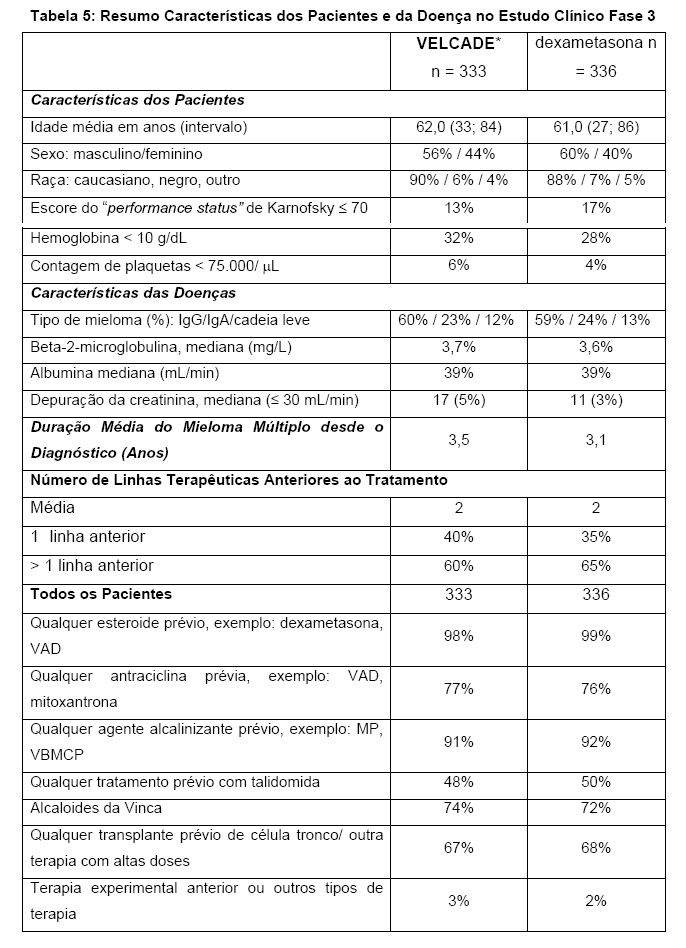
Pacientes que estavam no grupo de tratamento com VELCADE* receberam 8 ciclos de tratamento com duração de 3 semanas para cada ciclo e 3 ciclos de tratamento com duração de 5 semanas para cada ciclo. Dentro do tratamento com cada ciclo de duração de 3 semanas, VELCADE* 1,3 mg/m2/dose foi administrado isolado via injeção intravenosa em bolus duas vezes por semana, durante duas semanas nos dias 1, 4, 8 e 11 seguido de um período de 10 dias de descanso (compreendido entre o dia 12 e o dia 21). Dentro do tratamento com cada ciclo de duração de 5 semanas, VELCADE* 1,3 mg/m2/dose foi administrado isolado via injeção intravenosa em bolus uma vez semanalmente durante 4 semanas nos dias 1, 8, 15 e 22 seguido de um período de 13 dias de descanso (compreendido entre o dia 23 e o dia 35).
Pacientes que estavam no grupo de tratamento com dexametasona receberam 4 ciclos de tratamento com duração de 5 semanas seguido de 5 ciclos de tratamento com duração de 4 semanas. Dentro de cada ciclo do tratamento com duração de 5 semanas, dexametasona 40 mg/dia via oral foi administrada uma vez ao dia nos dias 1 ao 4 seguido de um período de 24 dias de descanso (compreendido entre o dia 21 e o dia 35). Dentro de cada ciclo do tratamento com duração de 5 semanas, dexametasona 40 mg/dia via oral foi administrada uma vez ao dia nos dias 1 ao 4, 9 ao 12 e 17 ao 20 seguido de um período de 15 dias de descanso (compreendido entre o dia 5 e o dia 28). Para pacientes que apresentaram a progressão da doença, durante o uso de dexametasona, documentada foi oferecida uma dose padrão de VELCADE* e agendado um estudo de acompanhamento.
Seguindo uma análise interina pré-planejada de tempo de progressão, o braço de dexametasona foi interrompido e para todos pacientes selecionados para o uso de dexametasona foi oferecido VELCADE*, independente do status da doença. Neste momento o estudo foi encerrado e uma análise estatística foi elaborada. Em decorrência deste encerramento precoce do estudo, a duração média do acompanhamento para pacientes sobreviventes (n=534) está limitado a 8,3 meses.
No braço de VELCADE*, 34% dos pacientes receberam pelo menos uma dose de VELCADE* em todos os 8 ciclos com duração de 3 semanas de terapia e 13% receberam pelo menos uma dose em todos os 11 ciclos. O número médio de doses de VELCADE* durante todo o estudo foi 22, com uma faixa de 1 a 44. No braço de dexametasona, 40% dos pacientes receberam pelo menos uma dose em todos os 4 ciclos com duração de 5 semanas de terapia, e 6% receberam pelo menos uma dose em todos os 9 ciclos.
O tempo de finalização de análises e taxas de respostas de estudos clínicos fase 3 estão apresentados na Tabela 6. Resposta e progressão foram avaliados considerando os critérios do grupo europeu para sangue e transplante de medula. A resposta completa requer < 5 % células plasmáticas na medula, 100% de redução em M-proteína e um resultado negativo para teste de imunofixação. A resposta parcial requer ≥ 50% de redução na concentração sérica da proteína do mieloma e ≥ 90% de redução da concentração da proteína do mieloma na urina em pelo menos duas ocasiões para um mínimo de pelo menos 6 semanas ao longo da doença óssea estável e níveis normais de cálcio. Foi verificado que todos os dados se encontram em acordo com os critérios para a resposta completa, incluindo 100% de redução em M-proteína pela eletroforese proteica, mas a M-proteína pode ser detectada pelo teste de imunofixação.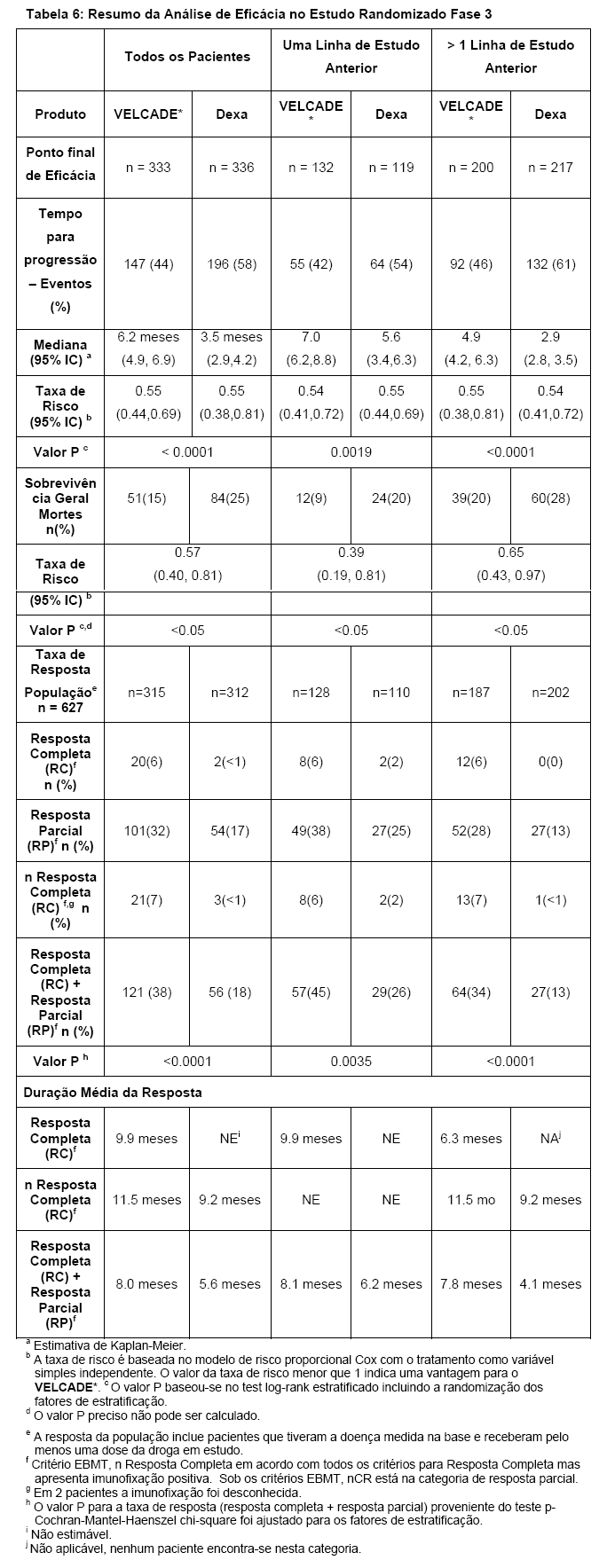
REFERÊNCIAS
1. http://ctep.info.nih.gov/reporting/ctc.html.
2. Bladé J et al.. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and hematopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998 102(5): 1115-23.
3. Salmon SE, Haut A, Bonnet JD, Amare M, Weick JK, Durie BG et al. Alternating combination chemotherapy and levamisole improves survival in multiple myeloma: a Southwest Oncology Group Study. Journal of Clinical Oncology 1983; 1(8): 453-461.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
Mecanismo de ação
O bortezomibe é um inibidor reversível da atividade do tipo quimiotripsina do proteassoma 26S em células de mamíferos. O proteassoma 26S é um complexo proteico grande que degrada proteínas ubiquitinadas. A via da ubiquitina-proteassoma representa um papel essencial na regulação da concentração intracelular de proteínas específicas, mantendo, desta forma, a homeostase intracelular. A inibição do proteassoma 26S impede esta proteólise desejada, o que pode afetar as cascatas múltiplas de sinalização dentro da célula. Esta interrupção dos mecanismos normais de homeostasia pode levar à morte celular. Os experimentos demonstraram que o bortezomibe é citotóxico para uma variedade de tipos de células neoplásicas in vitro. O bortezomibe causa um retardo no crescimento tumoral in vivo em modelos tumorais não clínicos, incluindo mieloma múltiplo.
Propriedades Farmacocinéticas
Farmacocinética
Após a administração intravenosa em bolus de 1,0 mg/ m2 e 1,3 mg/m2 em onze pacientes com mieloma múltiplo, a média da concentração plasmática máxima de bortezomibe foi, respectivamente, de 57 e 112 ng/mL após a primeira dose. Em doses subsequentes, a média observada da concentração plasmática máxima variou de 67 a 106 ng/mL para dose de 1,0 mg/ m2 e 89 a 120 ng/mL para a dose de 1,3 mg/ m2. A meia-vida média de eliminação de bortezomibe após o regime de múltiplas doses variou de 40 a 193 horas. O bortezomibe é eliminado mais rapidamente após a primeira dose do que após as doses subsequentes. As médias totais de depuração corporal foram de 102 e 112 L/h após a primeira dose de 1,0 mg/ m2 e 1,3 mg/ m2, respectivamente, e variou de 15 a 32 L/h após doses subsequentes de 1,0 mg/ m2 e 1,3 mg/ m2, respectivamente.
Distribuição
O volume médio de distribuição de bortezomibe variou de 1659 a 3294 Litros após a primeira dose ou após a administração de repetidas doses de 1,0 mg/ m2 ou 1,3 mg/ m2 em pacientes com mieloma múltiplo. Isto sugere que o bortezomibe se distribui amplamente através dos tecidos periféricos. A ligação do bortezomibe às proteínas plasmáticas foi em média 83% na faixa de concentração de 100-1000 ng/mL.
Metabolismo
Estudos in vitro com microssomas hepáticos humanos e isoenzimas do citocromo P450 humano indicam que o bortezomibe é metabolizado primariamente por oxidação via isoenzimas 3A4, 2C19 e 1A2 do citocromo P450. O metabolismo do bortezomibe pelas enzimas CYP 2D6 e 2C9 é mínimo. A principal via metabólica é remoção de um átomo de boro para formar dois metabólitos sem boro que subsequentemente, sofrem hidroxilação para diversos metabólitos. Os metabólitos sem boro do bortezomibe são inativos como inibidores do proteassoma 26S. Dados agrupados do plasma de 8 pacientes aos 10 minutos e aos 30 minutos após a administração indicam que os níveis plasmáticos de metabólitos são baixos em comparação ao fármaco-mãe.
Eliminação
As vias de eliminação do bortezomibe não foram caracterizadas em seres humanos
Populações Especiais
Idade, Sexo e Raça: os efeitos da idade, sexo e raça sobre a farmacocinética do bortezomibe não foram avaliados.
Insuficiência Hepática: O efeito da insuficiência hepática (para definição de insuficiência hepática, veja Tabela 10) na farmacocinética do bortezomibe foi avaliado em 51 pacientes no tratamento com doses de bortezomibe variando de 0,5 a 1,3 mg/m2. Quando comparado aos pacientes com função hepática normal, a insuficiência hepática leve não altera a AUC da dose-normalizada de bortezomibe. Contudo, os valores médios de AUC da dose-normalizada foram aumentados em aproximadamente 60% em pacientes com insuficiência hepática moderada ou grave. Baixas doses iniciais são recomendadas em pacientes com insuficiência hepática moderada ou grave, e esses pacientes devem ser cautelosamente monitorados (veja Tabela 10).
Insuficiência Renal: Um estudo farmacocinético foi conduzido em pacientes com diferentes graus de insuficiência renal. Os pacientes foram classificados de acordo com seus valores de depuração de creatinina (CrCL) dentro dos seguintes grupos: Normal (CrCL≥60mL/min/1,73m2, n=12), Intermediário (CrCL≥40-59mL/min/1,73m2, n=10), Moderado (CrCL=20-39 mL/min/1,73 m2, n=9) e Grave (CrCL < 20mL/min/1,73m2, n=3). Um grupo de pacientes em diálise que recebeu a dose após a diálise também foi incluído no estudo (n=8). Os pacientes receberam dose intravenosa de 0,7-1,3mg/m2 de bortezomibe 2 vezes por semana. A exposição ao bortezomibe (dose-normalizada AUC e Cmáx) foi comparável entre todos os grupos.
Contraindicações.
VELCADE* é contraindicado em pacientes com hipersensibilidade ao bortezomibe, boro ou manitol.
Advertências e precauções.
VELCADE* deve ser administrado sob a supervisão de médico com experiência no uso de tratamento antineoplásico.
Ocorreram casos fatais de administração inadvertida de VELCADE* pela via intratecal. VELCADE* deve ser administrado somente pela via intravenosa. VELCADE* NÃO DEVE SER ADMINISTRADO PELA VIA INTRATECAL.
Geralmente, o perfil de segurança de pacientes tratados com VELCADE em monoterapia foi similar ao observado em pacientes tratados com VELCADE combinado com melfalano e prednisona.
Atenção: Este medicamento contém Açúcar (manitol), portanto, deve ser usado com cautela em portadores de Diabetes.
Neuropatia Periférica
O tratamento com VELCADE* causa neuropatia periférica (NP) que é predominantemente sensitiva. Entretanto, foram relatados casos de neuropatia motora grave com ou sem neuropatia sensitiva periférica.
Pacientes com sintomas preexistentes (dormência, dor ou sensação de queimação nos pés ou mãos) e/ou sinais de neuropatia periférica podem apresentar piora da neuropatia periférica (incluindo Grau ≥3) durante o tratamento com VELCADE*. Os pacientes devem ser monitorados quanto aos sintomas de neuropatia, como sensação de queimação, hiperestesia, hipoestesia, parestesia, desconforto, dor neuropática ou fraqueza. Pacientes que apresentarem piora ou aparecimento de neuropatia periférica podem exigir alteração no esquema de tratamento com VELCADE*.
Após o ajuste de doses, a melhora ou resolução da neuropatia periférica
foi relatada em 51% dos pacientes com neuropatia periférica Grau ≥2 no estudo fase 3 com agente único de VELCADE* versus dexametasona. A melhora ou resolução da neuropatia periférica foi relatada em 73% dos pacientes que descontinuaram o medicamento devido a neuropatia periférica Grau 2 ou que apresentaram neuropatia periférica Grau ≥3 nos estudos fase 2.
Hipotensão
Em estudos fase 2 e 3 com agente único, a incidência de hipotensão (postural, ortostática e hipotensão inespecífica) foi de 11 a 12%. Estes eventos são observados ao longo do tratamento. Recomenda-se cautela ao tratar pacientes com história de síncope, pacientes recebendo medicamentos sabidamente associados com hipotensão e pacientes desidratados. A conduta na hipotensão ortostática/postural deve incluir ajuste da medicação anti-hipertensiva, hidratação ou administração de mineralocorticoides e/ou simpatomiméticos.
Alterações Cardíacas
Desenvolvimento agudo ou exacerbação de insuficiência cardíaca congestiva e/ou início de redução da fração de ejeção do ventrículo esquerdo têm sido relatados, incluindo relatos em pacientes com pouco ou nenhum risco de redução da fração de ejeção do ventrículo esquerdo. Pacientes com fatores de risco ou com doença cardíaca preexistente devem ser cuidadosamente monitorados. Em estudo fase 3 com agente único de VELCADE* versus dexametasona, a incidência de qualquer alteração cardíaca que aparecem com o tratamento foi de 15% e 13%, respectivamente. A incidência de eventos de insuficiência cardíaca (edema pulmonar agudo, insuficiência cardíaca, insuficiência cardíaca congestiva, choque cardiogênico e edema de pulmão) foi similar nos grupos de VELCADE* e dexametasona, 5% e 4%, respectivamente. Houve casos isolados de prolongação do intervalo QT em estudos clínicos; a causalidade não foi estabelecida.
Eventos Hepáticos
Têm sido relatados casos raros de insuficiência hepática aguda em pacientes recebendo medicações concomitantes múltiplas e com sérias condições médicas de base. Outros eventos adversos relatados incluem aumento das enzimas hepáticas,
hiperbilirrubinemia e hepatite. Estas alterações podem ser reversíveis com a descontinuação do VELCADE*. Há informações limitadas relacionadas à reexposição nestes pacientes.
Transtorno Pulmonar
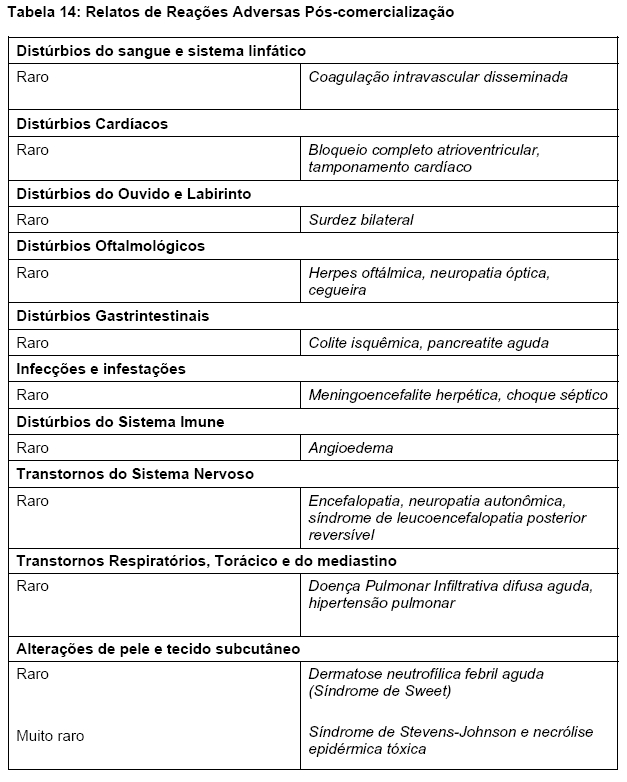
Houve casos raros relatados de doença pulmonar infiltrante difusa aguda de etiologia desconhecida tais como pneumonite, pneumonia intersticial, infiltração pulmonar, Síndrome do Desconforto Respiratório Agudo (ARDS) em pacientes recebendo VELCADE*. Alguns desses eventos têm sido fatais. Uma proporção mais elevada desses efeitos foi relatada no Japão. Na ocorrência de um evento pulmonar ou na piora de sintomas pulmonares já existentes, uma rápida avaliação diagnóstica deve ser realizada e os pacientes tratados apropriadamente.
Num estudo clínico, dois pacientes que receberam altas doses de citarabina (2g/m2 por dia) por infusão contínua com daunorubicina e VELCADE* para recaída de leucemia mieloide aguda morreram com ARDS precocemente durante o tratamento.
Exames laboratoriais
O resultado do hemograma completo deve ser frequentemente monitorado durante o tratamento com VELCADE*.
Trombocitopenia
VELCADE*está associado com trombocitopenia. As plaquetas tiveram seu nível mais baixo no dia 11 de cada ciclo de tratamento com VELCADE* e normalmente recuperaram seu nível basal no próximo ciclo. O padrão cíclico de redução e recuperação da contagem de plaquetas permaneceu consistente ao longo do período de estudo de 8 ciclos de duas doses semanais e não houve evidência de trombocitopenia cumulativa. A média das contagens mais baixas de plaquetas foi aproximadamente 40% da condição basal. A gravidade da trombocitopenia relacionada à contagem de plaquetas antes do tratamento está na Tabela 11 para estudos de fase 3 com agente único. No estudo fase 3 de VELCADE* versus dexametasona, a incidência de eventos de sangramento significativo (≥Grau 3) foi similar em ambos braços VELCADE* (4%) e dexametasona (5%). A contagem de plaquetas deve ser monitorada antes de cada dose de VELCADE*. O tratamento deve ser interrompido quando a contagem de plaquetas for < 25.000/mL e reiniciado com dose reduzida. Existem relatos de hemorragia gastrintestinal e intracerebral associadas com a trombocitopenia induzida por VELCADE*. Transfusão deve ser considerada.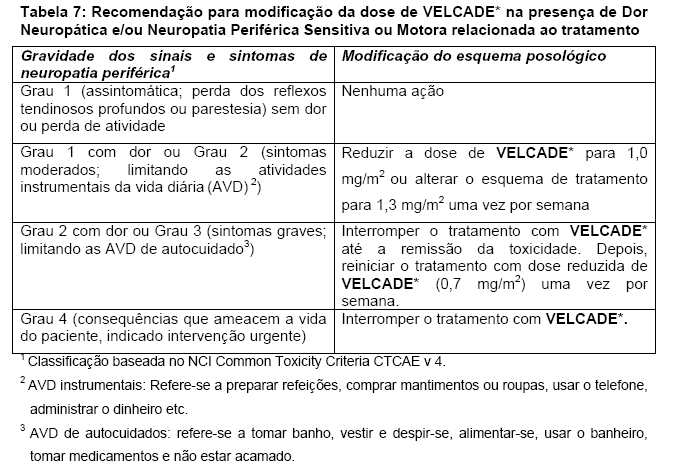
Eventos Adversos Gastrintestinais
O tratamento com VELCADE* pode causar náusea, diarreia, constipação e vômito que exigem, algumas vezes, uso de antieméticos e medicamentos antidiarreicos. A reposição de líquidos e eletrólitos deve ser realizada para evitar a desidratação. Uma vez que alguns pacientes em tratamento com VELCADE* podem apresentar vômito e/ou diarreia, os pacientes devem ser orientados como proceder para evitar a desidratação. Os pacientes devem ser instruídos para procurar o médico se apresentarem sintomas de vertigem, tontura ou desmaios.
Síndrome de Lise Tumoral
Uma vez que VELCADE* é um agente citotóxico e pode matar células malignas rapidamente, as complicações da Síndrome de Lise Tumoral podem ocorrer. Os pacientes sob risco de Síndrome de Lise Tumoral são aqueles com carga tumoral alta antes do tratamento. Estes pacientes devem ser monitorados de perto e as precauções apropriadas devem ser tomadas.
Pacientes com Insuficiência Hepática
O bortezomibe é metabolizado pelas enzimas hepáticas e sua exposição é aumentada em pacientes com insuficiência hepática moderada ou grave. Esses pacientes devem ser tratados com doses iniciais reduzidas de VELCADE* e monitorados com relação à toxicidade.
Síndrome de Leucoencefalopatia Posterior Reversível (SLPR)
Foram relatados casos de Síndrome de Leucoencefalopatia Posterior Reversível (SLPR) em pacientes recebendo VELCADE*. SLPR é um distúrbio neurológico raro, reversível, que pode se apresentar com convulsões, hipertensão, cefaleia, letargia, confusão mental, cegueira, entre outros distúrbios visuais e neurológicos. Exames de imagem do cérebro, preferencialmente RMN (Ressonância Magnética Nuclear) são usados para confirmar o diagnóstico. Em pacientes com SLPR em desenvolvimento, descontinue VELCADE*. A segurança em reiniciar o tratamento com VELCADE* em pacientes com histórico de SLPR não é conhecida.
Carcinogênese, Mutagênese, Prejuízo da Fertilidade
Não foram conduzidos estudos de carcinogenicidade com bortezomibe.
O bortezomibe demonstrou atividade clastogênica (aberrações cromossômicas estruturais) em teste in vitro de aberrações cromossômicas usando células de ovário de hamster Chinês. O bortezomibe não foi genotóxico no teste in vitro de mutagenicidade (teste de Ames) e no teste in vivo de micronúcleos em camundongos.
Não foram realizados estudos de fertilidade com bortezomibe, mas foi realizada avaliação dos tecidos reprodutivos nos estudos de toxicidade geral. No estudo de toxicidade de 6 meses em rato, foram observados efeitos degenerativos no ovário em doses ≥ 0,3 mg/m2 (um quarto da dose clínica recomendada) e alterações degenerativas nos testículos ocorreram com 1,2 mg/m2. VELCADE* pode ter um potencial efeito sobre a fertilidade masculina ou feminina.
Achados de Toxicidade em Animais
Toxicidade Cardiovascular
Estudos em macacos mostraram que a administração de doses aproximadamente o dobro da dose clínica recomendada resultaram em aumento da frequência cardíaca seguido de significante hipotensão progressiva, bradicardia e morte 12-14 horas após a administração. Doses ≥ 1,2 mg/m2 induziram alterações proporcionais à dose nos parâmetros cardíacos. O bortezomibe distribuiu-se para a maioria dos tecidos, incluindo o miocárdio. Em um estudo de toxicidade de dose repetida em macaco também foram observadas hemorragia, inflamação e necrose miocárdica.
Administração Crônica
Em estudos em animais em dose e esquema posológico similar ao recomendado para pacientes (duas vezes por semana, durante duas semanas, seguido de uma semana de descanso), os sinais de toxicidade observados incluíram anemia grave e trombocitopenia, toxicidade gastrintestinal, neurológica e do sistema linfático. Efeitos neurotóxicos em estudos animais incluíram edema axonal e degeneração em nervos periféricos, raízes espinhais dorsais e tratos da medula espinhal. Adicionalmente, hemorragia multifocal e necrose no cérebro, olho e coração foram observadas.
Gravidez (Categoria D) e Lactação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. O médico deverá ser informado imediatamente em caso de suspeita de gravidez.
Mulheres em idade fértil devem evitar a gravidez durante o tratamento com VELCADE*.
O bortezomibe não foi teratogênico em estudos não-clínicos de toxicidade sobre o desenvolvimento em ratos e coelhos na maior dose testada [0,075 mg/kg (0,5 mg/m2) em ratos e 0,05 mg/kg (0,6 mg/m2) em coelhos] quando administrado durante a organogênese. Estas doses são aproximadamente a metade da dose clínica de 1,3 mg/m2 com base na área de superfície corporal.
Coelhas prenhes que receberam 0,05 mg/kg (0,6 mg/m2) de bortezomibe durante a organogênese apresentaram perda pós-implantação significante e número reduzido de fetos vivos. Os fetos vivos destas ninhadas também apresentaram reduções significantes no peso fetal. A dose é aproximadamente metade da dose clínica de 1,3 mg/m2 com base na área de superfície corporal.
Não foram conduzidos estudos de transferência placentária de bortezomibe. Não existem estudos controlados em mulheres grávidas. Se VELCADE* for utilizado durante a gestação ou se a paciente engravidar durante o tratamento, ela deve ser informada sobre o potencial risco para o feto.
As pacientes devem ser orientadas sobre o uso de medidas contraceptivas eficazes e para evitar a amamentação durante o tratamento com VELCADE*.
Lactação
Não existem dados sobre a excreção de bortezomibe no leite humano. Uma vez que muitos fármacos são excretados no leite humano e devido ao potencial para reações adversas graves em lactentes devido a VELCADE*, as mulheres devem ser alertadas para não amamentar durante o tratamento com VELCADE*.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Uma vez que VELCADE* pode estar associado à fadiga, tontura, síncope, hipotensão ortostática/postural, diplopia ou visão turva, os pacientes devem ser orientados para não dirigir veículos ou operar máquinas se houver ocorrência de qualquer destes sintomas.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Não foram observadas diferenças gerais em segurança e efetividade entre pacientes com idade ≥ 65 anos e pacientes mais novos recebendo VELCADE*; entretanto, uma maior sensibilidade de alguns pacientes mais velhos não pode ser afastada.
Interações medicamentosas.
Estudos in vitro e animal ex vivo indicam que o bortezomibe é um inibidor fraco das isoenzimas 1A2, 2C9, 2C19, 2D6 e 3A4 do citocromo P450. Baseado na ação limitada (7%) do CYP2D6 sobre o metabolismo do bortezomibe, não é esperado que o CYP2D6 afete a disposição global do bortezomibe.
Um estudo de interação medicamentosa avaliando o efeito de cetoconazol, potente inibidor do CYP3A4, na farmacocinética de VELCADE* demonstrou um aumento na média de AUC de bortezomibe de 35%, baseado em dados de 12 pacientes. Portanto, os pacientes devem ser monitorados quando ocorrer administração concomitante de bortezomibe com potentes inibidores do CYP3A4 (como por exemplo: cetoconazol e ritonavir).
Em um estudo de interação medicamentosa avaliando o efeito do omeprazol, potente inibidor do CYP2C19, na farmacocinética de VELCADE* não foi demonstrada alteração significativa na farmacocinética de bortezomibe, baseado em dados provenientes de 17 pacientes.
Um estudo de interação medicamentosa avaliando o efeito de rifampicina, um potente indutor do CYP3A4, na farmacocinética de VELCADE* demonstrou uma redução média de AUC de bortezomibe de 45%, baseado nos dados obtidos de 6 pacientes. Portanto, o uso concomitante de VELCADE* com indutores potentes do CYP3A4 não é recomendado, já que a eficácia pode ser reduzida. Exemplos de indutores do CYP3A4 são rifampicina, carbamazepina, fenitoína, fenobarbital e Erva-de-São-João. No mesmo estudo de interação medicamentosa, foi avaliado o efeito de dexametasona, um indutor fraco de CYP3A4. Baseado nos dados obtidos de 7 pacientes, não houve efeito significativo na farmacocinética de bortezomibe.
Um estudo de interação droga-droga avaliando o efeito de melfalano-prednisona sobre VELCADE* mostrou um aumento de 17% na AUC média de bortezomibe baseado em dados de 21 pacientes. Isto não é considerado clinicamente relevante. Pacientes que estão recebendo tratamento concomitante com VELCADE* e fármacos inibidores ou indutores da enzima 3A4 do citocromo P450 devem ser monitorados de perto no que se refere a sinais de toxicidade ou eficácia reduzida. Durante os estudos clínicos, foram relatadas hipoglicemia e hiperglicemia em pacientes diabéticos recebendo hipoglicemiantes orais. Pacientes em tratamento com agentes antidiabéticos orais e que recebem VELCADE* podem necessitar monitoramento da glicemia e ajuste da dose da medicação antidiabética. Os pacientes devem ser orientados sobre o uso de medicações concomitantes que podem estar associadas à neuropatia periférica, tais como amiodarona, antivirais, isoniazida, nitrofurantoína ou estatinas, ou com redução da pressão arterial.
Interações com Exames de Laboratório
Não são conhecidas.
Cuidados de armazenamento.
ARMAZENAGEM
As embalagens de VELCADE* devem ser mantidas em temperatura ambiente (entre 15°C e 30°C), protegidas da luz.
Posologia e modo de usar.
Antes de usar, o conteúdo do frasco-ampola de VELCADE* com 1,0 mg de bortezomibe deve ser diluído com 1 mL de solução salina (0,9%).
O conteúdo do frasco-ampola de VELCADE* com 3,5 mg de bortezomibe deve ser diluído com 3,5 mL de solução salina (0,9%).
A solução resultante deve ser clara e incolor. O medicamento reconstituído pode ser administrado em até 8 horas se estiver a uma temperatura inferior a 25°C, podendo permanecer em uma seringa por até 3 horas nesta mesma temperatura. Não pode ser armazenado a uma temperatura maior que 30°C.
VELCADE* deve ser administrado em injeção intravenosa em bolus (3-5 segundos), através de catéter intravenoso periférico ou central, seguido por lavagem com solução de cloreto de sódio 0,9%.
Ocorreram casos fatais de administração inadvertida de VELCADE* pela via intratecal. VELCADE* deve ser administrado somente pela via intravenosa. VELCADE* NÃO DEVE SER ADMINISTRADO PELA VIA INTRATECAL
POSOLOGIA
Monoterapia
Dose recomendada
A dose recomendada de VELCADE* é de 1,3 mg/m2/dose administrada em 3 a 5 segundos como injeção intravenosa em bolus, 2 vezes por semana durante 2 semanas (dias 1, 4, 8 e 11), seguido por período de repouso de 10 dias (dias 12-21).Este período de 3 semanas é considerado como um ciclo de tratamento. Deve ser observado intervalo de pelo menos 72 horas entre as doses consecutivas de VELCADE*. Para extensão do tratamento além de 8 ciclos, VELCADE* pode ser administrado no esquema padrão ou no esquema de manutenção de uma vez por semana por 4 semanas (Dias 1, 8, 15 e 22), seguido por um período de repouso de 13 dias (Dias 23 a 35).
Em estudos clínicos, pacientes com resposta completa confirmada (RC) receberam 2 ciclos adicionais de VELCADE*. Recomenda-se que pacientes que respondem ao VELCADE* recebam até 8 ciclos de tratamento.
Modificação da Dose e Reinício do Tratamento
O tratamento com VELCADE* deve ser interrompido ao início de qualquer evidência de toxicidade não hematológica de Grau 3 ou hematológica de Grau 4, excluindo neuropatia. Após a remissão dos sintomas de toxicidade, o tratamento com VELCADE* pode ser reiniciado com dose 25% menor (1,3 mg/m2/dose reduzida para 1,0 mg/m2/dose; 1,0 mg/m2/dose reduzida para 0,7 mg/m2/dose). A tabela a seguir contém a recomendação para modificação da dose em pacientes que apresentarem dor neuropática e/ou neuropatia sensitiva periférica relacionada ao VELCADE* (Tabela 7). Pacientes com neuropatia grave preexistente devem ser tratados com VELCADE* somente após avaliação cuidadosa do risco-benefício.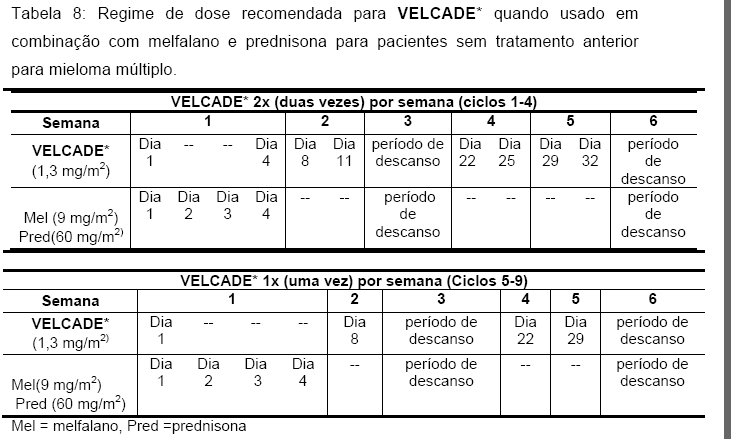
Obs.: A redução da dose de VELCADE*, recomendada quando da ocorrência de dor neuropática e/ou neuropatia sensitiva periférica relacionada ao tratamento, pode levar à redução da eficácia do tratamento.
Terapia combinada
Dose recomendada
VELCADE* (bortezomibe) é administrado em 3 a 5 segundos como injeção intravenosa em bolus, em combinação com melfalano e prednisona, por 9 ciclos de 6-semanas de tratamento. Nos ciclos 1-4,VELCADE* é administrado 2 (duas) vezes por semana (dias 1,4,8,11,22,25,29 e 32). Nos ciclos 5-9, VELCADE* é administrado uma vez por semana (dias 1, 8, 22 e 29).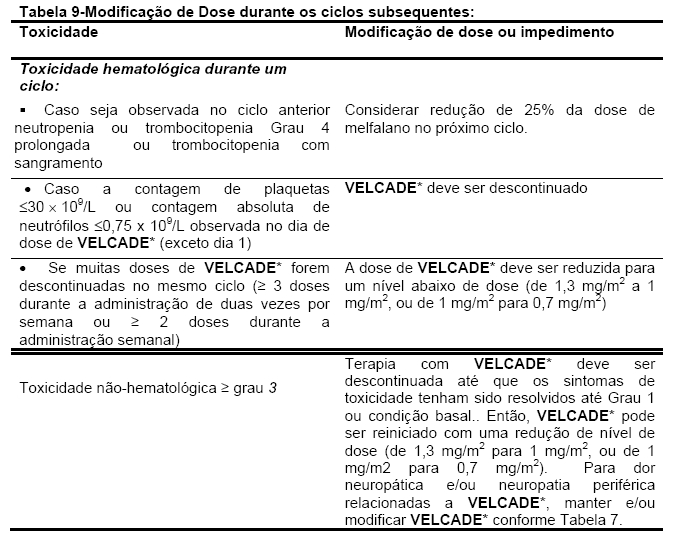
Guia de Gerenciamento de dose para Terapia Combinada
Modificação de dose e reiniciação quando VELCADE* é administrado em combinação com melfalano e prednisona.
Antes de inicar um novo ciclo de terapia:
• Contagem de plaquetas deve ser ≥70 x 109/L e a contagem absoluta de neutrófilos deve ser ≥ 1,0 x 109/L.
• Toxicidade não-hematológica deve ser resolvida até Grau 1 ou condição basal.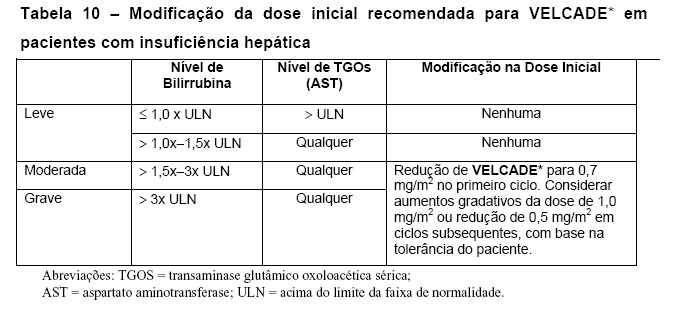
Pacientes com insuficiência renal
A farmacocinética de VELCADE* não é influenciada pela gravidade da insuficiência renal. Desta forma, não é necessário ajuste da dose de VELCADE* em pacientes com insuficiência renal. Uma vez que a diálise pode reduzir a concentração de VELCADE*, o medicamento deve ser administrado após o procedimento de diálise.
Pacientes com insuficiência hepática
Pacientes com insuficiência hepática leve não requerem ajuste de dose inicial e devem ser tratados de acordo com a posologia recomendada de VELCADE*. Pacientes com insuficiência hepática moderada ou grave devem iniciar o tratamento com VELCADE* utilizando uma dose reduzida de 0,7 mg/m2 por injeção durante o primeiro ciclo e subsequentes aumentos gradativos da dose para 1,0 mg/m2 ou reduções de dose para 0,5 mg/m2 podem ser consideradas, com base na tolerância do paciente (veja Tabela 10).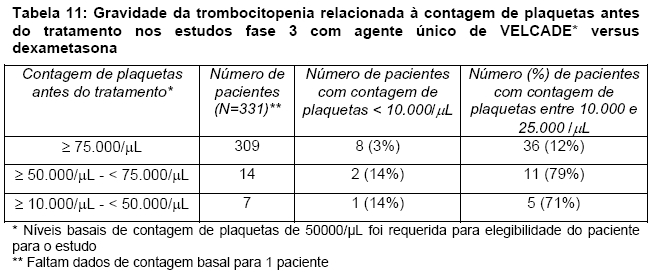
Reações adversas.
Atenção: Este é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis para comercialização, efeitos indesejáveis e não conhecidos podem ocorrer. Neste caso, informe seu médico.
Resumo dos Estudos Clínicos de VELCADE* em pacientes com mieloma múltiplo recidivado/refratário:
A segurança e eficácia de VELCADE* foi avaliada em 3 estudos com a dose recomendada de 1,3 mg/m2, incluindo um estudo fase 3, randomizado, comparativo, versus dexametasona de 669 pacientes com mieloma múltiplo refratário ou recidivo que já haviam recebido de 1 a 3 linhas terapêuticas anteriores (M34101-039); um estudo fase 2, braço único, aberto, multicêntrico com 202 pacientes que haviam recebido pelo menos 2 terapias anteriores e demonstraram progressão da doença na terapia mais recente (M34101-025); e um estudo clínico fase 2, dose-resposta em mieloma múltiplo recidivado em pacientes que tiveram progressão ou recidiva da doença após terapia de primeira linha com VELCADE* 1,0 mg/m2 ou 1,3 mg/m2 (M34100-024).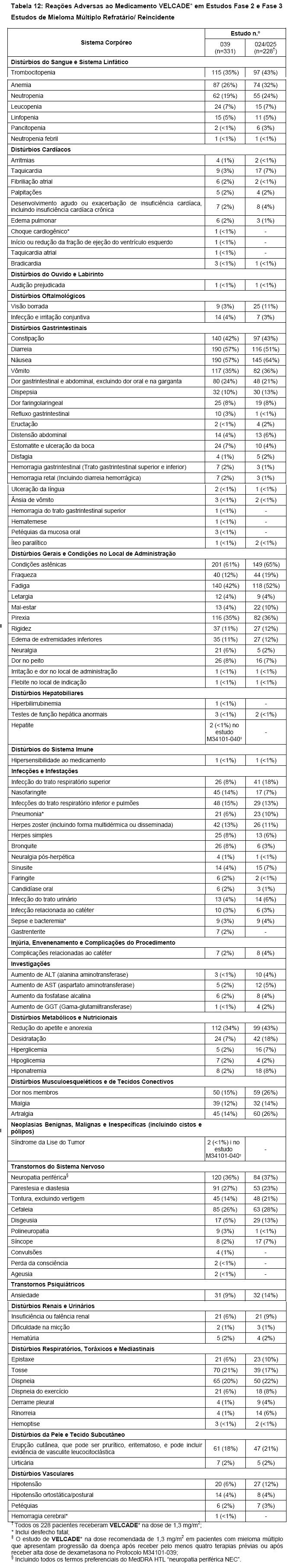
Resumo de Estudos Clínicos em pacientes com mieloma múltiplo sem tratamento anterior:
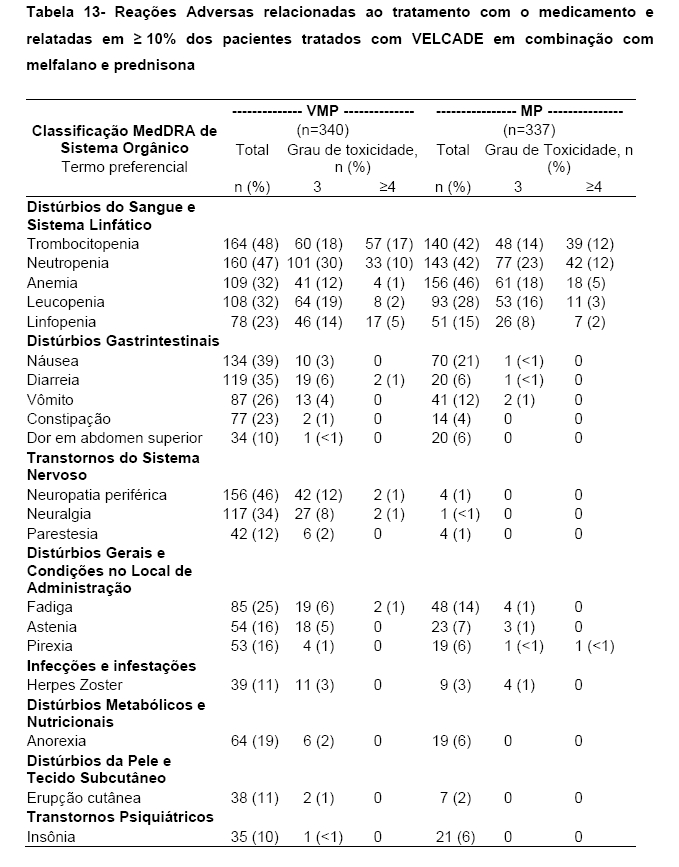
A Tabela 13 a seguir, descreve dados de segurança de 340 pacientes com mieloma múltiplo sem tratamento anterior que receberam VELCADE (1.3 mg/m2) em combinação com melfalano (9 mg/m2<