UPTRAVI
JANSSEN-CILAG
selexipague
Anti-hipertensivo pulmonar.
Apresentações.
Comprimidos revestidos de 200 mcg, 400 mcg, 600 mcg, 800 mcg, 1000 mcg, 1200 mcg, 1400 mcg ou 1600 mcg:
embalagem com 60 comprimidos ou 140 comprimidos (somente na concentração 200 mcg).
USO ORAL. USO ADULTO.
Composição.
Uptravi® 200 mcg e Uptravi® 1400 mcg:
Cada comprimido revestido contém 200 mcg ou 1400 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro amarelo, cera de carnaúba.
Uptravi® 400 mcg:
Cada comprimido revestido contém 400 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro vermelho, cera de carnaúba.
Uptravi® 600 mcg e Uptravi® 1200 mcg:
Cada comprimido revestido contém 600 mcg ou 1200 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro vermelho, óxido de ferro preto, cera de carnaúba.
Uptravi® 800 mcg:
Cada comprimido revestido contém 800 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro amarelo, óxido de ferro preto, cera de carnaúba.
Uptravi® 1000 mcg:
Cada comprimido revestido contém 1000 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro vermelho, óxido de ferro amarelo, cera de carnaúba.
Uptravi® 1600 mcg:
Cada comprimido revestido contém 1600 mcg de selexipague. Excipientes: manitol, amido de milho, hidroxipropilcelulose, estearato de magnésio, hipromelose, propilenoglicol, dióxido de titânio, óxido de ferro vermelho, óxido de ferro amarelo, óxido de ferro preto, cera de carnaúba.
Informações técnicas.
1. INDICAÇÕES
Uptravi® é indicado para o tratamento de longo prazo da hipertensão arterial pulmonar (HAP, grupo I da OMS) para retardar a ocorrência de eventos de morbimortalidade em pacientes adultos com classe funcional (CF) II-III. Uptravi® pode ser utilizado em terapia combinada sequencial com antagonistas do receptor de endotelina (ARE) e/ou inibidores da fosfodiesterase 5 (PDE-5I), ou em monoterapia para pacientes que não sejam candidatos a estas terapias.
A eficácia foi demonstrada em uma população com HAP incluindo HAP idiopática e hereditária, HAP associada à doença do tecido conjuntivo, e HAP associada à cardiopatia congênita simples reparada.
2. RESULTADOS DE EFICÁCIA
Segurança e eficácia clínica
Eficácia em pacientes com HAP
O efeito do selexipague na progressão da HAP foi demonstrado em um estudo Fase 3, multicêntrico, de longo prazo (duração máxima da exposição de aproximadamente 4,2 anos), duplo-cego, controlado por placebo, de grupos paralelos, evento dirigido, em 1.156 pacientes com HAP sintomática (Classe funcional [CF] I - IV OMS). Os pacientes foram randomizados para placebo (N = 582) ou selexipague (N = 574) duas vezes ao dia. A dosagem foi aumentada em intervalos semanais por incrementos de 200 mcg administrados duas vezes ao dia, para determinar a dosagem de manutenção individualizada (200 - 1600 mcg duas vezes ao dia).
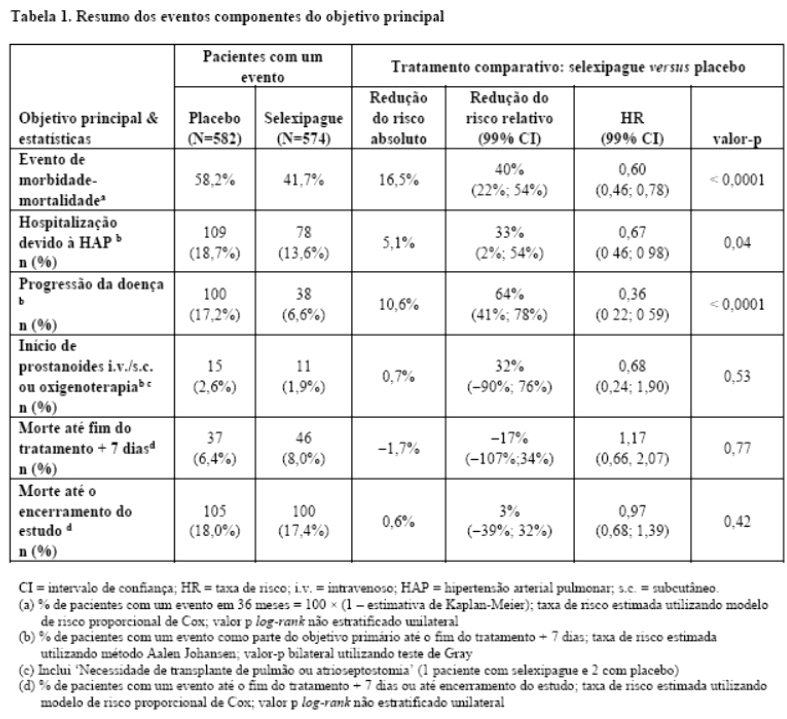
O desfecho primário do estudo foi o tempo para a ocorrência do primeiro evento de morbidade ou mortalidade até o fim do tratamento, definido por: a) morte (por todas as causas), b) hospitalização por HAP, c) piora da HAP resultando na necessidade de transplante de pulmão ou atriosseptostomia com balão, d) início de terapia parenteral prostanoide ou oxigenoterapia crônica, ou e) outros eventos de progressão da doença baseados em uma redução de 15% da distância de caminhada de 6 minutos (DTC6) basal mais a piora da classe funcional (para pacientes com classe funcional pela OMS/New York Heart Association [NYHA] modificada II-III no início do tratamento) ou necessidade de terapia específica adicional para HAP (para pacientes com classe funcional pela OMS/New York Heart Association [NYHA] modificada III-IV no início do tratamento).
Todos os eventos foram confirmados por um comitê de julgamento independente, cego em relação à alocação do tratamento.
A idade mediana foi de 48,1 anos (intervalo 18 - 80 anos de idade), com a maioria dos indivíduos sendo caucasianos (65,0%) e mulheres (79,8%). 17,9% dos pacientes tinham 65 anos de idade ou mais e 1,1% dos pacientes tinham 75 anos de idade ou mais. Aproximadamente 1%, 46%, 53% e 1% dos pacientes eram CF I, II, III e IV da OMS, respectivamente, no início do tratamento.
HAP idiopática ou hereditária foi a etiologia mais comum na população do estudo (58%), seguido por HAP devido a doenças do tecido conjuntivo (29%), HAP associada à cardiopatia congênita com shunts reparadores (10%), e HAP associada a outras etiologias (medicamentos e toxinas [2%] e HIV [1%]).
No início do estudo, a maioria dos pacientes envolvidos (80%) estavam sendo tratados com uma dosagem estável de uma terapia especifica para HAP, com um ARE (15%), ou com um inibidor PDE-5 (32%), ou com ambos ARE e inibidor PDE-5 (33%).
A duração mediana global do tratamento duplo-cego foi de 63,7 semanas para o grupo placebo e 70,7 semanas para o grupo com selexipague. Entre os pacientes no grupo de selexipague, 23% atingiram uma dose de manutenção na faixa de dose de 200-400 mcg; 31% atingiram uma dose de manutenção na faixa de 600-1000 mcg e 43% atingiram uma dose de manutenção na faixa de 1200-1600 mcg.
O tratamento com selexipague 200 - 1600 mcg duas vezes ao dia resultou em uma redução de 40% (razão de risco [HR] 0,60; 99% CI: 0,46, 0,78; log-rank valor-p unilateral < 0,0001) na ocorrência de eventos de morbidade ou mortalidade até 7 dias após a última dose em comparação ao placebo (Figura 1). O efeito benéfico do selexipague foi atribuído principalmente a uma redução na hospitalização por HAP e a uma redução em outros eventos de progressão da doença (tabela 1).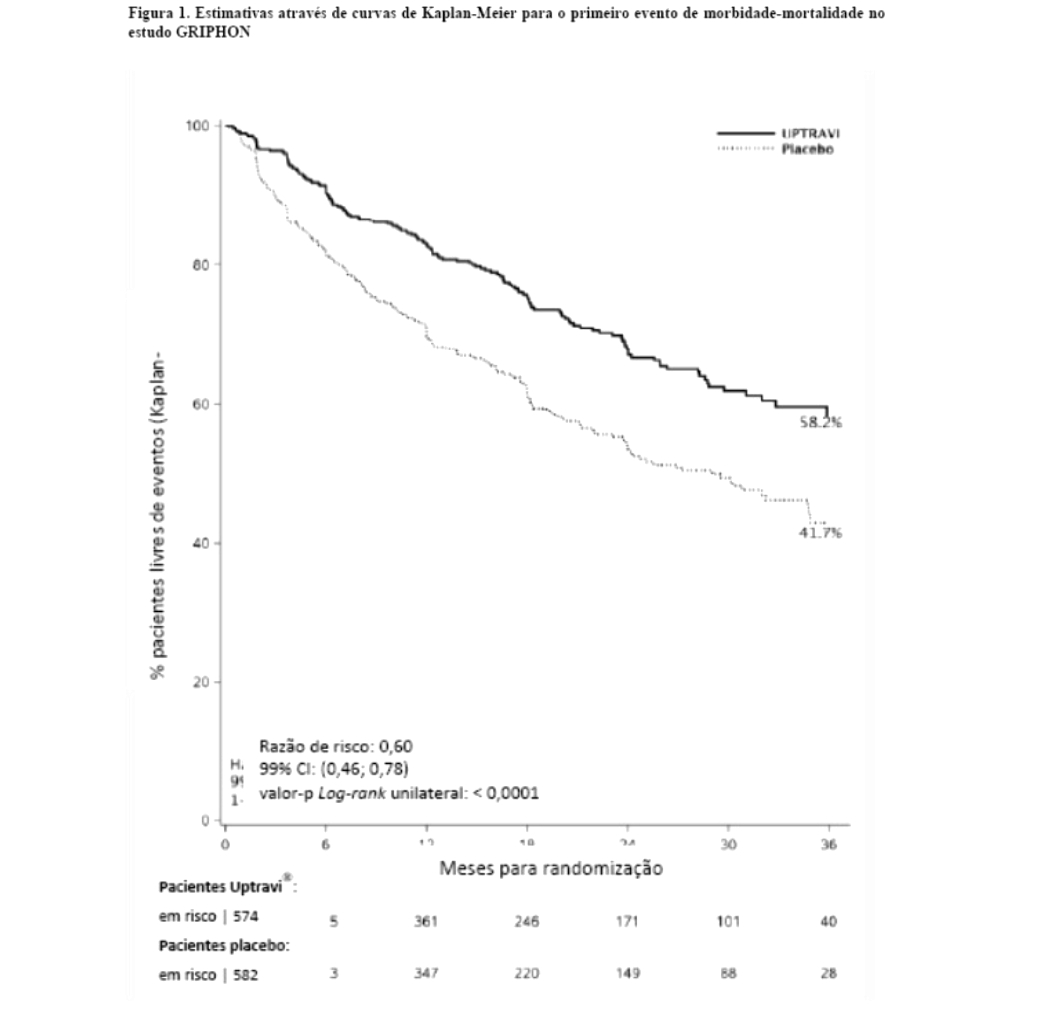
Desfecho sintomático - distância de caminhada de 6 minutos (DTC6)
A capacidade de exercício foi avaliada como um desfecho secundário. A DTC6 mediana no período basal foi de 376 m (faixa de 90-482 m) e 369 m (faixa de 50-515 m) nos pacientes dos grupos com selexipague e placebo, respectivamente. A alteração mediana absoluta na DTC6 medida no vale (isto é, aproximadamente 12 horas após a dose) na Semana 26 em relação ao valor basal foi de 4 metros para Uptravi® e de -9 metros para placebo. Isto resultou em um aumento mediano corrigido por placebo na DTC6 de 12 metros (IC 99%: 1,24 m, valor p unilateral = 0,0027). No subgrupo de pacientes sem terapia concomitante específica para HAP, o efeito do tratamento medido no vale foi de 34 metros (IC 99%: 10,63 m).
A qualidade de vida foi avaliada em um subgrupo de pacientes do estudo GRIPHON usando o questionário CAMPHOR (Cambridge Pulmonary Hypertension Outcome Review). Não houve efeito significativo do tratamento na Semana 26 em relação ao período basal.
Referências bibliográficas
1. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R, Rubin LJ, Di Scala L, Tapson V, Adzerikho I, Liu J, Moiseeva O, Zeng X, Simonneau G, McLaughlin VV; GRIPHON Investigators - N Engl J Med. 2015 Dec 24;373(26):2522-33. doi: 10.1056/NEJMoa1503184
Segurança pré-clínica
Nos estudos de toxicidade de doses repetidas em roedores, uma forte redução na pressão sanguínea como resultado da farmacologia exacerbada induziu sinais clínicos transitórios e reduziu o consumo de alimento e o ganho de peso. Em cães adultos e jovens, o intestino e o osso/medula óssea foram identificados como os principais órgãos alvo após o tratamento com selexipague. Um retardo no fechamento das placas de crescimento epifisárias femorais e/ou tibiais foi observado em cães jovens. O NOAEL para este efeito não foi estabelecido. Em cães com idade inferior a 1 ano, intussuscepção devido aos efeitos relacionados à prostaciclina na motilidade intestinal foi observada esporadicamente. As margens de segurança adaptadas para a potência do receptor IP para o metabólito ativo foram 2 vezes (com base na exposição total) em relação à exposição terapêutica humana. Estes achados não ocorreram em estudos de toxicidade em camundongos e ratos. Devido à sensibilidade espécie-específica em cães de desenvolver intussuscepção e à margem de segurança, este resultado é considerado não relevante em humanos adultos.
Ossificação óssea aumentada e alterações relacionadas na medula óssea em estudos com cães são considerados decorrentes da ativação dos receptores EP4 em cães. Uma vez que os receptores EP4 em humanos não são ativados pelo selexipague ou seu metabólito ativo, este efeito é espécie-específico e, portanto, não relevante em humanos.
Com base na evidência geral dos estudos de genotoxicidade conduzidos, o selexipague e seu metabólito ativo não são genotóxicos.
Em estudos de 2 anos sobre carcinogenicidade, o selexipague causou uma incidência aumentada de adenomas da tireoide em camundongos e adenomas de célula de Leydig em ratos. Os mecanismos são específicos para roedores. Tortuosidade na arteríola retinal foi observada após dois anos de tratamento apenas em ratos. Do ponto de vista mecânico, o efeito é considerado induzido pela vasodilatação de longa duração e por alterações subsequentes na hemodinâmica ocular. Achados histopatológicos adicionais do selexipague foram observados apenas em exposições suficientemente em excesso à máxima exposição humana, indicando pouca relevância em humanos.
O selexipague não foi teratogênico em ratos e coelhos (margens de exposição 13 vezes maior para selexipague e 43 vezes maior para o metabólito ativo, com base na exposição total). As margens de segurança para potenciais efeitos relacionados ao receptor IP na reprodução foram de 20 para a fertilidade e de 5 e 1 (com base na exposição livre) para o desenvolvimento embriofetal em ratos e coelhos, respectivamente, quando adaptados para diferenças na potência receptora. No estudo de desenvolvimento pré- / pós-natal em ratos, selexipague induziu efeitos na função reprodutiva maternal e dos filhotes.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Os efeitos vásculo-protetores da prostaciclina (PGI2) são mediados pelo receptor da prostaciclina (receptor IP). A redução na expressão dos receptores IP e a redução da síntese da prostaciclina contribuem para a fisiopatologia da HAP.
O selexipague é um agonista do receptor IP, seletivo, oral e é estruturalmente e farmacologicamente distinto da prostaciclina e seus análogos. O selexipague é hidrolisado por carboxilesterase para produzir seu metabólito ativo, que é aproximadamente 37 vezes mais potente que o selexipague. O selexipague e seu metabólito ativo são agonistas do receptor IP com alta afinidade, com uma alta seletividade para o receptor IP versus outros receptores prostanoides (EP1-EP4, DP, FP e TP). A seletividade contra EP1, EP3, FP e TP é importante porque estes são receptores contráteis descritos no trato gastrintestinal e vasos sanguíneos. A seletividade contra EP2, EP4 e DP1 é importante, uma vez que estes receptores mediam efeitos imunológicos depressivos.
A estimulação do receptor IP pelo selexipague e seu metabólito ativo leva à vasodilatação, assim como a efeitos antiproliferativos e antifibróticos. O selexipague melhora as variáveis hemodinâmicas e previne a remodelação cardíaca e pulmonar em modelos de HAP em ratos. Nestes ratos com HAP, a vasodilatação pulmonar e periférica em resposta correlacionada ao selexipague indica que a vasodilatação periférica reflete a eficácia farmacodinâmica pulmonar. O selexipague não causa dessensibilização do receptor IP in vitro nem taquifilaxia em modelos em ratos.
Os pacientes com HAP apresentam graus variáveis de expressão do receptor IP. Diferenças na dose de manutenção do selexipague entre indivíduos podem estar relacionadas às diferenças nos níveis de expressão do receptor IP.
Farmacodinâmica
Eletrofisiologia cardíaca
Em um estudo minucioso do intervalo QT em voluntários saudáveis, a administração de doses repetidas de 800 microgramas e 1600 microgramas de selexipague duas vezes ao dia não demonstrou um efeito de repolarização (intervalo QTc) ou condução cardíaca (intervalos PR e QRS) e apresentou um efeito acelerador leve na frequência cardíaca.
Hemodinâmica pulmonar
Um estudo clínico de fase 2, duplo-cego, controlado por placebo, avaliou as variáveis hemodinâmicas após 17 semanas de tratamento em pacientes com HAP classe funcional II-III da OMS e recebendo concomitantemente ARE (antagonista do receptor de endotelina) e/ou inibidores da PDE-5 (fosfodiesterase-5). Os pacientes titulando selexipague até uma dose tolerada individual (incrementos de 200 microgramas duas vezes ao dia até uma dose de 800 microgramas duas vezes ao dia; N=33) atingiram uma redução média estatisticamente significativa na resistência vascular pulmonar de 30,3% (intervalo de confiança 95% [CI]: - 44,7%, - 12,2%; P=0,0045) e um aumento no índice cardíaco (efeito médio do tratamento) de 0,48 L/min/m2, (CI 95%: 0,13, 0,83) comparado com o placebo (N=10).
Farmacocinética
A farmacocinética do selexipague e seu metabólito ativo foi estudada principalmente em voluntários saudáveis. A farmacocinética do selexipague e seu metabólito ativo, ambos após administração de dose única e doses múltiplas, foi dose-proporcional até uma dose única de 800 microgramas e doses múltiplas de até 1800 microgramas duas vezes ao dia. Após a administração de doses múltiplas, a condição de estado de equilíbrio do selexipague e do seu metabólito ativo foi atingida dentro de 3 dias. Após a administração de doses múltiplas, não ocorreu nenhum acúmulo no plasma, tanto do composto principal quanto do metabólito ativo.
Em voluntários saudáveis, a variabilidade intersujeitos na exposição (área sob a curva durante um intervalo de dose) no estado de equilíbrio foi de 43% e 39% para o selexipague e o metabólito ativo, respectivamente. A variabilidade intrasujeito na exposição foi de 24% e 19% para o selexipague e o metabólito ativo, respectivamente.
A exposição ao selexipague e ao metabólito ativo no estado de equilíbrio em pacientes com HAP e em indivíduos saudáveis foi similar. A farmacocinética do selexipague e do metabólito ativo em pacientes com HAP não foi influenciada pela gravidade da doença e não alterou com o tempo.
Absorção
O selexipague é rapidamente absorvido e é hidrolisado por carboxilesterase no fígado para seu metabólito ativo.
As concentrações plasmáticas máximas observadas para o selexipague e seu metabólito ativo após a administração oral são atingidas em 1-3 horas e 3-4 horas, respectivamente.
A biodisponibilidade absoluta de selexipague é de aproximadamente 49%.
Na presença de alimentos, a exposição ao selexipague após a administração de dose única de 400 microgramas foi aumentada em 10% em indivíduos caucasianos e reduzida em 15% em japoneses, enquanto a exposição ao metabólito ativo foi reduzida em 27% (indivíduos caucasianos) e 12% (japoneses). Os indivíduos reportaram um número maior de eventos adversos após a administração do medicamento em jejum do que após uma refeição.
Distribuição
O selexipague e seu metabólito ativo são altamente ligados às proteínas plasmáticas (aproximadamente 99% no total, e na mesma extensão à albumina e alfa 1 glicoproteína ácida).
O volume de distribuição de selexipague em estado de equilíbrio é de 11,7L.
Biotransformação
O selexipague é hidrolisado ao seu metabólito ativo no fígado e no intestino por carboxilesterase. O metabolismo oxidativo catalisado principalmente por CYP2C8 e, a uma menor extensão, por CYP3A4, leva à formação de produtos hidroxilados e dealquilados. UGT1A3 e UGT2B7 estão envolvidos na glicuronidação do metabólito ativo. Com exceção do metabólito ativo, nenhum dos metabólitos circulantes no plasma humano excede 3% do total do material relacionado ao fármaco. Tanto em voluntários saudáveis quanto em pacientes com HAP, a exposição ao metabólito ativo em estado de equilíbrio, após a administração oral, é de aproximadamente 3 a 4 vezes maior que o composto principal.
Eliminação
A eliminação do selexipague se dá predominantemente via metabolismo, com meia-vida terminal média de 0,8 - 2,5 h. O metabólito ativo apresenta meia-vida de 6,2 - 13,5h. A depuração total do selexipague no organismo é de 17,9 L/h. A excreção em voluntários saudáveis se deu por completo 5 dias após a administração e ocorreu principalmente pelas fezes (contabilizando 93% da dose administrada) comparada com 12% pela urina.
Populações especiais
Não foi observado nenhum efeito clinicamente relevante na farmacocinética do selexipague e seu metabólito ativo com relação ao gênero, raça, idade ou peso corporal em voluntários saudáveis e em pacientes com HAP.
Insuficiência Renal
Um aumento de 1,4 - 1,7 na exposição (concentração plasmática máxima e área sob a curva de tempo-concentração plasmática) ao selexipague e seu metabólito ativo foi observado em pacientes com insuficiência renal grave (taxa de filtração glomerular estimada < 30 mL/min/1,73 m2).
Insuficiência hepática
Em indivíduos com insuficiência hepática leve (Child-Pugh classe A) ou moderada (Child-Pugh classe B), após a administração de dose única de 400 microgramas de selexipague, a exposição ao selexipague foi 2 e 4 vezes maior, respectivamente, quando comparado com voluntários saudáveis. A exposição ao metabólito ativo permaneceu quase inalterada em indivíduos com insuficiência hepática leve e dobrou em indivíduos com insuficiência hepática moderada. Apenas dois indivíduos com insuficiência hepática grave (Child-Pugh classe C) receberam tratamento com selexipague. A exposição ao selexipague e seu metabólito ativo nestes dois indivíduos foi similar àquela observada em indivíduos com insuficiência hepática moderada (Child-Pugh classe B).
Com base no modelo farmacocinético dos dados de um estudo em indivíduos com comprometimento hepático, espera-se que a exposição ao metabólito ativo em estado de equilíbrio, em indivíduos com comprometimento hepático moderado (Child-Pugh classe B), após o regime de dose de uma vez ao dia, seja semelhante àquela em indivíduos saudáveis recebendo um regime de dose de duas vezes ao dia. A exposição ao selexipague em estado de equilíbrio em indivíduos com comprometimento hepático moderado durante o regime de dose de uma vez ao dia é prevista como aproximadamente duas vezes àquela observada em indivíduos saudáveis recebendo o regime de duas vezes ao dia.
4. CONTRAINDICAÇÕES
Uptravi® está contraindicado em caso de hipersensibilidade à substância ativa ou a qualquer um dos excipientes.
Uptravi® é contraindicado com uso concomitante de inibidores fortes de CYP2C8 (ex. genfibrozila) (ver Interações Medicamentosas).
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipotensão
Uptravi® possui propriedades vasodilatadoras que podem resultar na diminuição da pressão sanguínea. Antes de prescrever Uptravi®, os médicos devem considerar se os pacientes possuem certas condições subjacentes que possam ser afetadas negativamente por seus efeitos vasodilatadores (por exemplo: pacientes com terapia anti-hipertensiva ou com hipotensão de repouso, hipovolemia, obstrução grave da via de saída do ventrículo esquerdo ou disfunção autonômica).
Hipertireoidismo
Hipertireoidismo foi observado com Uptravi®. Na presença de sintomas de hipertireoidismo, testes de função da tireoide são recomendados, conforme clinicamente indicado.
Doença Pulmonar Veno-oclusiva
Casos de edema pulmonar foram relatados com vasodilatadores (principalmente prostaciclinas) quando utilizados em pacientes com doença pulmonar veno-oclusiva. Consequentemente, se sinais de edema pulmonar ocorrerem quando Uptravi® for administrado em pacientes com HAP, a possibilidade de doença pulmonar veno-oclusiva deve ser considerada. Se confirmado, o tratamento com Uptravi® deve ser descontinuado.
Gravidez e lactação
Gravidez
Não há dados sobre o uso de selexipague em mulheres grávidas. Estudos em animais não indicam efeitos nocivos diretos ou indiretos em relação à toxicidade reprodutiva. Selexipague e seu principal metabólito demonstraram potência receptora a prostaciclina (IP) 20 a 80 vezes menor in vitro para espécies animais usadas em testes de toxicidade reprodutiva, em comparação com seres humanos. Portanto, as margens de segurança para potenciais efeitos mediados pelo receptor IP na reprodução são menores do que para efeitos não relacionados à IP.
Uptravi® não é recomendado durante a gravidez e em mulheres em idade fértil que não utilizam métodos contraceptivos.
Categoria de risco na gravidez: B. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Lactação
Não se sabe se o selexipague ou seus metabólitos são excretados no leite materno.
Em ratos, selexipague e seus metabólitos são excretados no leite (vide "Características Farmacológicas").
A amamentação não é recomendada durante o tratamento com Uptravi®.
Populações especiais
População Pediátrica ( < 18 anos)
A segurança e a eficácia de Uptravi® em crianças não foram estabelecidas. Não existem dados disponíveis sobre o uso de Uptravi® em pacientes pediátricos. Portanto, a administração de selexipague a pacientes pediátricos não é recomendada. Estudos em animais indicaram um risco aumentado de intussuscepção intestinal, mas a relevância desses achados para humanos é desconhecida.
Idosos (≥ 65 anos)
Nenhum ajuste no regime posológico é necessário em pacientes idosos.
Devido à experiência limitada com Uptravi® em pacientes com mais de 75 anos, o uso nesta população deve ser feito com cautela.
Insuficiência renal
Em pacientes com insuficiência renal grave (taxa de filtração glomerular estimada < 30 mL/min/1,73 m2) devem ser tomadas precauções durante o ajuste de dose. Não há experiência com Uptravi® em pacientes em diálise, portanto Uptravi® não deve ser utilizado nesses pacientes.
Insuficiência hepática
Não há experiência clínica com selexipague em pacientes com insuficiência hepática grave (Child-Pugh classe C); portanto, Uptravi® não deve ser administrado a esses pacientes.
Recomenda-se o regime de dose de uma vez ao dia a pacientes com comprometimento hepático moderado (Child-Pugh classe B) devido ao aumento na exposição ao selexipague e ao seu metabólito ativo nesta população.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não há estudos sobre o efeito do selexipague sobre a capacidade de conduzir e utilizar máquinas. No entanto, Uptravi® pode ter uma influência pequena na habilidade de dirigir e operar máquinas. A situação clínica do paciente e o perfil de eventos adversos de selexipague (como dor de cabeça e hipotensão) devem ser levados em consideração.
6. INTERAÇÕES MEDICAMENTOSAS
Estudos in vitro
O selexipague é hidrolisado ao seu metabólito ativo através da carboxilesterase (vide "Características farmacológicas"). O selexipague e seu metabólito ativo são submetidos ao metabolismo oxidativo principalmente por CYP2C8 e, a uma menor extensão, por CYP3A4. A glicuronidação de seu metabólito ativo é catalisada pela UGT1A3 e UGT2B7. O selexipague e seu metabólito ativo são substratos da OATP1B1 e OATP1B3. O selexipague é substrato do P-gp e o metabólito ativo é um substrato da proteína transportadora resistente ao câncer de mama (BCRP).
O selexipague e seu metabólito ativo não inibem nem induzem as enzimas do citocromo P450 ou proteínas transportadoras em concentrações clinicamente relevantes.
Estudos in vivo
Anticoagulantes e inibidores de agregação plaquetária: O selexipague é um inibidor da agregação plaquetária in vitro. Em estudos controlados por placebo, de fase 3, em pacientes com HAP, não foi detectado aumento no risco de sangramento com o selexipague quando comparado ao placebo, incluindo quando o selexipague foi administrado com anticoagulantes (tais como a heparina e anticoagulantes cumarínicos) ou inibidores da agregação plaquetária. Em estudos com voluntários saudáveis, selexipague (400 microgramas duas vezes ao dia) não alterou a exposição à S-varfarina (substrato da CYP2C9) ou R-varfarina (substrato do CYP3A4) após dose única de 20 mg de varfarina. O selexipague não influenciou o efeito farmacodinâmico da varfarina na relação normalizada internacional. A farmacocinética do selexipague e seu metabólito ativo não foram afetados pela varfarina.
Lopinavir/ritonavir: Na administração duas vezes ao dia de 400/100 mg de lopinavir/ritonavir, um inibidor forte da CYP3A4, OATP (OATP1B1 e OATP1B3) e inibidor de P-gp, a exposição ao selexipague aumentou aproximadamente duas vezes, enquanto a exposição ao metabólito ativo de selexipague não foi alterada. Considerando que o metabólito ativo é responsável pela maior parte do efeito farmacológico do selexipague, este efeito não é considerado clinicamente significativo. Como um inibidor forte da CYP3A4 não afeta a farmacocinética do metabólito ativo, indicando que a via do CYP3A4 não é importante na eliminação do metabólito ativo, não é esperado nenhum efeito dos indutores da CYP3A4 na farmacocinética do metabólito ativo. Com base nesses dados, também não se espera um efeito clinicamente significativo na inibição dos transportadores (OATP1B1, OATP1B3 e P-gp).
Genfibrozila: Na administração duas vezes ao dia de 600 mg de genfibrozila, um forte inibidor de CYP2C8, a exposição ao selexipague aumentou aproximadamente duas vezes, ao passo que a exposição ao metabólito ativo aumentou aproximadamente 11 vezes. A administração concomitante de Uptravi® com fortes inibidores de CYP2C8 (por exemplo, genfibrozila) é contraindicada (vide "Contraindicações").
Clopidogrel: a administração concomitante de selexipague com clopidogrel (300 mg como dose de ataque ou dose de manutenção de 75 mg uma vez ao dia), um inibidor moderado de CYP2C8, não teve efeito relevante na exposição ao selexipague e aumentou a exposição ao metabólito ativo em aproximadamente 2,2 a 2,7 vezes após dose de ataque e dose de manutenção, respectivamente (vide "Posologia e Modo de usar").
Rifampicina: Na administração uma vez ao dia de 600 mg de rifampicina, um indutor das enzimas CYP2C8 e UGT, a exposição ao selexipague não foi alterada, ao passo que a exposição ao metabólito ativo foi reduzida pela metade. Pode ser necessário ajuste de dose de Uptravi®.
Midazolam: No estado estacionário após o ajuste de dose crescente para 1600 mg de selexipague duas vezes ao dia, não foi observada nenhuma alteração na exposição ao midazolam, um substrato do CYP3A4 intestinal e hepático-sensível, ou seu metabólito, 1-hidroximidazolam. A administração concomitante de selexipague com substratos CYP3A4 não requer ajuste de dose.
Inibidores da UGT1A3 e UGT2B7: o efeito de fortes inibidores de UGT1A3 e UGT2B7 na exposição do selexipague ou seu metabólito ativo não foi estudado. A administração concomitante pode resultar em um aumento significativo de selexipague ou de seu metabólito ativo.
Contraceptivos hormonais: Não foram conduzidos estudos específicos de interação fármaco-fármaco com contraceptivos orais. Uma vez que selexipague não afeta a exposição ao substrato da CYP3A4, midazolam e R-varfarina, ou à S-varfarina, substrato da CYP2C9, não se espera redução na eficácia dos contraceptivos hormonais.
Interações Farmacodinâmicas
Podem ocorrer reduções na pressão sanguínea quando Uptravi® for administrado com diuréticos, agentes anti-hipertensivos ou outros vasodilatadores.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Uptravi® deve ser mantido em temperatura ambiente (entre 15 e 30°C).
Prazo de validade: 36 meses a partir da data de fabricação.
Número de lote, datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas e organolépticas
Uptravi® 200 mcg comprimidos revestidos
Comprimido revestido redondo, amarelo claro, com o número '2' gravado em um dos lados.
Uptravi® 400 mcg comprimidos revestidos
Comprimido revestido redondo, vermelho, com o número '4' gravado em um dos lados.
Uptravi® 600 mcg comprimidos revestidos
Comprimido revestido redondo, lilás claro, com o número '6' gravado em um dos lados.
Uptravi® 800 mcg comprimidos revestidos
Comprimido revestido redondo, verde, com o número '8' gravado em um dos lados.
Uptravi® 1.000 mcg comprimidos revestidos
Comprimido revestido redondo, laranja, com o número '10' gravado em um dos lados.
Uptravi® 1.200 mcg comprimidos revestidos
Comprimido revestido redondo, roxo escuro, com o número '12' gravado em um dos lados.
Uptravi® 1.400 mcg comprimidos revestidos
Comprimido revestido redondo, amarelo escuro, com o número '14' gravado em um dos lados.
Uptravi® 1.600 mcg comprimidos revestidos
Comprimido revestido redondo, marrom, com o número '16' gravado em um dos lados.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
Posologia
Ajuste de dose individualizado
O objetivo é atingir uma dose individualizada adequada para cada paciente (dose de manutenção individualizada).
A dose inicial recomendada de Uptravi® é de 200 microgramas administrado duas vezes ao dia, com diferença de aproximadamente 12 horas. A dose é aumentada em incrementos de 200 microgramas administrados duas vezes ao dia, geralmente em intervalos semanais, até que o paciente apresente eventos farmacológicos adversos que não possam mais ser tolerados ou medicamente gerenciados, ou até que a dose máxima de 1600 microgramas duas vezes ao dia seja atingida. Durante o ajuste de dose, recomenda-se não descontinuar o tratamento no caso de efeitos colaterais farmacologicamente esperados, uma vez que, geralmente, estes efeitos são transitórios ou gerenciáveis com tratamento sintomático (vide "Reações Adversas"). Se o paciente atingir uma dose que não pode mais ser tolerada, a dose deve ser reduzida para o nível de dosagem anterior.
Dose de Manutenção Individualizada
A maior dose tolerada atingida durante a fase de ajuste de dose deve ser mantida. Se, no decorrer do tempo, o tratamento tornar-se menos tolerável na dose administrada, deve ser considerado tratamento sintomático ou redução da dose para o próximo nível mais baixo.
Interrupções ou descontinuações
Se uma dose do medicamento for esquecida, ela deve ser administrada o quanto antes. A dose esquecida não deve ser administrada se já estiver quase na hora da próxima dose (dentro de aproximadamente 6 horas).
Se o tratamento for esquecido por 3 dias ou mais, Uptravi® deve ser reiniciado com uma dose menor e então ajustado. Existe experiência limitada com a descontinuação abrupta de Uptravi® em pacientes com HAP. Nenhuma evidência de efeito rebote agudo foi observada. No entanto, se for decido interromper o tratamento com Uptravi®, a retirada deve ser feita gradualmente, enquanto se introduz uma terapia alternativa.
Ajuste de dose com administração conjunta com inibidores moderados de CYP2C8
Quando administrado em conjunto com inibidores moderados de CYP2C8 (ex. clopidogrel, deferasirox e teriflunomida), reduzir a dose de Uptravi® para uma vez ao dia. Retornar a frequência de dose de duas vezes ao dia de Uptravi® quando a administração conjunta de inibidores moderados de CYP2C8 for interrompida (vide "Interações medicamentosas").
Método de administração
Os comprimidos revestidos devem ser ingeridos pela manhã e à noite, com um pouco de água.
Para aumentar a tolerabilidade, é recomendado que Uptravi® seja administrado com alimento.
Não há estudos sobre os efeitos de Uptravi® administrado por vias não recomendadas. Portanto, por segurança e para garantir a eficácia deste medicamento, a administração deve se dar somente por via oral.
O tratamento com Uptravi® deve continuar durante o período que o médico indicar.
Este medicamento não deve ser partido, aberto ou mastigado.
Populações especiais
População Pediátrica ( < 18 anos)
A segurança e a eficácia de Uptravi® em crianças não foram estabelecidas.
Não existem dados disponíveis sobre o uso de Uptravi® em pacientes pediátricos. Portanto a administração de selexipague a pacientes pediátricos não é recomendada. Estudos em animais indicaram um risco aumentado de intussuscepção intestinal, mas a relevância desses achados para humanos é desconhecida.
Uso em Idosos (≥ 65 anos)
Nenhum ajuste no regime posológico é necessário em pacientes idosos.
Devido à experiência limitada com Uptravi® em pacientes com mais de 75 anos, o uso nesta população deve ser feito com cautela.
Insuficiência renal
Nenhum ajuste no regime posológico é necessário em pacientes com insuficiência renal leve a moderada.
Nenhuma alteração de dosagem inicial é requerida em pacientes com insuficiência renal grave. Deve-se ter cautela durante o ajuste de dose em pacientes com insuficiência renal grave (taxa de filtração glomerular estimada < 30 mL/min/1,73 m2). Não há experiência do uso de Uptravi® em pacientes submetidos à diálise.
Insuficiência hepática
Nenhum ajuste no regime posológico é necessário em pacientes com insuficiência hepática leve (isto é, Child-Pugh classe A).
Recomenda-se o regime de dose de uma vez ao dia a pacientes com comprometimento hepático moderado (Child-Pugh classe B) devido ao aumento na exposição ao selexipague e ao seu metabólito ativo nesta população.
Não há experiência clínica do uso de Uptravi® em pacientes com insuficiência hepática grave (Child-Pugh classe C).
9. REAÇÕES ADVERSAS
As reações adversas ao fármaco mais comumente relatadas relacionadas aos efeitos farmacológicos de Uptravi® são cefaleia, diarreia, náusea e vômito, dor na mandíbula, mialgia, dor nas extremidades, rubor e artralgia. Estas reações são mais frequentes durante a fase de ajuste de dose. A maioria destas reações é de intensidade leve a moderada.
A segurança do selexipague foi avaliada em estudos de fase 3 de longo prazo, controlados por placebo, envolvendo 1.156 pacientes com HAP sintomática. A duração mediana de tratamento foi de 76,4 semanas (mediana 70,7 semanas) para os pacientes recebendo selexipague versus 71,2 semanas (mediana 63,7 semanas) para os pacientes recebendo placebo. A exposição ao selexipague foi de até 4,2 anos.
As reações adversas associadas ao selexipague durante o período completo de tratamento neste estudo são apresentadas na tabela abaixo. A frequência é reportada de acordo com o Conselho das Organizações Internacionais de Ciências Médicas - CIOMS: muito comum (≥ 1/10), comum (≥ 1/100 a < 1/10), incomum (≥ 1/1000 a < 1/100), raro (≥ 1/10.000 a < 1/1000), e muito raro ( < 1/10.000).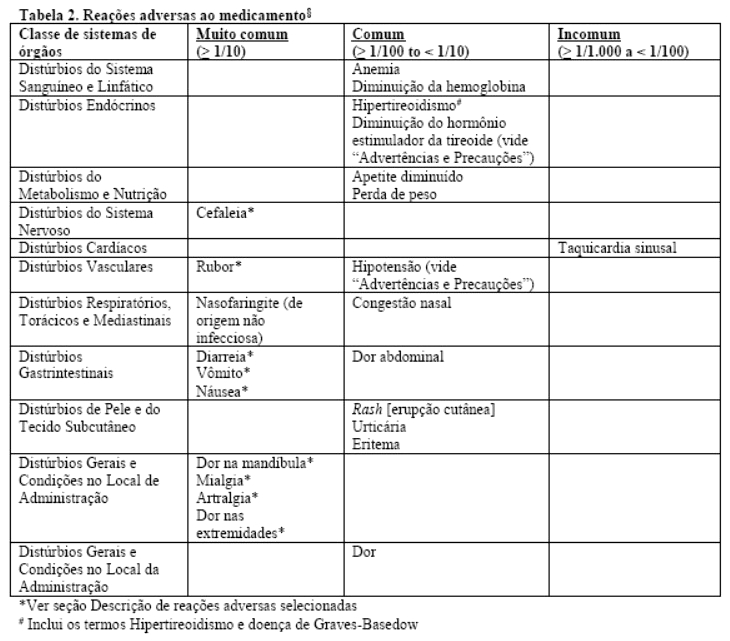
Descrição de reações adversas selecionadas
Efeitos farmacológicos associados ao ajuste de dose e manutenção do tratamento
As reações adversas associadas ao modo de ação de selexipague foram observadas com frequência, em particular durante a fase de titulação de dose individualizada, e estão tabuladas abaixo. Estes efeitos geralmente são transitórios ou gerenciáveis com tratamento sintomático.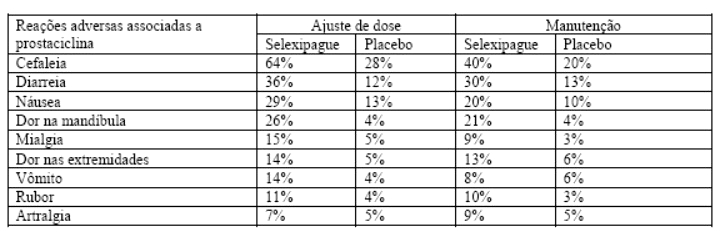
Aumento da frequência cardíaca
No estudo de fase 3 controlado por placebo em pacientes com HAP, observou-se um aumento transitório na frequência cardíaca média de 3-4 bpm 2-4 horas pós-dose. As investigações de eletrocardiograma mostraram taquicardia sinusal em 11,3% dos pacientes no grupo selexipague em comparação com 8,8% no grupo placebo
Anormalidades laboratoriais
Hemoglobina
Em um estudo de fase 3, controlado por placebo, em pacientes com HAP, alterações absolutas médias na hemoglobina nas visitas regulares comparadas ao período basal variaram de -0,34 a -0,02 g/dL no grupo recebendo selexipague comparado com - 0,05 a 0,25 g/dL no grupo recebendo placebo. Uma redução, a partir do período basal, na concentração de hemoglobina para menos de 10 g/dL foi relatada em 8,6% dos pacientes tratados com selexipague e 5,0% dos pacientes tratados com placebo.
Testes de função da tireoide
Em um estudo de fase 3, controlado por placebo, em pacientes com HAP, hipertireoidismo ou doença de Graves-Basedow foram relatados em 1,6% dos pacientes no grupo selexipague, comparado a nenhum caso no grupo placebo (ver Advertências e Precauções). Na maioria das visitas dos pacientes do grupo recebendo selexipague foi observada uma redução na média do hormônio estimulante da tireoide (TSH) (de até 0,3 UM/L, a partir de uma mediana basal de 2.5 UM/L). No grupo placebo, uma pequena alteração nos valores médios foi aparente. Não houve alteração na média tri-iodotironina ou tiroxina em ambos os grupos.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique ao Sistema de Notificação de Eventos Adversos a Medicamentos - VIGIMED, disponível em http://portal.anvisa.gov.br/vigimed, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Casos isolados de superdose até 3200 mcg foram reportados. Náusea leve e transitória foi a única consequência relatada. Em caso de superdose, devem ser tomadas as medidas de suporte habituais, conforme requerido.
Selexipague e seu metabólito ativo são altamente ligados às proteínas; sendo assim, é pouco provável que a diálise seja efica