VABYSMO
ROCHE
faricimabe
Tratamento da degeneração macular (DMRIn).
Apresentações.
Solução injetável 120 mg/ml
Cada cartucho contém 1 frasco-ampola com 0,24 ml de solução e 1 agulha com filtro para retirada do conteúdo do frasco.
VIA INTRAVÍTREA
USO ADULTO
Composição.
Princípio ativo: cada frasco-ampola contém 28,8 mg de faricimabe em 0,24 ml de solução. Isso fornece uma quantidade utilizável para administrar uma dose única de 0,05 ml que contém 6 mg de faricimabe.
Excipientes: histidina, ácido acético, metionina, cloreto de sódio, sacarose, polissorbato 20 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Vabysmo™ é indicado para o tratamento de pacientes adultos com:
- Degeneração macular relacionada à idade (DMRI) neovascular (exsudativa ou úmida)
- Deficiência visual devido ao edema macular diabético (EMD)
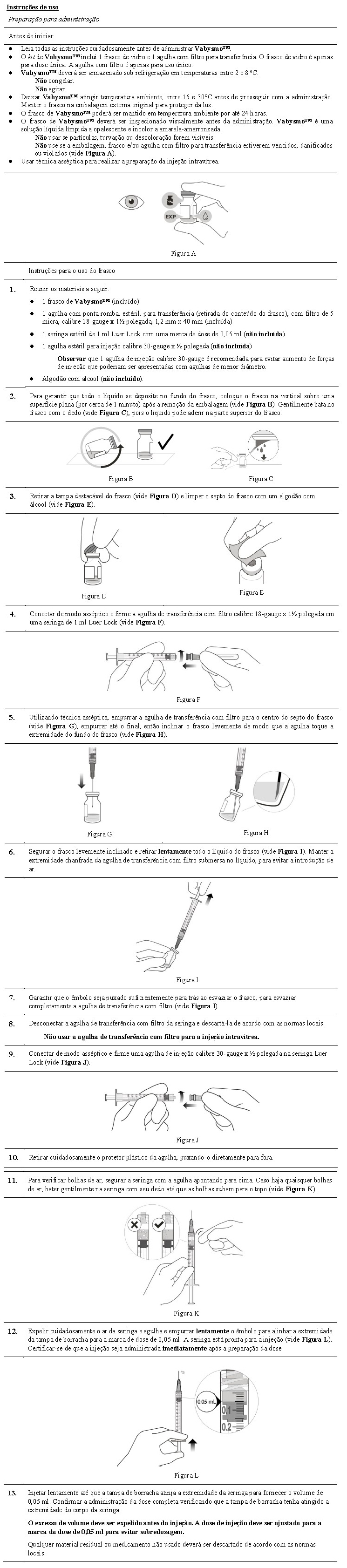
2. RESULTADOS DE EFICÁCIA
Tratamento da degeneração macular relacionada à idade neovascular (DMRIn)
A segurança e a eficácia de Vabysmo™ foram avaliadas em dois estudos randomizados, multicêntricos, duplos-cegos, controlados por comparador ativo, de dois anos em pacientes com DMRIn, TENAYA e LUCERNE. Foram incluídos 1.329 pacientes nesses estudos, e 1.326 pacientes receberam, no mínimo, uma dose (664 com Vabysmo™). A idade dos pacientes variou de 50 a 99 anos, com média de 75,9 anos.
Em ambos os estudos, os pacientes foram randomizados em uma proporção de 1:1 a um dos dois grupos de tratamento:
- Vabysmo™ 6 mg administrado até a cada 16 semanas (Q16W) após quatro doses mensais iniciais.
- Aflibercepte 2 mg administrado a cada oito semanas (Q8W) após três doses mensais iniciais.
Após as primeiras quatro doses mensais (semanas 0, 4, 8 e 12), os pacientes randomizados ao braço Vabysmo™ receberam administração a cada 16 semanas (Q16W), a cada 12 semanas (Q12W) ou a cada 8 semanas (Q8W), com base em uma avaliação da atividade da doença nas semanas 20 e 24 e uso de critérios objetivos visuais e anatômicos pré-especificados, bem como avaliação clínica do(a) médico(a) responsável pelo tratamento. Os pacientes permaneceram nesses intervalos fixos de administração até a semana 60 sem terapia suplementar.
O desfecho primário de eficácia foi a alteração média da melhor acuidade visual corrigida (BCVA), em relação ao valor medido na linha de base, quando a média foi realizada nas visitas das semanas 40, 44 e 48, conforme medida pela pontuação de letras do Estudo de Tratamento Precoce de Retinopatia Diabética (Early Treatment Diabetic Retinopathy Study - ETDRS). Em ambos os estudos, os pacientes tratados com Vabysmo™ até Q16W apresentaram alteração média desde o valor basal comparável na BCVA, assim como os pacientes tratados com aflibercepte Q8W.
A proporção de pacientes em cada intervalo de tratamento diferente na semana 48 nos estudos TENAYA e LUCERNE, respectivamente, foi:
- Q16W, 46% e 45%
- Q12W, 34% e 33%
- Q8W, 20% e 22%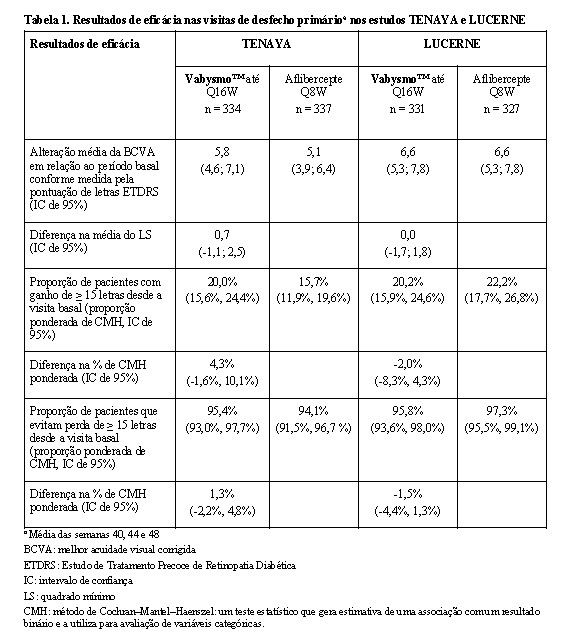
Em ambos os estudos TENAYA e LUCERNE, melhoras desde o valor basal da BCVA e da espessura do subcampo central (CST) na semana 60 foram comparáveis entre os dois braços de tratamento e consistentes com as melhoras observadas na semana 48.
Os resultados de eficácia em todos os subgrupos avaliáveis (por exemplo, idade, sexo, raça, acuidade visual basal, tipo de lesão, tamanho da lesão) em cada estudo e na análise agrupada foram consistentes com os resultados nas populações gerais.
Em ambos os estudos, Vabysmo™ até Q16W demonstrou melhoras clinicamente significativas desde a visita basal até a semana 48 na pontuação composta do Questionário de Função Visual do Instituto Nacional do Olho (National Eye Institute Visual Function Questionnaire - NEI VFQ-25) que foram comparáveis a aflibercepte Q8W. Os pacientes nos braços Vabysmo™ nos estudos TENAYA e LUCERNE atingiram melhora de ≥ 4 pontos desde o valor basal na pontuação composta do NEI VFQ-25 na semana 48.
A incidência de eventos adversos oculares no olho do estudo foi de 38,3% e 37,2% e de eventos adversos não oculares foi de 52,1% e 54,8%, até a Semana 48 nos braços faricimabe e aflibercepte, respectivamente (vide itens "5. Advertências e Precauções" e "9. Reações Adversas").
Tratamento do edema macular diabético (EMD)
A segurança e a eficácia de Vabysmo™ foram avaliadas em dois estudos randomizados, multicêntricos, duplos-cegos, controlados por comparador ativo por dois anos (YOSEMITE e RHINE) em pacientes com EMD. Foram incluídos 1.891 pacientes nos dois estudos com 1.622 (86%) pacientes completando os estudos até a semana 100. Um total de 1.887 pacientes foram tratados com, no mínimo, uma dose até a semana 56 (1.262 com Vabysmo™). A idade dos pacientes variou de 24 a 91 anos, com média de 62,2 anos. A população geral incluiu pacientes não tratados anteriormente com anti-VEGF (78%) e pacientes tratados anteriormente com um inibidor de VEGF antes da participação no estudo (22%). Em ambos os estudos, os pacientes foram randomizados em proporção de 1:1:1 a um dos três regimes de tratamento:
- Vabysmo™ 6 mg Q8W após as 6 primeiras doses mensais.
- Administração de dose ajustável de Vabysmo™ 6 mg até Q16W em intervalos de 4, 8, 12 ou 16 semanas após as 4 primeiras doses mensais.
- Aflibercepte 2 mg Q8W após as 5 primeiras doses mensais.
No braço de administração ajustável Q16W, o intervalo de administração pôde ser aumentado em etapas de 4 semanas ou pôde ser reduzido em etapas de 4 ou 8 semanas, com base na avaliação objetiva automatizada de critérios visuais e anatômicos pré-especificados de atividade da doença.
Resultados
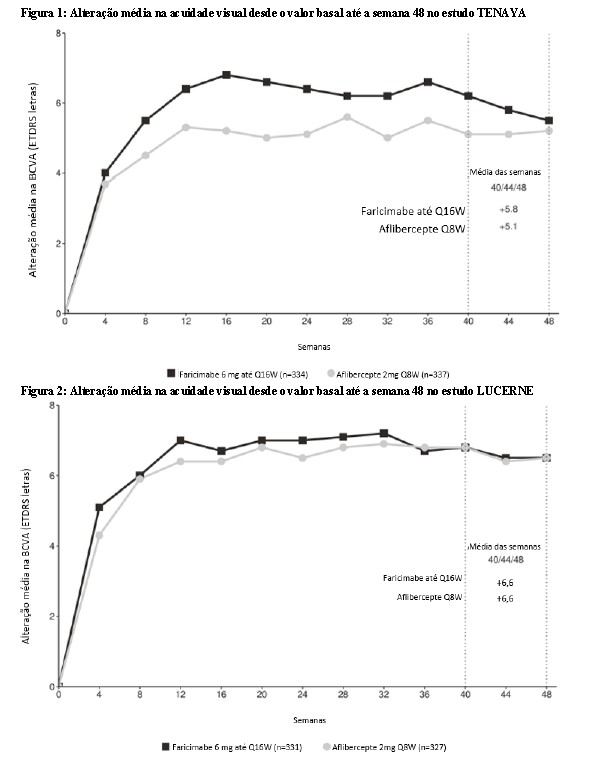
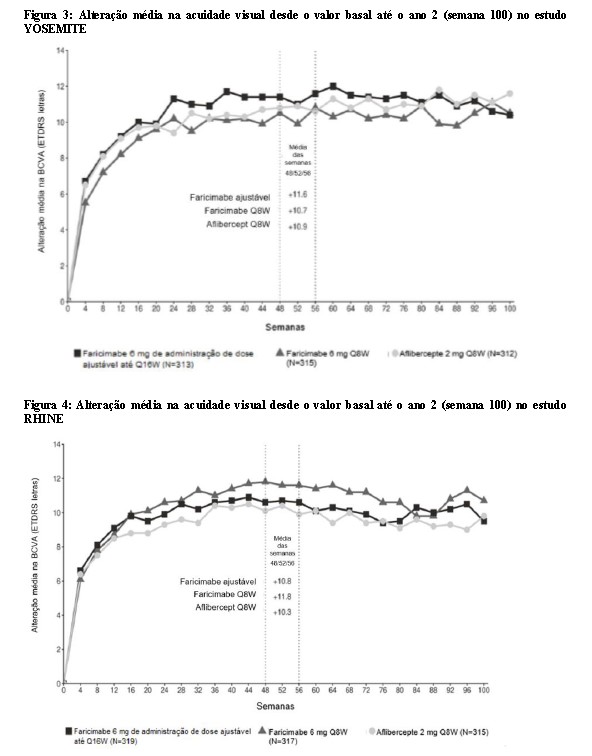
Ambos os estudos demonstraram eficácia no desfecho primário, definido como a alteração média desde o valor basal da BCVA no ano 1 (média das visitas das semanas 48, 52 e 56), medida pela Pontuação de Letras ETDRS. Em ambos os estudos, pacientes tratados com Vabysmo™ até Q16W apresentaram uma alteração média desde o valor basal comparável na BCVA, assim como os pacientes tratados com aflibercepte Q8W no ano 1, e os aumentos de visão foram mantidos durante o ano 2. Os resultados detalhados de ambos os estudos são ilustrados na Tabela 2, Figura 3 e Figura 4 a seguir.
Após 4 doses mensais iniciais, os pacientes no braço de dosagem ajustável de Vabysmo™ até Q16W poderiam ter recebido entre o mínimo de 6 e o máximo de 21 injeções totais até a semana 96. Na semana 52, 74% e 71% dos pacientes no braço de administração de dose ajustável de Vabysmo™ até Q16W atingiram intervalo de administração de Q16W ou Q12W nos estudos YOSEMITE e RHINE, respectivamente (53% e 51% em Q16W, 21% e 20% em Q12W). Desses pacientes, 75% e 84% mantiveram a dosagem ≥ Q12W sem redução do intervalo abaixo de Q12W até a semana 96; dos pacientes em Q16W na semana 52, 70% e 82% dos pacientes mantiveram a dosagem de Q16W sem redução do intervalo até a semana 96 nos estudos YOSEMITE e RHINE, respectivamente. Na semana 96, 78% dos pacientes no braço de dosagem ajustável de Vabysmo™ até Q16W alcançaram um intervalo de dosagem Q16W ou Q12W em ambos os estudos (60% e 64% em Q16W, 18% e 14% em Q12W). Dos pacientes, 4% e 6% foram estendidos para Q8W e permaneceram em intervalos de dosagem ≤ Q8W até a semana 96; 3% e 5% receberam apenas dosagem Q4W nos estudos YOSEMITE e RHINE, respectivamente.
Os resultados detalhados das análises dos estudos YOSEMITE e RHINE são listados na Tabela 2 e Figuras 3 e 4 a seguir.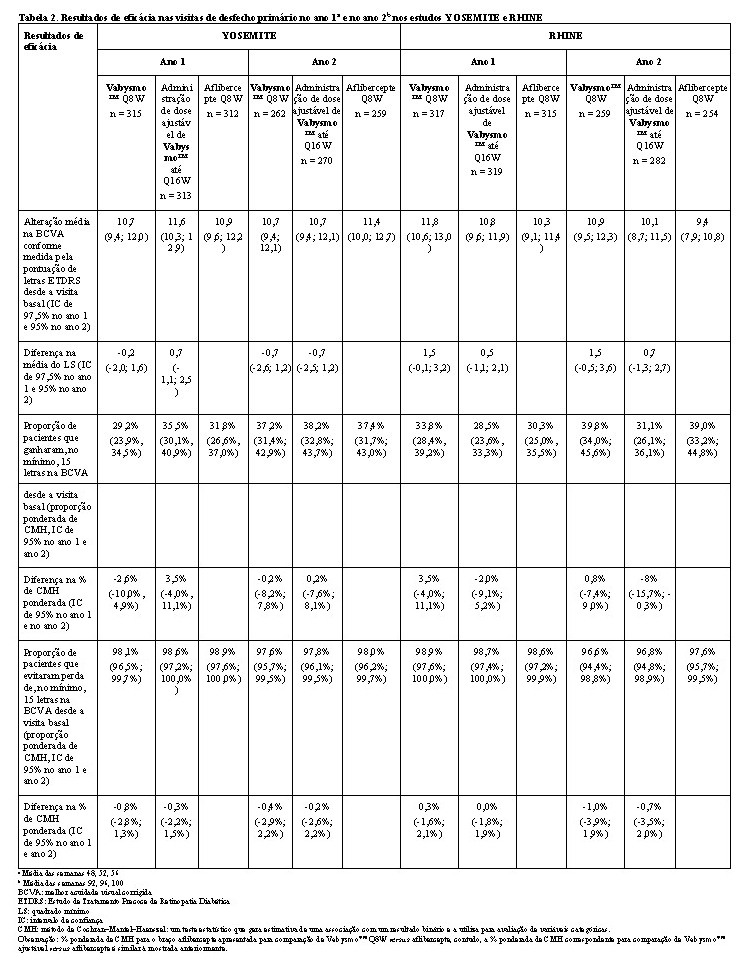
Os resultados de eficácia em pacientes não tratados anteriormente com anti-VEGF antes da participação no estudo e em todos os demais subgrupos avaliáveis (por exemplo, por idade, sexo, raça, HbA1c basal, acuidade visual basal) em cada estudo foram consistentes com os resultados nas populações gerais.
Em todos os estudos, a administração de dose ajustável de Vabysmo™ Q8W e até Q16W mostrou melhoras no desfecho de eficácia pré-especificado de mudança média desde a visita basal até a semana 52 nas pontuações compostas do NEI VFQ -25 que foram comparáveis a aflibercepte Q8W e ultrapassaram o limite de 4 pontos. Vabysmo™ Q8W e dose ajustável até Q16W também demonstraram melhorias clinicamente significativas no desfecho de eficácia pré-especificado de mudança da linha de base até a semana 52 nas pontuações de atividades próximas, atividades à distância e de direção do NEI VFQ-25, que foram comparáveis ao aflibercepte Q8W. A magnitude dessas mudanças corresponde a um ganho de 15 letras no BCVA. Proporções comparáveis de pacientes tratados com Vabysmo™ Q8W, dose ajustável de Vabysmo™ até Q16W e aflibercepte Q8W apresentaram melhora clinicamente significativa de ≥ 4 pontos desde o valor basal até a semana 52 na pontuação composta de NEI VFQ -25, um desfecho de eficácia pré-especificado. Esses resultados foram mantidos na semana 100.
Um resultado chave adicional de eficácia em estudos de EMD foi a alteração na Escala de Severidade da Retinopatia Diabética do Estudo de Tratamento Precoce de Retinopatia Diabética (ETDRS-DRSS) desde a visita basal até a semana 52. Dos 1.891 pacientes incluídos nos estudos YOSEMITE e RHINE, respectivamente, 708 e 720 pacientes foram avaliáveis quanto aos desfechos de DR (retinopatia diabética).
As pontuações ETDRS-DRSS variaram de 10 a 71 na visita basal.
A maior parte dos pacientes, aproximadamente 60%, apresentou DR não proliferativa moderada a severa (DRSS 43/47/53) na visita basal.
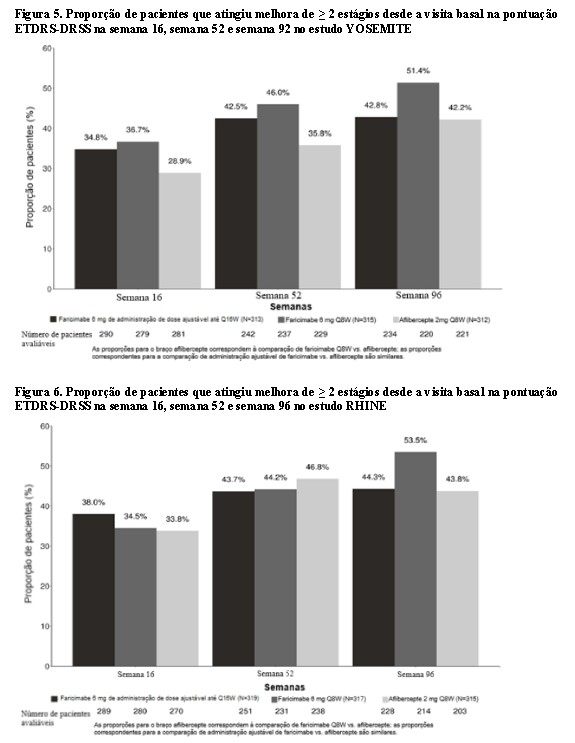
Na semana 52, a proporção de pacientes com melhora em ≥ 2 estágios na ETDRS-DRSS foi de 43% a 46% nos braços Vabysmo™ Q8W e Vabysmo™ ajustável até Q16W em ambos os estudos, em comparação a 36% e 47% nos braços aflibercepte Q8W dos estudos YOSEMITE e RHINE, respectivamente. Os resultados na semana 96 foram de 43% a 54% nos braços Vabysmo™ Q8W e Vabysmo™ ajustável até Q16W em ambos os estudos, em comparação com 42% e 44% nos braços aflibercepte Q8W dos estudos YOSEMITE e RHINE, respectivamente.
Foram observados resultados comparáveis entre os braços de tratamento em ambos os estudos nas proporções de pacientes com melhora em ≥ 3 estágios na ETDRS-DRSS desde o valor basal na semana 52, e esses resultados foram mantidos na semana 96.
Os resultados das análises de melhora de ≥ 2 estágios e ≥ 3 estágios na ETDRS-DRSS desde a visita basal na semana 52 e na semana 96 são apresentados na Tabela 3 a seguir. A proporção de pacientes com melhora de ≥ 2 estágios na ETDRS-DRSS na visita basal, semanas 16,semana 52 e na semana 96, é mostrada nas Figuras 5 e 6 a seguir.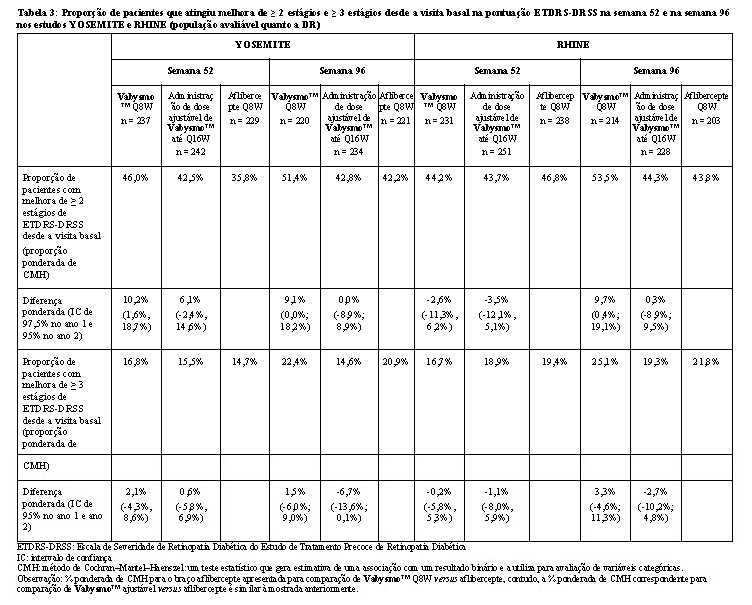
As proporções de pacientes com novo diagnóstico de DR proliferativa (definido por ETDRS-DRSS de 61 ou pior) desde a visita basal até a semana 96 foram comparáveis entre pacientes tratados com Vabysmo™ Q8W, administração de dose ajustável de Vabysmo™ até Q16W e aflibercepte Q8W nos estudos YOSEMITE e RHINE. Quase nenhum paciente necessitou de vitrectomia (0 a 4 por grupo) ou fotocoagulação panretiniana (PRP) (1 a 2 por grupo) durante os dois anos de duração dos estudos.
Os efeitos do tratamento da DR no subgrupo de pacientes não tratados anteriormente com anti-VEGF antes da participação no estudo foram comparáveis aos observados na população avaliável geral quanto a DR. Os efeitos do tratamento em subgrupos avaliáveis (por exemplo, por idade, sexo, raça, HbA1c basal e acuidade visual basal) em cada estudo foram geralmente consistentes com os resultados na população geral.
Os efeitos do tratamento nos subgrupos por severidade de DR na visita basal foram diferentes e mostraram as maiores melhoras de ≥ 2 estágios na DRSS entre pacientes com DR não proliferativa moderadamente severa e severa, com, aproximadamente, 90% dos pacientes atingindo melhoras. Esses resultados foram comparáveis entre os braços de estudo e comparáveis nas populações gerais e não tratadas anteriormente com anti-VEGF.
A incidência de eventos adversos oculares no olho do estudo foi de 49,7%; 49,2% e 45,4% e de eventos adversos não oculares foi de 73,0%; 74,2% e 75,7% até a Semana 100, nos braços de faricimabe Q8W, faricimabe até Q16W e aflibercepte Q8W, respectivamente (vide itens "5. Advertências e Precauções" e "9. Reações Adversas").
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
Faricimabe é um anticorpo de imunoglobulina G1 (IgG1) biespecífica humanizada que atua na inibição de duas vias distintas pela neutralização da angiopoietina-2 (Ang-2) e do fator de crescimento endotelial vascular A (VEGF-A).
Ang-2 causa instabilidade vascular pela promoção de desestabilização endotelial, perda de pericitos e angiogênese patológica, potencializando assim extravasamento e inflamação vascular. Ela também sensibiliza os vasos sanguíneos à atividade de VEGF-A, e isso resulta em desestabilização vascular adicional. Ang-2 e VEGF-A aumentam, de modo sinérgico, a permeabilidade vascular e estimulam a neovascularização.
Por meio da inibição dupla de Ang-2 e VEGF-A, faricimabe reduz a permeabilidade e a inflamação vasculares, inibe a angiogênese patológica e restaura a estabilidade vascular.
Farmacodinâmica
Foi observada redução desde o valor basal nas concentrações oculares livres de Ang-2 e VEGF-A desde o dia 7 e por todo o intervalo de tratamento (na maioria dos pacientes) nos quatro estudos clínicos de fase III.
DMRIn
Nos estudos fase III em paciente com DMRIn (TENAYA e LUCERNE), critérios objetivos visuais e anatômicos pré-especificados e também a avaliação clínica do(a) médico(a) responsável pelo tratamento foram utilizados para orientar decisões de tratamento nos momentos de avaliação da atividade da doença (semanas 20 e 24).
Foram observadas reduções na espessura média do subcampo central (CST) desde a visita basal até a semana 48 com Vabysmo™ e foram comparáveis às observadas com aflibercepte. A redução média de CST desde a visita basal até as visitas de desfecho primário (média nas semanas 40 - 48) foi de -137 mm e -137 mm para Vabysmo™ administrado até cada 16 semanas (Q16W) versus -129 mm e -131 mm com aflibercepte nos estudos TENAYA e LUCERNE, respectivamente.
Houve efeito comparável de Vabysmo™ e aflibercepte na redução de fluido intrarretiniano (IRF), fluido sub-retiniano (SRF) e descolamento epitelial pigmentar (PED). Nas visitas de desfecho primário (min-máx, semanas 40 - 48), a proporção de pacientes nos estudos TENAYA e LUCERNE com ausência de IRF, respectivamente, foi: 76%-82% e 78% - 85% em Vabysmo™ versus 74% - 85% e 78% - 84% em aflibercepte; ausência de SRF: 70% - 79% e 66% - 78% em Vabysmo™ versus 66% - 78% e 62% - 76% em aflibercepte; ausência de PED: 3% - 8% e 3% -6% em Vabysmo™ versus 8% - 10% e 7% - 9% em aflibercepte.
Na semana 48, houve alteração comparável na área total de lesão por neovascularização coroidal (CNV) desde o valor basal nos braços de tratamento (0,0 mm2 e 0,4 mm2 em Vabysmo™ versus 0,4 mm2 e 1,0 mm2 em aflibercepte, nos estudos TENAYA e LUCERNE, respectivamente). Houve redução comparável na área de extravasamento de CNV desde o valor basal entre os braços de tratamento (-3,8 mm2 e -3,2 mm2 com Vabysmo™ e -3,0 mm2 e -2,2 mm2 com aflibercepte nos estudos TENAYA e LUCERNE, respectivamente).
EMD
Nos estudos fase III em pacientes com EMD (YOSEMITE e RHINE), os parâmetros anatômicos relacionados a edema macular foram parte das avaliações de atividade da doença que orientaram as decisões de tratamento.
As reduções na CST média desde o valor basal foram numericamente maiores em pacientes tratados com Vabysmo™ a cada 8 semanas (Q8W) e administração de dose ajustável de Vabysmo™ até Q16W, em comparação a aflibercepte (Q8W) nas semanas 4 a 100, nos estudos YOSEMITE e RHINE. Maiores proporções de pacientes em ambos os braços Vabysmo™ atingiram ausência de IRF e ausência de EMD (definida como alcance de CST abaixo de 325 mm), conforme medida por tomografia de coerência óptica de domínio espectral (SD-OCT) com o tempo em ambos os estudos, em comparação ao braço aflibercepte. Foram observadas reduções comparáveis em SRF entre ambos os braços de tratamento com Vabysmo™ e aflibercepte com o tempo em ambos os estudos.
A redução média de CST desde o valor basal até as visitas de desfecho primário (média nas semanas 48 - 56) foi de 207 mm e 197 mm em pacientes tratados com Vabysmo™ Q8W e administração ajustável de Vabysmo™ até Q16W, em comparação a 170 mm em pacientes tratados com aflibercepte Q8W no estudo YOSEMITE. Os resultados foram de 196 mm, 188 mm e 170 mm, respectivamente, no estudo RHINE. Essas reduções médias de CST foram mantidas durante o ano 2. A proporção de pacientes com ausência de EMD nas visitas de desfecho primário (min. - máx., semanas 48 - 56) foi de 77% - 87% e 80% - 82% em pacientes tratados com Vabysmo™ Q8W e administração ajustável de Q16W, em comparação a 64% - 71% em pacientes tratados com aflibercepte Q8W no estudo YOSEMITE. Os resultados foram de 85% - 90%, 83% - 87% e 71% - -77%, respectivamente, no estudo RHINE. Esses resultados foram mantidos durante o ano 2.
Na semana 16, a proporção de pacientes com ausência de IRF foi numericamente maior em pacientes que recebem Vabysmo™ Q8W ou administração ajustável de Vabysmo™ até Q16W versus administração de aflibercepte Q8W em ambos os estudos (YOSEMITE: 16% e 22% versus 13%; RHINE: 20% e 20% versus 13%). As proporções de pacientes com ausência de IRF nas visitas de desfecho primário (min. - máx., semanas 48 - 56) foi de 42% - 48% e 34% - 43% em pacientes tratados com Vabysmo™ Q8W e administração ajustável de Vabysmo™ até Q16W, em comparação a 22% - 25% em pacientes tratados com aflibercepte Q8W no estudo YOSEMITE. Os resultados foram de 39% - 43%, 33% - 41% e 23% - 29%, respectivamente, no estudo RHINE.
Propriedades farmacocinéticas
Absorção
Vabysmo™ é administrado via intravítrea (IVT) para exercer efeitos locais no olho. Não houve estudos clínicos realizados com outras vias de administração.
Com base na análise farmacocinética populacional (que incluiu DMRIn e EMD n = 2.246), estima-se que as concentrações plasmáticas máximas de faricimabe livre (não ligado a VEGF-A e Ang-2) (Cmáx) ocorram, aproximadamente, dois dias após a administração. A média (± SD) da Cmáx plasmática é estimada em 0,23 (0,07) mg/ml e 0,22 (0,07) mg/ml, respectivamente, em pacientes com DMRIn e EMD / DR. Após administrações repetidas, as concentrações plasmáticas médias de faricimabe livre são estimadas em 0,002 - 0,003 mg/ml para a dosagem Q8W.
Faricimabe apresentou farmacocinética proporcional à dose (com base na Cmáx e AUC) no intervalo de doses de 0,5 mg - 6 mg. Não houve acúmulo aparente de faricimabe no humor vítreo ou no plasma após a administração mensal.
Distribuição
As concentrações plasmáticas máximas livres de faricimabe são previstas como sendo, aproximadamente, 600 e 6.000 vezes menores que no humor aquoso e vítreo, respectivamente, e abaixo da afinidade de ligação para VEGF e Ang-2. Portanto, os efeitos farmacodinâmicos sistêmicos são improváveis, adicionalmente embasados pela ausência de alterações significativas na concentração plasmática livre de VEGF e Ang-2 após tratamento com faricimabe em estudos clínicos.
A análise de farmacocinética populacional demonstrou um efeito da idade e peso corporal na farmacocinética ocular ou sistêmica de faricimabe, respectivamente. Ambos os efeitos foram considerados não clinicamente significativos; não é necessário ajuste de dose.
Metabolismo
O metabolismo de faricimabe não foi estudado diretamente, uma vez que os anticorpos monoclonais são eliminados principalmente por catabolismo. É esperado que o faricimabe seja catabolizado nos lisossomos em pequenos peptídeos e aminoácidos, que podem ser excretados por via renal, de maneira semelhante à eliminação de IgG endógena.
Eliminação
O perfil de concentração-tempo de faricimabe no plasma diminuiu em paralelo com os perfis de concentração-tempo vítreo e aquoso. A meia vida ocular média estimada, e a meia vida sistêmica aparente é de 7,5 dias após a administração intravítrea.
Farmacocinética em populações especiais
População pediátrica
A segurança e a eficácia de Vabysmo™ em pacientes pediátricos não foram estabelecidas.
População geriátrica
Nos quatro estudos clínicos de fase III, aproximadamente 60% (1.149/1.929) dos pacientes randomizados ao tratamento com Vabysmo™ tinham ≥ 65 anos de idade. A análise farmacocinética populacional mostrou um efeito da idade na farmacocinética ocular de faricimabe. O efeito foi considerado não clinicamente significativo. Não é necessário ajuste de dose em pacientes com 65 anos ou mais (vide item "8. Posologia e Modo de Usar").
Insuficiência renal
Não foram realizados estudos específicos que envolviam pacientes com insuficiência renal tratados com Vabysmo™. A análise farmacocinética de pacientes em todos os estudos clínicos, dos quais 64% tinham insuficiência renal (leve 38%, moderada 24% e grave 2%), não revelou diferenças em relação à farmacocinética sistêmica de faricimabe após administração intravítrea de Vabysmo™. Não é necessário ajuste de dose em pacientes com insuficiência renal (vide item "8. Posologia e Modo de Usar").
Insuficiência hepática
Não foram realizados estudos específicos que envolviam pacientes com insuficiência hepática tratados com Vabysmo™. No entanto, nenhuma consideração especial é necessária nessa população, porque o metabolismo ocorre via proteólise e não depende da função hepática.
Não é necessário ajuste de dose em pacientes com insuficiência hepática (vide item "8. Posologia e Modo de Usar").
Outros
A farmacocinética sistêmica de Vabysmo™ não foi influenciada pela raça nos estudos clínicos, que foram conduzidos com amostras compostas predominantemente por indivíduos brancos e asiáticos. O sexo não demonstrou possuir uma influência clinicamente relevante na farmacocinética sistêmica de Vabysmo™. Nenhum ajuste de dose é necessário.
Segurança não clínica
Carcinogenicidade
Não foram realizados estudos para estabelecer o potencial carcinogênico de Vabysmo™.
Genotoxicidade
Não foram realizados estudos para estabelecer o potencial mutagênico de Vabysmo™.
Comprometimento da fertilidade
Embora os componentes anti-VEGF e anti-Ang2 possam significar risco potencial para a reprodução baseado no mecanismo teórico, a exposição sistêmica decorrente do tratamento intravítreo sugere que esse risco pode ser insignificante. Nenhum efeito sobre a fertilidade foi observado em um estudo de 6 meses em macacos cynomolgus que receberam Vabysmo™.
Toxicidade reprodutiva
Foi demonstrado que a inibição do VEGF causa malformações, reabsorção embriofetal e diminuição do peso fetal. A inibição do VEGF também demonstrou afetar o desenvolvimento folicular, a função do corpo lúteo e a fertilidade. Não estão disponíveis estudos específicos que abordem os efeitos da inibição da Ang-2 na gravidez. Com base em informações não clínicas, a inibição de Ang-2 pode levar a efeitos comparáveis à inibição de VEGF. A exposição sistêmica após a administração ocular de Vabysmo™ é muito baixa.
Nenhum efeito nos órgãos reprodutivos foi observado em um estudo com macacos cynomolgus de 6 meses que receberam Vabysmo™. Nenhum efeito sobre a gravidez ou fetos foi observado em um estudo de desenvolvimento embriofetal em macacas cynomolgus grávidas que receberam 5 injeções intravenosas (IV) semanais de Vabysmo™, começando no dia 20 de gestação com 1 mg/kg ou 3 mg/kg. A exposição sérica (Cmáx) em macacos na dose do nível sem efeitos adversos observáveis (No Observed Adverse Effect Level - NOAEL) de 3 mg/kg foi 500 vezes maior do que a exposição em humanos com uma dose de 6 mg administrada por injeção intravítrea uma vez a cada 4 semanas.
Em macacas cynomolgus grávidas, as injeções IV de faricimabe, resultando em exposição sérica (Cmáx) superior a 500 vezes a exposição humana máxima, não provocaram toxicidade no desenvolvimento ou teratogenicidade e não tiveram efeito sobre o peso ou a estrutura da placenta, embora, com base em seu efeito farmacológico, faricimabe deve ser considerado como potencialmente teratogênico e embrio/fetotóxico.
4. CONTRAINDICAÇÕES
Vabysmo™ é contraindicado a pacientes com infecções oculares ou perioculares, ativas ou suspeitas.
Vabysmo™ é contraindicado a pacientes com inflamação intraocular ativa.
Vabysmo™ é contraindicado a pacientes com hipersensibilidade conhecida a faricimabe ou a qualquer um dos excipientes. As reações de hipersensibilidade podem se manifestar como erupção cutânea, prurido, urticária, eritema ou inflamação intraocular grave.
5. ADVERTÊNCIAS E PRECAUÇÕES
Geral
Para melhorar a rastreabilidade dos medicamentos biológicos, o nome e o número do lote do produto administrado deverão ser registrados claramente.
Reações relacionadas à injeção intravítrea
Injeções intravítreas, e isso inclui aquelas com Vabysmo™, foram associadas à endoftalmite, à inflamação intraocular, ao descolamento de retina regmatógeno e à laceração retiniana. As técnicas assépticas adequadas de injeção deverão ser sempre utilizadas ao administrar Vabysmo™. Os pacientes deverão ser instruídos a relatar quaisquer sintomas, como dor, perda de visão, fotofobia, visão turva, moscas volantes ou vermelhidão, sugestivos de endoftalmite ou qualquer dos eventos mencionados sem atraso, para permitir o tratamento imediato e adequado. Pacientes com maior frequência de injeções podem ter maior risco de complicações relacionadas ao procedimento.
Foi observado aumento temporário na pressão intraocular (PIO) no período de 60 minutos após a injeção intravítrea, isso inclui aquelas com Vabysmo™. É necessário que haja precaução especial em pacientes com glaucoma mal controlado (não injetar Vabysmo™ enquanto a PIO for ≥ 30 mmHg). Em todos os casos, a PIO e a perfusão do nervo óptico deverão ser monitoradas e tratadas adequadamente.
Efeitos sistêmicos
Foram relatados eventos adversos sistêmicos, que incluem eventos tromboembólicos arteriais, após a injeção intravítrea de inibidores de fator de crescimento endotelial vascular (VEGF), e há risco teórico de que possam estar relacionados à inibição de VEGF. Nos estudos clínicos com faricimabe em pacientes com DMRIn e EMD, uma baixa taxa de incidência de eventos tromboembólicos arteriais foi observada. Existem dados limitados sobre a segurança do tratamento com faricimabe em pacientes com EMD com hipertensão arterial (≥ 140/90 mmHg) e doença vascular, e em pacientes com DMRIn com ≥ 85 anos de idade.
Imunogenicidade
Como com toda proteína terapêutica, há potencial para resposta imune com Vabysmo™. Os pacientes deverão ser instruídos a informar seu(sua) médico(a) sobre quaisquer sinais ou sintomas de inflamação intraocular, como perda de visão, dor ocular, aumento da sensibilidade à luz, moscas volantes ou piora da vermelhidão ocular, que podem ser um sinal clínico atribuído à hipersensibilidade.
Tratamento bilateral
A segurança e a eficácia de Vabysmo™ administrado em ambos os olhos concomitantemente não foram estudadas.
O tratamento bilateral pode causar reações adversas oculares bilaterais e/ou potencialmente levar a um aumento na exposição sistêmica, o que pode aumentar o risco de reações adversas sistêmicas. Até que os dados para uso bilateral estejam disponíveis, este é um risco teórico para o faricimabe.
Uso concomitante de outro anti-VEGF
Não há dados disponíveis sobre o uso concomitante de Vabysmo™ com medicamentos anti-VEGF no mesmo olho.
Faricimabe não deve ser administrado concomitantemente com outros medicamentos anti-VEGF (sistêmicos ou oculares).
Suspensão do tratamento
O tratamento deverá ser suspenso em pacientes com:
- Descolamento de retina regmatógeno, buraco macular estágio 3 ou 4, ruptura da retina. O tratamento não deverá ser retomado até que um reparo adequado tenha sido realizado.
- Redução relacionada ao tratamento na melhor acuidade visual corrigida (BCVA) de ≥ 30 letras em comparação à última avaliação de acuidade visual. O tratamento não deverá ser retomado antes do próximo tratamento agendado.
- Pressão intraocular de ≥ 30 mmHg
- Hemorragia sub-retiniana envolvendo o centro da fóvea ou, se o tamanho da hemorragia for ≥ 50%, da área total da lesão.
- Cirurgia intraocular realizada ou planejada nos 28 dias anteriores ou seguintes. O tratamento não deverá ser retomado antes do próximo tratamento agendado.
Laceração do epitélio pigmentar da retina
Os fatores de risco associados ao desenvolvimento de laceração do epitélio pigmentar da retina após terapia anti-VEGF para DMRIn incluem um descolamento epitelial pigmentar grande e/ou elevado. Ao iniciar a terapia com Vabysmo™, recomenda-se cautela em pacientes com esses fatores de risco para lacerações do epitélio pigmentar da retina.
Populações com dados limitados
Há apenas experiência limitada no tratamento de pacientes com DMRIn com ≥ 85 anos, com EMD com diabetes tipo I, HbA1c acima de 10%, pacientes com risco elevado de retinopatia diabética proliferativa (DR), hipertensão arterial (≥ 140/90 mmHg) e doença vascular, intervalos de dosagem mantidos menores que Q8W, ou DMRIn e EMD com infecções sistêmicas ativas. A informação de segurança sobre intervalos entre administrações continuados de 8 semanas ou menos é limitada, podendo estes estar associados a um maior risco de reações adversas oculares e sistêmicas, incluindo reações adversas graves. Também não há experiência de tratamento com Vabysmo™ em pacientes diabéticos com hipertensão não controlada. Essa ausência de informação deverá ser considerada pelo(a) médico(a) ao tratar esses pacientes.
Uso em populações especiais
Homens e mulheres com potencial reprodutivo
Fertilidade
Não foram realizados estudos reprodutivos ou de fertilidade. Nenhum efeito nos órgãos reprodutivos ou na fertilidade foi observado em um estudo de 6 meses em macacos cynomolgus que receberam Vabysmo™. A inibição do VEGF demonstrou afetar o desenvolvimento folicular, a função do corpo lúteo e a fertilidade. Com base no mecanismo de ação dos inibidores de VEGF e Ang-2, há risco potencial para a capacidade reprodutiva feminina e para o desenvolvimento embriofetal, no entanto, o risco é considerado baixo por causa da baixa exposição sistêmica após administração ocular (vide item "3. Características Farmacológicas").
Contracepção
Pacientes do sexo feminino com potencial reprodutivo deverão utilizar contracepção durante o tratamento com Vabysmo™ e por, no mínimo, 3 meses após a última dose de Vabysmo™.
Gravidez
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Não existem dados sobre a utilização de Vabysmo™ em mulheres grávidas. A exposição sistêmica ao faricimabe é baixa após administração ocular, mas devido ao seu mecanismo de ação (ou seja, inibição do VEGF), o faricimabe deve ser considerado como potencialmente teratogênico e embrio/fetotóxico (vide item "3. Características Farmacológicas").
Nenhum efeito adverso foi observado em um estudo em macacas cynomologus grávidas que receberam Vabysmo™ por via intravenosa durante o período de organogênese em doses que atingiram mais de 500 vezes a exposição humana sistêmica prevista de Vabysmo™ após o tratamento de um único olho (vide item "3. Características Farmacológicas").
Não se sabe se Vabysmo™ pode atravessar a placenta ou causar danos ao feto quando administrado a mulheres grávidas. Com base no mecanismo de ação dos inibidores de VEGF e Ang-2, existe risco potencial para a capacidade reprodutiva feminina e para o desenvolvimento embriofetal. Embora a exposição sistêmica após a administração ocular seja muito baixa, Vabysmo™ não deve ser usado durante a gravidez, a menos que o benefício potencial à paciente supere o risco potencial para o feto.
Parto e trabalho de parto
O uso seguro de VabysmoTM durante o parto e trabalho de parto não foi estabelecido.
Lactação
Não se sabe se Vabysmo™ é excretado no leite materno. Não foram realizados estudos para avaliar o impacto do VabysmoTM na produção de leite ou na sua presença no leite materno. Uma vez que muitos medicamentos são excretados no leite humano com potencial de absorção e danos ao crescimento e desenvolvimento infantil, deve-se ter cuidado quando VabysmoTM é administrado a mulheres a amamentar. Os benefícios da amamentação para o desenvolvimento e a saúde devem ser considerados juntamente com a necessidade clínica da mãe de utilizar VabysmoTM e quaisquer efeitos adversos potenciais do VabysmoTM sobre a criança amamentada.
Uso pediátrico
A segurança e a eficácia de Vabysmo™ em pacientes pediátricos não foram estabelecidas.
Uso geriátrico
Nos quatro estudos clínicos de fase III, aproximadamente 60% (1.149 / 1.929) dos pacientes randomizados ao tratamento com Vabysmo™ tinham ≥ 65 anos de idade. Nenhuma diferença significativa na eficácia ou na segurança de Vabysmo™ foi observada com o aumento da idade nesses estudos (vide itens 3. Características Farmacológicas e 8. Posologia e Modo de usar).
Comprometimento renal
Não é necessário ajuste de dose em pacientes com comprometimento renal (vide itens "3. Características Farmacológicas" e "8. Posologia e Modo de usar").
Comprometimento hepático
A segurança e a eficácia Vabysmo™ em pacientes com comprometimento hepático não foram estabelecidas (vide itens "3. Características Farmacológicas" e "8. Posologia e Modo de usar").
Abuso e dependência do medicamento
Não há evidências de que Vabysmo™ tenha potencial para causar abuso e dependência da droga.
Capacidade de conduzir veículos ou operar máquinas
Vabysmo™ poderá causar influência mínima na capacidade de dirigir e utilizar máquinas causada por possíveis distúrbios visuais temporários após a injeção intravítrea e exame ocular associado. Os pacientes não deverão dirigir ou operar máquinas até recuperação suficiente da função visual.
Outras
Até o momento, não há informações de que Vabysmo™ (faricimabe) possa causar doping.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação medicamentosa com Vabysmo™.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenamento
Conservar sob refrigeração (temperatura entre 2 e 8 °C). Não congelar. Não agitar.
Manter o frasco na embalagem externa original para proteger da luz.
Antes do uso, o frasco não aberto de Vabysmo™ deverá ser mantido em temperatura ambiente, entre 15 e 30°C, por até 24 horas. Certificar-se de que a injeção seja administrada imediatamente após a preparação da dose.
Prazo de validade
Este medicamento não deve ser utilizado após a data de validade ("Val:") presente na embalagem.
Este medicamento possui prazo de validade de 30 meses a partir da data de fabricação.
Número de lote e data de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
Vabysmo™ é uma solução estéril, sem conservante, límpida a opalescente, incolor a amarela amarronzada.
Vabysmo™ deve ser inspecionado visualmente após a remoção do refrigerador e antes da administração. Se partículas, turvação ou descoloração forem visíveis, o frasco não deve ser usado.
O conteúdo do frasco e a agulha de transferência com filtro são estéreis e de uso único. Não use se a embalagem, frasco e/ou agulha de transferência com filtro estiverem danificados ou vencidos.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto e o descarte em lixo doméstico deve ser evitado.
Quaisquer medicamentos não utilizados ou resíduos devem ser eliminados de acordo com os requerimentos locais.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Vabysmo™ deverá ser administrado por um(a) médico(a) qualificado(a) e experiente em injeções intravítreas. Cada frasco deverá ser utilizado apenas para o tratamento de um único olho.
Posologia
Degeneração macular relacionada à idade neovascular (DMRIn)
A dose recomendada para Vabysmo™ é de 6 mg (0,05 ml) administrada por injeção intravítrea a cada 4 semanas (mensalmente) para as primeiras 4 doses.
Posteriormente, recomenda-se uma avaliação da atividade da doença com base nos resultados anatômicos e/ou visuais, 20 e/ou 24 semanas após o início do tratamento, para que o tratamento possa ser individualizado. Em pacientes sem atividade da doença, deve ser considerada a administração de faricimabe a intervalos de 16 semanas (4 meses). Em pacientes com atividade da doença, deve considerar-se o tratamento a cada 8 semanas (2 meses) ou 12 semanas (3 meses). Existem dados de segurança limitados sobre intervalos de tratamento de 8 semanas ou menos entre as injeções. O monitoramento entre as visitas de administração deve ser programado com base no estado do paciente e segundo o critério do médico, mas não há necessidade de monitoramento mensal entre as injeções.
Edema macular diabético (EMD)
A dose recomendada para Vabysmo™ é de 6 mg (0,05 ml) administrada por injeção intravítrea a cada 4 semanas (mensalmente) para as primeiras 4 doses.
Posteriormente, o tratamento é individualizado utilizando uma abordagem de tratamento e extensão (treat-and-extend). Com base na avaliação do médico sobre os resultados anatômicos e/ou visuais do paciente, o intervalo entre doses pode ser estendido até cada 16 semanas (4 meses), em incrementos de até 4 semanas. Se os resultados anatômicos e/ou visuais se alterarem, o intervalo de tratamento deve ser ajustado em conformidade, e deve ser implementada uma redução do intervalo se os resultados anatômicos e/ou visuais se deteriorarem (vide item "2. Resultados de Eficácia"). Não foram estudados intervalos entre injeções inferiores a 4 semanas. O monitoramento entre as visitas de administração deve ser progr