VEKLURY
GILEAD
rendesivir
Antiviral para o tratamento de COVID-19.
Apresentações.
Veklury® é apresentado na forma farmacêutica de pó liofilizado para solução injetável. Após a reconstituição, a concentração é de 5 mg/mL.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO COM IDADE ≥ 28 DIAS E PESANDO ≥ 3 KG
Composição.
Cada frasco contém 100 mg de rendesivir. Após reconstituição, cada frasco contém 5 mg/mL de solução de rendesivir.
Excipientes: éter sulfobutílico sódico betaciclodextrina, ácido clorídrico, hidróxido de sódio.
Cada frasco-ampola contém 3 g de éter sulfobutílico sódico betaciclodextrina.
Informações técnicas.
1. INDICAÇÕES
Veklury® é indicado para o tratamento da doença causada pelo coronavírus de 2019 (COVID-19) em:
• pacientes adultos e pediátricos com idade ≥ 28 dias e pesando ≥ 3 kg com pneumonia que requerem administração suplementar de oxigênio (oxigênio de baixo ou alto fluxo, ou outra ventilação não invasiva no início do tratamento).
• pacientes adultos e pediátricos pesando ≥ 40 kg que não requerem administração suplementar de oxigênio e que apresentam risco aumentado de progredir para COVID-19 grave.
Para mais informações vide itens 2. Resultados de eficácia e 3. Características farmacológicas.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos em pacientes com COVID-19
Estudo NIAID ACTT-1 (CO-US-540-5776)
Um estudo clínico randomizado, duplo-cego e controlado por placebo avaliou a administração de 200 mg de rendesivir uma vez por dia, no primeiro dia do tratamento, seguido de 100 mg de rendesivir, uma vez por dia, durante um período de até 9 dias (um total de até 10 dias de tratamento administrado por via intravenosa) em pacientes adultos hospitalizados com COVID-19 com evidência de comprometimento do trato respiratório inferior. O estudo incluiu 1.062 pacientes hospitalizados: 159 (15%) pacientes com doença leve/moderada (15% em ambos os grupos de tratamento) e 903 (85%) pacientes com doença grave (85% em ambos os grupos de tratamento). A doença leve/moderada foi definida como SpO2 > 94% e taxa respiratória < 24 incursões/minuto sem administração suplementar de oxigênio; doença grave foi definida como SpO2 ≤ 94% em ar ambiente, uma taxa respiratória ≥ 24 incursões//minuto, e uma necessidade de oxigenação, ou a necessidade de ventilação mecânica. Um total de 285 pacientes (26,8%) (n=131 recebendo rendesivir) estavam em ventilação mecânica ou oxigenação por membrana extracorpórea (ECMO). Os pacientes foram randomizados numa proporção 1:1, estratificados por gravidade da doença no momento da inclusão, para receberem rendesivir (n=541) ou placebo (n=521), mais tratamento padrão.
A idade média no início do estudo era de 59 anos (36% dos pacientes com 65 anos ou mais); 64% dos pacientes eram do sexo masculino; 53% eram caucasianos; 21% eram negros e 13% eram asiáticos. As comorbidades mais frequentes foram hipertensão (51%), obesidade (45%), e diabetes mellitus tipo 2 (31%); a distribuição das comorbidades foi similar entre os dois grupos de tratamento.
Aproximadamente 38,4% (208/541) dos pacientes receberam um ciclo de tratamento de 10 dias com rendesivir.
O desfecho primário foi o tempo de recuperação até 29 dias após a randomização, definida como alta hospitalar (com ou sem limitações da atividade e com ou sem necessidade de oxigênio no domicílio) ou hospitalização, mas sem necessidade de administração suplementar de oxigênio e sem necessitar de assistência médica contínua. A mediana do tempo de recuperação foi de 10 dias no grupo de rendesivir comparativamente com 15 dias no grupo do placebo (razão de taxa de recuperação 1,29 [IC de 95%: 1,12-1,49]; p < 0,001).
Não foi observada diferença no tempo de recuperação entre os pacientes com doença leve/moderada no momento da inclusão do estudo (n=159). A mediana do tempo de recuperação foi de 5 dias no grupo do rendesivir e 7 dias no grupo placebo (razão de taxa de recuperação 1,10 [IC de 95%: 0,8-1,53]); as chances de melhora na escala ordinal no grupo de rendesivir no dia 15, quando comparadas com o grupo placebo, foram as seguintes: razão de probabilidade 1,2; [IC de 95%, 0,7-2, 2, p= 0,562].
Entre os pacientes com doença grave no momento da inclusão do estudo (n=903), a mediana do tempo de recuperação foi de 12 dias no grupo de rendesivir e de 19 dias no grupo do placebo (razão de taxa de recuperação 1,34 [IC de 95%: 1,14-1,58]; p < 0.001; Tabela 1); as chances de melhora na escala ordinal no grupo de rendesivir no Dia 15, quando comparadas com o grupo placebo, foram as seguintes: razão de probabilidade 1,6; [IC de 95%, 1,3-2,0].
No geral, as chances de melhora na escala ordinal foram maiores no grupo rendesivir no dia 15, quando comparadas com o grupo do placebo (razão de probabilidade, 1,6; [IC de 95%, 1,3-1,9], p < 0,001).
A mortalidade no dia 29 na população em geral foi de 11,6% para o grupo de rendesivir e 15,4% para o grupo placebo (razão de risco, 0,73; [IC de 95% 0,52- 1,03; p = 0,07). Uma análise post-hoc da mortalidade no dia 29 pela escala ordinal é relatada na Tabela 1.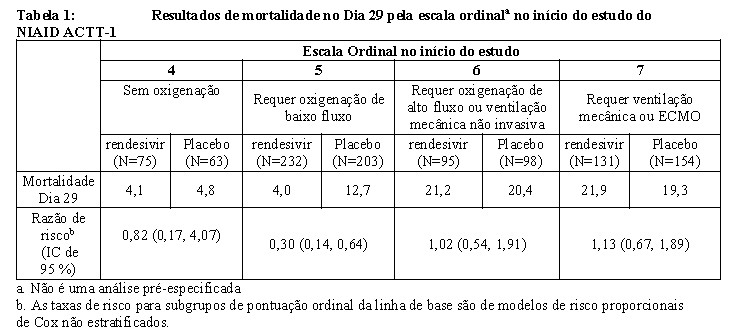
Estudo GS-US-540-5773 em pacientes com COVID-19 grave
Um ensaio clínico multicêntrico, aberto e randomizado (Estudo 5773) em pacientes com, pelo menos, 12 anos de idade com infecção por SARS-CoV-2 confirmada, saturação de oxigênio ≤ 94% em ar ambiente e evidência radiológica de pneumonia, comparou 200 pacientes que receberam rendesivir durante 5 dias com 197 pacientes que receberam rendesivir durante 10 dias. Todos os pacientes receberam 200 mg de rendesivir no dia 1 e 100 mg, uma vez por dia, nos dias consecutivos, além do tratamento padrão. O objetivo primário foi o estado clínico no dia 14 avaliado com uma escala ordinal de 7 pontos, a variar desde alta hospitalar a níveis crescentes de oxigênio e suporte ventilatório até à morte.
A probabilidade de melhoria no dia 14 para os pacientes randomizados para um tratamento de 10 dias com rendesivir, comparativamente com os pacientes randomizados para um tratamento de 5 dias, foi de 0,67 (razão de probabilidade); [IC de 95%: 0,46 1,98]. Foram observados desequilíbrios estatisticamente significativos no estado clínico basal neste estudo. Depois do ajuste das diferenças entre grupos no início do estudo, a probabilidade de melhoria no dia 14 foi de 0,75 (razão de probabilidade); [IC de 95%: 0,51 1,12]. Além disso, não existiram diferenças estatisticamente significativas nas taxas de recuperação ou taxas de mortalidade nos grupos de 5 dias e de 10 dias, após o ajuste das diferenças entre grupos no início do estudo. A mortalidade aos 28 dias por todas as causas foi de 12% vs.14% nos grupos de tratamento de 5 e de 10 dias, respetivamente.
Estudo GS US 540 9012 em pacientes com COVID-19 confirmada e risco aumentado de progressão da doença
Um ensaio clínico randomizado, duplo-cego, controlado com placebo e multicêntrico para avaliar o tratamento com rendesivir em contexto de ambulatório em 562 pacientes incluindo 8 adolescentes (12 anos de idade ou mais e pesando ao menos 40 kg) com COVID-19 confirmada e, pelo menos, um fator de risco de progressão da doença para hospitalização. Os fatores de risco de progressão da doença foram: idade ≥ 60 anos, doença pulmonar crônica, hipertensão, doença cardiovascular ou cerebrovascular, diabetes mellitus, obesidade, estado imunocomprometido, doença renal crônica leve ou moderada, doença hepática crônica, câncer atual ou doença de células falciformes. Os pacientes vacinados foram excluídos do estudo.
Os pacientes tratados com rendesivir receberam 200 mg no dia 1 e 100 mg uma vez por dia nos dias subsequentes durante um total de 3 dias de tratamento administrado por via intravenosa. Os pacientes foram randomizados de um modo 1:1, estratificados por residência em instalações de cuidados especializados (sim/não), idade ( < 60 versus ≥ 60 anos) e região (EUA versus fora dos EUA), para receber rendesivir (n=279; 276 adultos e 3 adolescentes) ou placebo (n=283; 278 adultos e 5 adolescentes), além do tratamento padrão.
No início do estudo, a idade média era de 50 anos (com 30% dos pacientes com idade igual ou superior a 60); 52% eram do sexo masculino, 80% eram caucasianos, 8% eram negros, 2% eram asiáticos, 44% eram hispânicos ou latinos; o índice de massa corporal mediano era de 30,7 kg/m2. As comorbidades mais frequentes eram diabetes mellitus (62%), obesidade (56%) e hipertensão (48%). A duração mediana (Q1, Q3) dos sintomas antes do tratamento era de 5 (3,6) dias; a carga viral mediana era de 6,3 log10 cópias/mL no início do estudo. As características demográficas e da doença no início do estudo estavam equilibradas entre os grupos de tratamento de rendesivir e de placebo. A análise exploratória post-hoc de amostras de biomarcadores opcionais mostrou que 14,8% dos pacientes eram sorologicamente positivos no início do estudo e 37,7% eram sorologicamente negativos (47,5% não consentiram com a coleta opcional de biomarcadores).
O objetivo primário foi a proporção de pacientes com hospitalização relacionada com a COVID-19 (definida como, pelo menos, 24 horas de cuidados agudos) ou mortalidade aos 28 dias por todas as causas. Ocorreram eventos (hospitalização relacionada com a COVID-19 ou mortalidade aos 28 dias por todas as causas) em 2 (0,7%) pacientes tratados com rendesivir, em comparação com 15 (5,3%) pacientes randomizados simultaneamente para o placebo, demonstrando uma redução de 87% na hospitalização relacionada com a COVID-19 ou mortalidade por todas as causas, em comparação com o placebo (razão de risco, 0,134 [IC de 95%: 0,031 a 0,586]; p = 0,0076). A redução de risco absoluto foi de 4,6% (IC de 95%: 1,8% a 7,5%). Não foram observadas mortes ao dia 28 em ambos os grupos (grupos de tratamento de rendesivir e de placebo). Seis dos 17 eventos de hospitalização ocorreram em pacientes com status sorológico basal conhecido (sorológico positivo: n=0 no grupo rendesivir e n=2 no grupo placebo; sorológico negativo: n=2 no grupo rendesivir e n=2 no grupo placebo). Onze dos 17 eventos de hospitalização ocorreram em pacientes com status sorológico inicial desconhecido no grupo placebo e nenhum no grupo rendesivir. Nenhuma conclusão pode ser feita sobre a eficácia nos subgrupos estratificados pelo status sorológico devido ao pequeno número de pacientes com a sorologia conhecida e baixas taxas gerais de eventos.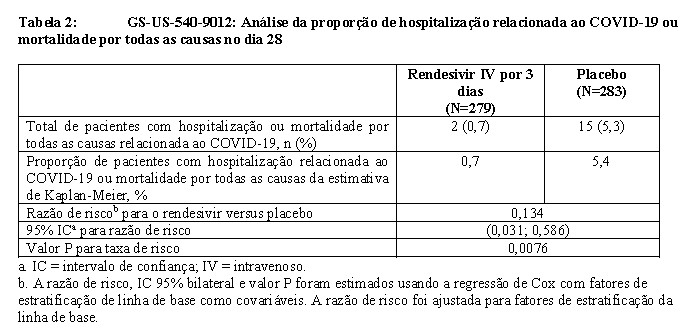
QT
Os atuais dados clínicos e não clínicos não sugerem um risco de prolongamento do intervalo QT, contudo este não foi totalmente estudado nos seres humanos.
População pediátrica
Estudo GS-US-540-5823 em pacientes pediátricos hospitalizados com COVID-19
Os objetivos primários e secundários deste estudo clínico aberto de fase 2/3 de braço único (Estudo GS-US-540-5823) foram avaliar a farmacocinética e segurança, e eficácia, respetivamente, por até 10 dias de tratamento com rendesivir em pacientes pediátricos. Um total de 53 pacientes pediátricos com pelo menos 28 dias de idade e pesando pelo menos 3 kg com infecção confirmada por SARS-CoV-2 e COVID-19 leve, moderada ou grave foi avaliada em cinco coortes: pacientes ≥ 12 anos e pesando ≥ 40 kg (n=12); pacientes < 12 anos e pesando ≥ 40 kg (n=5); pacientes ≥ 28 dias e pesando ≥ 20 a < 40 kg (n=12); pacientes ≥ 28 dias e pesando ≥ 12 a < 20 kg (n=12); e pacientes ≥ 28 dias e pesando ≥ 3 a < 12 kg (n=12). Pacientes pesando ≥ 40 kg receberam 200 mg de rendesivir no Dia 1, seguido de 100 mg de rendesivir uma vez ao dia nos dias subsequentes; pacientes com peso ≥ 3 kg a < 40 kg receberam rendesivir 5 mg/kg no Dia 1 seguido de rendesivir 2,5 mg/kg uma vez ao dia nos dias subsequentes. As avaliações ocorreram nos seguintes intervalos: Triagem; Dia 1 (início do estudo); Dias 2-10, ou até a alta, o que ocorrer primeiro; Acompanhamento no Dia 30 (±5). O tratamento com rendesivir foi interrompido em indivíduos que receberam alta do hospital antes da conclusão de 10 dias de tratamento.
No início do estudo, a idade mediana era de 7 anos (Q1, Q3: 2 anos, 12 anos); 57% eram do sexo feminino, 70% eram brancos, 30% eram negros, e 44% eram hispânicos ou latinos; peso médio foi de 25 kg (variação: 4 a 192 kg). Os pacientes neste estudo não foram vacinados. Um total de 12 pacientes (23%) estava em ventilação mecânica invasiva, 18 (34%) em ventilação não invasiva ou oxigênio de alto fluxo; 10 (19%) estavam em baixo fluxo de oxigênio; e 13 (25%) estavam em ar ambiente, no início do estudo. A duração mediana geral (Q1, Q3) dos sintomas e da hospitalização antes da primeira dose de rendesivir foi de 5 (3, 7) dias e 1 (1, 3) dia, respetivamente.
As análises descritivas dos resultados mostraram que o tratamento com rendesivir por até 10 dias resultou em uma mudança geral mediana (Q1, Q3) da linha de base no estado clínico (avaliado em uma escala ordinal de 7 pontos variando de morte [escore de 1] a suporte ventilatório e diminuindo os níveis de oxigênio até a alta hospitalar [pontuação de 7]) de +2,0 (1,0; 4,0) pontos no Dia 10.
A recuperação (definida como uma melhora de uma pontuação do estado clínico inicial de 2 a 5 para uma pontuação de 6 ou 7, ou uma melhora de uma pontuação inicial de 6 para uma pontuação de 7) foi relatada para 62% dos pacientes no Dia 10; a mediana (Q1, Q3) do tempo de recuperação foi de 7 (5, 16) dias.
No geral, 60% dos pacientes receberam alta no Dia 10 e 83% dos pacientes receberam alta no Dia 30. Três pacientes (6%) morreram durante o estudo.
Ver itens 3. Características farmacológicas e 8. Posologia e modo de usar, para informação sobre uso na população pediátrica.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Antivirais para uso sistémico, antivirais de ação direta, outros antivirais, código ATC: J05AB16.
Mecanismo de ação
O rendesivir é um pró-fármaco nucleotídeo que é metabolizado nas células hospedeiras para formar o metabólito nucleosídeo trifosfato farmacologicamente ativo. O trifosfato de rendesivir atua como análogo da adenosina trifosfato (ATP) e compete com o substrato natural de ATP pela incorporação nas cadeias de RNA nascentes pela RNA polimerase dependente do RNA do SARS-CoV-2, o que resulta na terminação prematura da cadeia durante a replicação do RNA viral.
Como mecanismo adicional, o trifosfato de rendesivir também pode inibir a síntese do RNA viral após a sua incorporação no molde de RNA viral, como resultado da continuação da leitura pela polimerase viral que pode ocorrer na presença de concentrações de nucleotídeos mais elevadas. Quando o nucleotídeo de rendesivir está presente no molde de RNA viral, a eficiência de incorporação do nucleotídeo natural complementar fica comprometida, inibindo assim a síntese de RNA viral.
Atividade antiviral
O rendesivir exibiu atividade in vitro contra um isolado clínico de SARS-CoV-2 em células epiteliais primárias das vias respiratórias humanas com uma concentração eficaz de 50% (EC50) de 9,9 nM após 48 horas de tratamento. Rendesivir inibiu a replicação de SARS-CoV-2 nas linhas celulares epiteliais de pulmão humano contínuas Calu-3 e A549-hACE2 com valores de EC50 de 280 nM após 72 horas de tratamento e 115 nM após 48 horas de tratamento, respetivamente. Os valores de EC50 de rendesivir contra o SARS-CoV-2 em células Vero foi de 137 nM às 24 horas e de 750 nM às 48 horas pós-tratamento. A atividade antiviral de rendesivir foi antagonizada por fosfato de cloroquina de forma dose-dependente quando os dois medicamentos foram co-incubados em concentrações clinicamente relevantes em células HEp-2 infetadas com o vírus sincicial respiratório (respiratory syncytial virus, VSR). Foram observados valores de EC50 de rendesivir mais elevados com o aumento das concentrações de fosfato de cloroquina. O aumento das concentrações de fosfato de cloroquina diminuiu a formação de trifosfato de rendesivir em A549-hACE2, HEp-2 e células epiteliais brônquicas humanas normais.
Com base em testes in vitro, o rendesivir manteve atividade antiviral semelhante (alteração < 2,5 vezes) contra isolados clínicos de variantes de SARS-CoV-2 contendo a substituição P323L na polimerase viral, incluindo as variantes Alfa (B.1.1.7), Beta (B. 1.351), Gamma (P.1), Epsilon (B.1.429), Kappa (B.1.617.1), Lambda (C.37), Iota (B.1.526) e Zeta (P.2) em comparação com isolados das linhagens anteriores de SARS-CoV-2 (linhagem A). Para os isolados clínicos das variantes Delta (B.1.617.2) e Omicron (B.1.1.529), o rendesivir manteve a atividade antiviral (alteração < 0,6 vezes). A atividade antiviral do rendesivir contra as variantes do SARS-CoV-2 é apresentada na Tabela 3.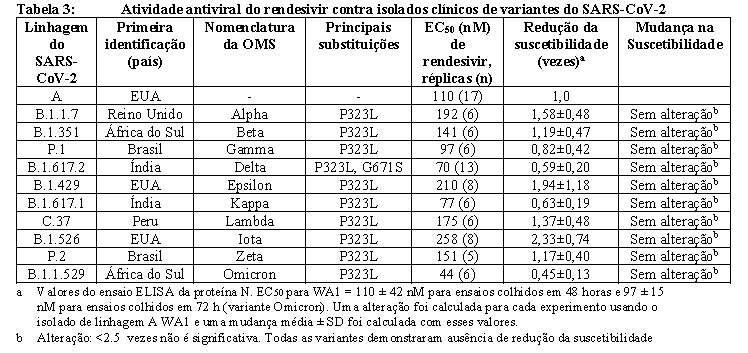
Resistência
Em cultura celular
Foram selecionados isolados de SARS-CoV-2 com suscetibilidade reduzida ao rendesivir em cultura celular. Numa seleção com GS-441524, o nucleosídeo precursor de rendesivir, surgiram pools de vírus expressando combinações de substituições de aminoácidos em V166A, N198S, S759A, V792I, C799F e C799R na RNA polimerase dependente de RNA viral, conferindo alterações da EC50 de 2,7 a 10,4 vezes. Quando introduzidas individualmente num vírus recombinante selvagem através de mutagênese direcionada no local, foi observada uma suscetibilidade 1,7 a 3,5 vezes menor ao rendesivir. Numa segunda seleção com rendesivir, utilizando um isolado de SARS-CoV-2 que contém a substituição P323L na polimerase viral, surgiu uma única substituição de aminoácido em V166L. Os vírus recombinantes com substituições em P323L apenas ou P323L+V166L em combinação exibiram alterações de 1,3 e 1,5 vezes na suscetibilidade ao rendesivir, respetivamente.
A determinação do perfil de resistência a rendesivir em culturas celulares utilizando o vírus responsável pela hepatite murina CoV de roedores identificou duas substituições (F476L e V553L) na RNA polimerase dependente de RNA viral em resíduos conservados em todos os CoVs que conferiram uma suscetibilidade 5,6 vezes menor ao rendesivir. A introdução das substituições correspondentes (F480L e V557L) no SARS-CoV resultou numa suscetibilidade 6 vezes menor ao rendesivir em culturas celulares e atenuou a patogênese do SARS-CoV num modelo de camundongo. Quando introduzidas individualmente num vírus recombinante de SARS-CoV-2, as substituições correspondentes em F480L e V557L conferiram, cada uma delas, uma suscetibilidade 2 vezes menor ao rendesivir.
Em Ensaios Clínicos
No Estudo GS-US-540-5823, entre os pacientes com dados de sequenciamento na linha de base e pós-linha de base disponíveis, foram observadas substituições na RNA polimerase (A656P e G670V) dependente de RNA viral em um dos 23 pacientes tratados com rendesivir. As substituições observadas não foram associadas à resistência ao rendesivir.
Propriedades farmacocinéticas
As propriedades farmacocinéticas de rendesivir foram estudadas em voluntários saudáveis. Não há dados farmacocinéticos disponíveis de pacientes com COVID-19.
Absorção
As propriedades farmacocinéticas de rendesivir e do metabólito circulante predominante GS-441524 foram avaliadas em indivíduos adultos saudáveis. Após a administração intravenosa do regime posológico de rendesivir para adultos, foi observada uma concentração plasmática máxima no final da infusão intravenosa, independentemente do nível da dose, que diminuiu de maneira rápida posteriormente com uma meia-vida aproximada de 1 hora. Foram observadas concentrações plasmáticas máximas de GS-441524 entre 1,5 e 2,0 horas após o início de uma infusão intravenosa de 30 minutos.
Distribuição
O rendesivir liga-se aproximadamente em 93% às proteínas plasmáticas humanas (dados ex-vivo) com a fração livre variando entre 6,4% e 7,4%. A ligação é independente da concentração do fármaco no intervalo de 1 a 10 mM, sem evidência de saturação da ligação de rendesivir. Após uma dose única de 150 mg de [14C]-rendesivir em indivíduos saudáveis, a razão da radioatividade de 14C entre o sangue e o plasma foi de aproximadamente 0,68 aos 15 minutos após o início da infusão intravenosa, tendo aumentado ao longo do tempo alcançando uma razão de 1,0 após 5 horas, indicando uma distribuição diferencial de rendesivir e dos respetivos metabólitos no plasma ou para componentes celulares do sangue.
Biotransformação
O rendesivir é extensivamente metabolizado gerando o análogo nucleosídeo trifosfato farmacologicamente ativo GS-443902 (formado no interior das células). A via de ativação metabólica envolve hidrólise por esterases, que originam a formação do metabólito intermediário GS-704277. A clivagem do fosforamidato seguida de fosforilação forma o trifosfato ativo GS-443902. A desfosforilação de todos os metabólitos fosforilados pode resultar na formação do metabolito nucleosídeo GS-441524 que não é refosforilado de modo eficiente. O estudo do balanço de massa em seres humanos também indica a presença de um metabólito principal atualmente não identificado (M27) no plasma.
Eliminação
Após uma dose única IV de 150 mg de [14C] -rendesivir, a recuperação total média da dose foi de 92%, composta por aproximadamente 74% e 18% recuperada na urina e nas fezes, respetivamente. A maior parte da dose de rendesivir recuperada na urina foi na forma de GS-441524 (49%), enquanto 10% foi recuperado na forma de rendesivir. Estes dados indicam que a depuração renal é a principal via de eliminação de GS-441524. As meias-vidas terminais medianas de rendesivir e GS-441524 foram de aproximadamente 1 e 27 horas, respetivamente.
Farmacocinética em populações especiais
Sexo, raça e idade
Diferenças farmacocinéticas baseadas no sexo, raça, idade, função renal e função hepática nas exposições de rendesivir foram avaliadas usando análise farmacocinética populacional. Sexo e raça não afetaram a farmacocinética do rendesivir e seus metabólitos (GS-704277 e GS-441524).
Pacientes pediátricos
Modelos farmacocinéticos populacionais para o rendesivir e seus metabólitos circulantes (GS-704277 e GS-441524), desenvolvidos usando dados agrupados de estudos em indivíduos saudáveis e em pacientes adultos e pediátricos com COVID-19, foram usados para estimar exposições farmacocinéticas em pacientes pediátricos com idade ≥ 28 dias a < 18 anos e pesando ≥ 3 kg (Estudo 5823). A média geométrica das exposições estimadas (AUCtau, Cmax e Ctau) para esses pacientes nas doses administradas foram maiores para o rendesivir (33% a 129%), GS-441524 (0% a 60%) e GS-704277 (37% a 124%) em comparação com pacientes adultos com COVID-19; entretanto, os aumentos não foram considerados clinicamente significativos.
Os parâmetros farmacocinéticos de dose múltipla de rendesivir e metabólitos em pacientes pediátricos com COVID-19 são fornecidos na Tabela 4.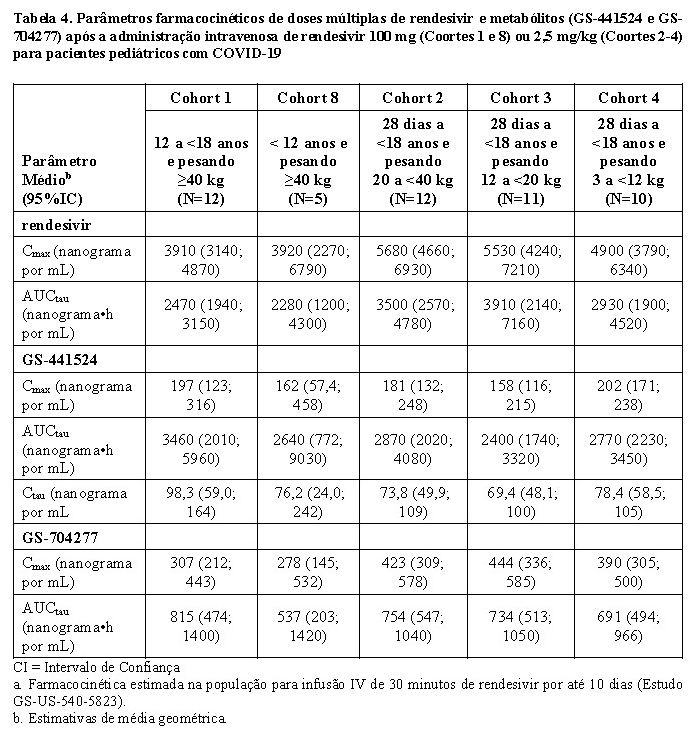
Comprometimento renal
A farmacocinética de rendesivir e GS-441524 em pacientes com comprometimento renal não foi avaliada. O rendesivir não é eliminado de forma inalterada na urina de maneira substancial, mas o seu principal metabólito GS-441524 é eliminado por via renal e os níveis de metabólito no plasma podem teoricamente aumentar em pacientes com função renal diminuída. O excipiente éter sulfobutílico sódico betaciclodextrina é eliminado por via renal e acumula-se em pacientes com função renal diminuída. Veklury® não deve ser utilizado em pacientes com Taxa de Filtração Glomerular TFGe < 30 mL/min.
Comprometimento hepático
A farmacocinética de rendesivir e GS-441524 em pacientes com comprometimento hepático não foi avaliada. Desconhece-se a função do fígado no metabolismo de rendesivir.
Interações
O potencial de interação de rendesivir como objeto da ação não foi estudado em relação à inibição da via hidrolítica (esterase). O risco de interação clinicamente relevante é desconhecido.
O rendesivir inibiu CYP3A4 in vitro (ver item 6. Interações medicamentosas). Em concentrações fisiologicamente relevantes (estado estacionário), rendesivir ou os seus metabólitos GS-441524 e GS-704277 não inibiram CYP1A2, 2B6, 2C8, 2C9, 2C19 e 2D6 in vitro. Contudo, rendesivir pode inibir transitoriamente o CYP2B6, 2C8, 2C9 e 2D6 no primeiro dia de administração. A relevância clínica desta inibição não foi estudada. O potencial de inibição tempo-dependente das enzimas CYP450 pelo rendesivir não foi estudado.
O rendesivir induziu o CYP1A2 e potencialmente o CYP3A4, mas não o CYP2B6 in vitro (ver item 6. Interações medicamentosas).
Os dados in vitro não indicam qualquer inibição clinicamente relevante de UGT1A1, 1A3, 1A4, 1A6, 1A9 ou 2B7 pelo rendesivir ou os seus metabolitos GS-441524 e GS-704277.
O rendesivir inibiu OATP1B1 e OATP1B3 in vitro (ver item 6. Interações medicamentosas). Não há dados disponíveis sobre a inibição de OAT1, OAT3 ou OCT2 pelo rendesivir.
Em concentrações fisiologicamente relevantes, rendesivir e os seus metabólitos não inibiram a gp-P nem BCRP in vitro.
Dados de segurança pré-clínica
Toxicologia
Após a administração intravenosa (in bolus lento) de rendesivir em macacos-rhesus e ratos, ocorreu toxicidade renal grave após curtas durações de tratamento. Em macacos-rhesus macho, a administração em níveis posológicos de 5, 10 e 20 mg/kg/dia durante 7 dias resultou, em todos os níveis de dose, no aumento médio do nitrogênio ureico (uréia) e no aumento médio da creatinina, atrofia tubular renal, basofilia e cilindros, bem como uma morte não planejada de um animal ao nível de dose 20 mg/kg/dia. Em ratos, a administração a níveis posológicos > 3 mg/kg/dia durante períodos até 4 semanas resultou em evidências indicativas de lesão e/ou disfunção renal. As exposições sistémicas (AUC) ao metabolito de rendesivir circulante predominante (GS-441524) foram 0,1 vezes (macacos a 5 mg/kg/dia) e 0,3 vezes (ratos a 3 mg/kg/dia) a exposição em seres humanos após a administração intravenosa com a dose humana recomendada (DHR). Foi demonstrada a presença de um metabólito principal não identificado (M27) no plasma humano ((ver item 3. Características farmacológicas). A exposição de M27 em macacos-rhesus e ratos não é conhecida. Portanto, é possível que os estudos em animais não forneçam informações sobre os potenciais riscos associados a este metabólito.
Carcinogênese
Não foram realizados estudos em animais a longo prazo para avaliar o potencial carcinogênico de rendesivir.
Mutagênese
O rendesivir não foi genotóxico numa bateria de ensaios, incluindo ensaios de mutagenicidade bacteriana, ensaios de aberração cromossômica utilizando linfócitos de sangue periférico humano e ensaios de micronúcleos em ratos in vivo.
Toxicidade reprodutiva
Foram observadas diminuições nos corpos-lúteos, nos números de locais de implantação e nos embriões viáveis, quando o rendesivir foi administrado diariamente por via intravenosa a uma dose tóxica sistemicamente (10 mg/kg/dia) em ratos fêmea 14 dias antes do acasalamento e durante a concepção; as exposições ao metabólito circulante predominante (GS-441524) foram 1,3 vezes a exposição em seres humanos com a DHR. Não houve quaisquer efeitos sobre o desempenho reprodutor feminino (acasalamento, fertilidade e concepção) com este nível de dose.
Em ratos e coelhos fêmea, rendesivir não demonstrou efeitos adversos no desenvolvimento embrionário ou fetal quando administrado em animais durante a gravidez em exposições sistémicas (AUC) ao metabolito de rendesivir circulante predominante (GS-441524) que foram até 4 vezes a exposição em seres humanos com a DHR.
Em ratos fêmea, não ocorreram efeitos adversos no desenvolvimento pré e pós-natal em exposições sistêmicas (AUC) ao metabólito de rendesivir circulante predominante (GS-441524) que foram semelhantes à exposição em seres humanos com a DHR.
Desconhece-se se o análogo nucleosídeo trifosfato ativo GS-443902 e o metabólito principal humano não identificado M27 se formam em ratos e coelhos. Portanto, é possível que os estudos de toxicidade reprodutiva não forneçam informações sobre os potenciais riscos associados a estes metabólitos.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em pacientes com histórico de hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados no item Composição.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipersensibilidade, incluindo reações relacionadas à infusão e reações anafiláticas
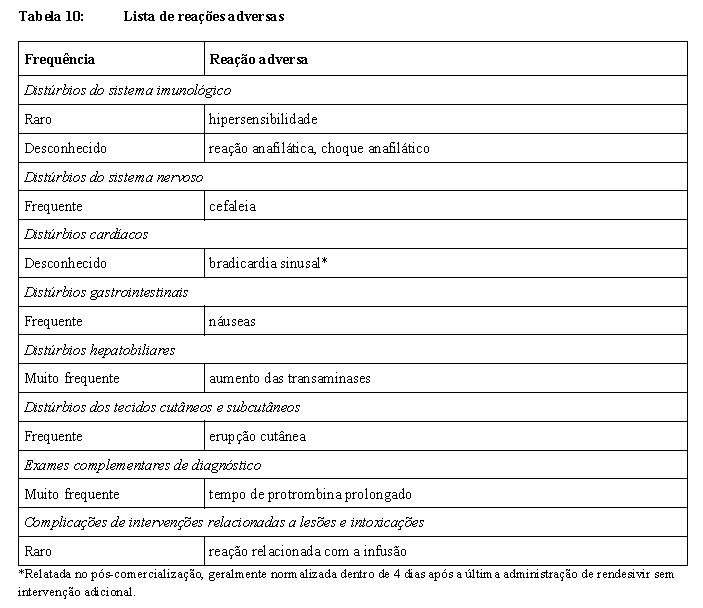
Foram observadas reações de hipersensibilidade, incluindo reações relacionadas à infusão e reações anafiláticas, durante e após a administração de rendesivir. Os sinais e sintomas podem incluir hipotensão, hipertensão, taquicardia, bradicardia, hipoxia, febre, dispneia, chiado no peito, angioedema, erupção cutânea, náuseas, vômitos, sudorese e calafrios. Taxas de infusão mais lentas, mantendo um tempo máximo de infusão de até 120 minutos, podem ser consideradas para prevenir potencialmente esses sinais e sintomas. Monitore a ocorrência de reações de hipersensibilidade nos pacientes durante e após a administração do rendesivir conforme clinicamente apropriado. Os pacientes que recebem rendesivir em contexto de ambulatório devem ser monitorados após a administração de acordo com a prática médica local. Rendesivir só pode ser administrado em ambientes em que os profissionais de saúde tenham acesso imediato aos medicamentos para tratar uma reação grave ou reação de hipersensibilidade, como anafilaxia, e a capacidade de ativar o sistema médico de emergência, conforme necessário. Caso ocorram sinais e sintomas de uma reação de hipersensibilidade clinicamente significativa, descontinue imediatamente a administração de rendesivir e inicie o tratamento apropriado.
Elevação das transaminases
Foram observadas elevações das transaminases nos ensaios clínicos com rendesivir, incluindo em voluntários saudáveis e pacientes com COVID-19. A função hepática deve ser determinada em todos os pacientes (incluindo pacientes ambulatoriais) antes de iniciar o tratamento com Veklury®, devendo também ser monitorada durante a administração conforme clinicamente apropriado. Não foram realizados estudos clínicos com rendesivir em pacientes com insuficiência hepática. Veklury®apenas deve ser utilizado em pacientes com insuficiência hepática se o potencial benefício superar o potencial risco.
• Veklury®não deve ser iniciado em pacientes com alanina aminotransferase (ALT) inicial ≥ 5 vezes o limite superior da normalidade (LSN).
• Veklury®deve ser descontinuado em pacientes que desenvolvam:
- ALT ≥ 5 vezes o LSN durante o tratamento com rendesivir. Veklury® poderá ser reiniciado quando a ALT for < 5 vezes o LSN.
OU
- Elevação da ALT acompanhada por sinais ou sintomas de inflamação hepática ou aumento da bilirrubina conjugada, da fosfatase alcalina ou da relação normalizada internacional (international normalised ratio, INR) (ver itens 3. Características farmacológicas e 9. Reações adversas).
Insuficiência renal
Em estudos em animais com ratos e macacos, foi observada toxicidade renal grave (ver item 3. Características farmacológicas). O mecanismo desta toxicidade renal não é totalmente compreendido. A relevância para os seres humanos não pode ser excluída.
A TFGe deve ser determinada em todos os pacientes (incluindo pacientes ambulatoriais) antes de se iniciar o tratamento com Veklury® e enquanto estiverem utilizando o rendesivir, conforme clinicamente apropriado. Veklury® não deve ser administrado em pacientes com TFGe < 30 ml/min.
Pacientes imunocomprometidos
Não é claro se a duração do tratamento de três dias é suficiente para eliminar o vírus em pacientes imunocomprometidos, nos quais ocorre uma produção viral prolongada. Existe um potencial risco de desenvolvimento de resistência. Estão disponíveis apenas dados limitados.
Excipientes
Veklury® contém éter sulfobutílico sódico betaciclodextrina, que é eliminado por via renal e se acumula em pacientes com função renal diminuída, o que poderá potencialmente afetar de forma adversa a função renal. Portanto, Veklury® não deve ser administrado em pacientes com TFGe < 30 mL/min (ver itens 3. Características farmacológicas e 8. Posologia e modo de usar).
Risco de redução da atividade antiviral quando coadministrado com cloroquina ou hidroxicloroquina
Não é recomendada a coadministração de rendesivir e fosfato de cloroquina ou sulfato de hidroxicloroquina com base em dados in vitro que demonstram um efeito antagonista da cloroquina na ativação metabólica intracelular e na atividade antiviral de rendesivir (ver itens 3. Características farmacológicas e 6. Interações medicamentosas).
Uso em Populações Especiais
Gravidez
Não existem dados ou os dados existentes sobre a utilização de rendesivir em mulheres grávidas são limitados. Os estudos com animais são insuficientes em relação à toxicidade reprodutiva (ver item 3. Características farmacológicas). Veklury® não deve ser utilizado durante a gravidez a não ser que o estado clínico da mulher justifique o tratamento com rendesivir.
As mulheres com potencial para engravidar devem utilizar métodos
Categoria B de risco na gravidez: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Amamentação
Desconhece-se se rendesivir é excretado no leite humano, ou quais os efeitos no lactente, ou os efeitos na produção de leite.
Em estudos com animais, o metabólito análogo de nucleosídeo GS-441524 foi detectado no sangue de filhotes de ratos amamentados por mães às quais se administrou rendesivir. Portanto, pode presumir-se a excreção de rendesivir e/ou metabolitos no leite de animais lactantes.
Devido ao potencial de transmissão viral a lactentes negativos para SARS-CoV-2 e às reações adversas do medicamento em lactentes, deve-se tomar uma decisão sobre interromper a amamentação ou descontinuar a terapia com rendesivir, considerando o benefício da amamentação para a criança e o benefício da terapia para a mulher.
Fertilidade
Não estão disponíveis dados em seres humanos sobre o efeito de rendesivir na fertilidade. Em ratos macho, não se observou qualquer efeito do tratamento com rendesivir no acasalamento ou na fertilidade. Contudo, em ratos fêmea observou-se um comprometimento na fertilidade (ver item 3. Características farmacológicas). Desconhece-se a relevância para os seres humanos.
Efeitos sobre a capacidade de conduzir e utilizar máquinas
Os efeitos de Veklury®sobre a capacidade de conduzir e utilizar máquinas são nulos ou desprezáveis.
Veklury® contém uma ciclodextrina
Este medicamento contém 3 g de éter sulfobutílico sódico betaciclodextrina em cada dose de 100 mg de Veklury® (6 g na dose inicial). Este componente é uma ciclodextrina emulsionante que ajuda o medicamento a dispersar-se pelo corpo.
Atenção diabéticos, contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos clínicos de interação com rendesivir. O potencial global de interações é atualmente desconhecido; os pacientes devem permanecer sob observação atenta durante os dias de administração de rendesivir. Devido ao antagonismo observado in vitro, a utilização concomitante de rendesivir com fosfato de cloroquina ou sulfato de hidroxicloroquina não é recomendada.
Efeitos de outros medicamentos no rendesivir
In vitro, rendesivir é um substrato de esterases em plasma e tecido, enzimas metabolizadoras de fármacos CYP2C8, CYP2D6 e CYP3A4, sendo também substrato dos transportadores polipeptídios de ânions orgânicos 1B1 (organic anion transporting polypeptides 1B1, OATP1B1) e dos transportadores da glicoproteína P (gp-P).
O potencial de interação de rendesivir com inibidores/indutores da via hidrolítica (esterase) ou de CYP2C8, 2D6 ou 3A4 não foi estudado. Desconhece-se o risco de interação clinicamente relevante. Os inibidores potentes podem resultar numa maior exposição a rendesivir. A utilização de indutores potentes (p. ex. rifampicina) pode diminuir a concentração plasmática de rendesivir e não é recomendada.
A dexametasona está descrita como um indutor moderado de CYP3A e gp-P. A indução depende da dose e ocorre após várias doses. Não é provável que a dexametasona tenha um efeito clinicamente significativo no rendesivir na medida em que rendesivir tem uma razão de extração hepática moderada-alta e é utilizado por um curto período no tratamento da COVID-19.
Efeitos de rendesivir em outros medicamentos
In vitro, rendesivir é um inibidor de CYP3A4, OATP1B1 e OATP1B3. A relevância clínica destas interações medicamentosas in vitro não foi estabelecida. O rendesivir poderá aumentar transitoriamente a concentração plasmática de medicamentos que são substratos de CYP3A ou OATP 1B1/1B3. Não há dados disponíveis. Contudo, pode sugerir-se que os medicamentos que são substratos de CYP3A4 ou substratos de OATP 1B1/1B3 devam ser administrados pelo menos 2 horas após rendesivir. O rendesivir induziu CYP1A2 e potencialmente CYP3A in vitro. A coadministração de rendesivir com substratos de CYP1A2 ou CYP3A4 com um índice terapêutico estreito pode levar à perda da sua eficácia.
A dexametasona é um substrato de CYP3A4 e, embora rendesivir iniba CYP3A4, devido à rápida depuração de rendesivir após a administração IV, não é provável que rendesivir tenha um efeito significativo na exposição a dexametasona.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Veklury® é um pó branco esbranquiçado a amarelo.
Armazenar em temperatura ambiente, entre 15°C e 30°C.
Solução reconstituída e diluída para infusão.
Após preparo, conservar a solução diluída para infusão de rendesivir por até 24 horas em temperatura ambiente (20°C a 25°C) ou até 48 horas sob refrigeração (2°C a 8°C).
Prazo de validade: 36 meses após a data de fabricação impressa na embalagem.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Os pacientes devem ser monitorados durante o tratamento com rendesivir (ver item 5. Advertências e Precauções).
Os pacientes que recebem rendesivir em contexto de ambulatório devem ser monitorados de acordo com a prática médica local. Utilizar sob condições nas quais o tratamento de reações de hipersensibilidade graves, incluindo anafilaxia, seja possível.
Realizar testes laboratoriais hepáticos e renais (TFGe) em todos os pacientes (incluindo pacientes ambulatoriais) antes ou ao iniciar o rendesivir, conforme clinicamente apropriado.
Determinar o tempo de protrombina em todos os pacientes (incluindo pacientes ambulatoriais) antes ou ao iniciar o rendesivir e monitorar enquanto estiver recebendo rendesivir, conforme clinicamente apropriado (ver seção 9. Reações Adversas).
Posologia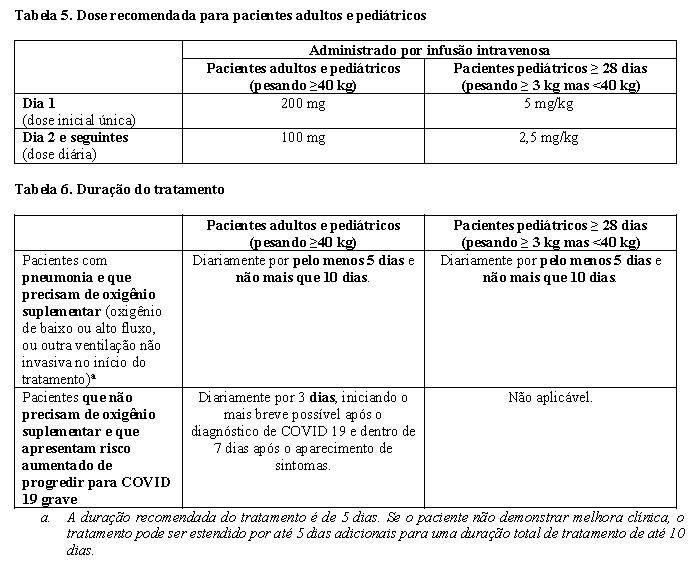
Populações especiais
Idosos
Não é necessário qualquer ajuste de dose de rendesivir em pacientes com mais de 65 anos (ver item 3. Características farmacológicas).