UPELIOR
RECORDATI
pasireotida
Tratamento da doença de Cushing.
Apresentações.
Solução injetável Upelior 0,3 mg/1 mL - embalagens contendo 60 ampolas de 1 mL. Upelior 0,6 mg/1 mL - embalagens contendo 60 ampolas de 1 mL. Upelior 0,9 mg/1 mL - embalagens contendo 60 ampolas de 1 mL.
VIA SUBCUTÂNEA
USO ADULTO
Composição.
Cada ampola com 1 mL de solução injetável de Upelior® 0,3 mg contém 0,3762 mg de diaspartato de pasireotida (equivalente a 0,3 mg de pasireotida base). Cada ampola com 1 mL de solução injetável de Upelior® 0,6 mg contém 0,7524 mg de diaspartato de pasireotida (equivalente a 0,6 mg de pasireotida base). Cada ampola com 1 mL de solução injetável de Upelior® 0,9 mg contém 1,1286 mg de diaspartato de pasireotida (equivalente a 0,9 mg de pasireotida base). Excipientes: manitol, ácido tartárico, hidróxido de sódio, água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Upelior® é um análogo da somatostatina indicado para o tratamento de pacientes adultos com Doença de Cushing, nos quais a cirurgia hipofisária não é uma opção ou não foi curativa.
2. RESULTADOS DE EFICÁCIA1,2,3
Um estudo de Fase III, multicêntrico e randomizado foi conduzido para avaliar a segurança e a eficácia de diferentes doses de Upelior® em um período de tratamento de doze meses com pacientes com Doença de Cushing persistente ou recorrente para os quais a cirurgia não era indicada ou que recusaram a cirurgia. O estudo incluiu 162 pacientes com cortisol livre urinário basal > 1,5 x LSN - limite superior da normalidade que foram randomizados em uma proporção de 1:1 para receber uma dose de 0,6 mg 2x/dia s.c. ou 0,9 mg 2x/dia s.c. de Upelior®. Após três meses de tratamento, os pacientes que tinham média de cortisol livre urinário de 24 horas ≤ 2x LSN e abaixo ou igual aos seus valores basais, continuaram o tratamento cego na dose randomizada até o 6° mês. Os pacientes que não satisfizeram esses critérios tiveram seu tratamento revelado e a dose foi aumentada em 0,3 mg 2x/dia. Após os 6 meses iniciais do estudo, os pacientes entraram em um período de tratamento adicional de 6 meses com rótulo aberto. Caso a resposta não fosse alcançada em 6 meses ou não fosse mantida durante o período de tratamento aberto, a dosagem podia ser aumentada em 0,3 mg s.c. 2x/dia. A dose máxima administrada aos pacientes foi de 1,2 mg s.c. 2x/dia. A dose podia ser diminuída em reduções de 0,3 mg 2x/dia a qualquer momento durante o estudo em caso de intolerância. O critério primário de eficácia foi a proporção de pacientes em cada braço que alcançaram normalização do cortisol livre urinário médio de 24 horas (cortisol livre urinário ≤ LSN) após 6 meses de tratamento e que não sofreram elevação da dose (com relação à dose randomizada) durante este período. Critérios secundários incluíram, entre outros, alterações com relação aos valores iniciais antes do início do tratamento: cortisol livre urinário de 24 horas, ACTH plasmático, níveis de cortisol sérico, sinais e sintomas da Doença de Cushing e qualidade de vida relacionada à saúde (QVRS) conforme medida pelo Cushing QoL4. Todas as análises foram conduzidas com base nos grupos de dose randomizada. Os dados demográficos basais foram bem equilibrados entre os dois grupos e consistentes com a epidemiologia da doença. A idade média dos pacientes foi de aproximadamente 40 anos, com uma predominância de mulheres (77,8%). A maioria dos pacientes tinha Doença de Cushing persistente ou reincidente (83,3%) e poucos pacientes (≤ 5%) de ambos os grupos de tratamento tinham recebido irradiação prévia na hipófise. As características basais foram equilibradas entre os dois grupos randomizados, exceto por marcantes diferenças no valor médio do cortisol livre urinário de 24 horas basal (1.156 nmol/24 hs para o grupo de 0,6 mg 2x/dia e 782 nmol/24 hs para o grupo de 0,9 mg 2x/dia); faixa normal de 30 a 145 nmol/24 hs.
Resultados
No 6° mês, a normalização dos níveis de cortisol livre urinário médios foi observada em 14,6% (IC 95% 7,0 a 22,3) e 26,3% (IC 95% 16,6 a 35,9) dos pacientes randomizados para pasireotida 0,6 mg 2x/dia e 0,9 mg 2x/dia, respectivamente. O estudo alcançou o objetivo primário de eficácia para o grupo de 0,9 mg 2x/dia, uma vez que o limite inferior para um Intervalo de Confiança de 95% é maior do que o limite pré-especificado de 15%. A resposta no braço de dose de 0,9 mg pareceu ser maior no caso de pacientes com média de cortisol livre urinário basal mais baixo (Tabela 1). A maioria dos que apresentaram resposta (55,6%) no 6° mês também o fizeram no 12° mês. A taxa de respostas no 12° mês foi comparável com a do 6° mês, com 13,4% e 25,0% no grupo de 0,6 mg 2x/dia e 0,9 mg 2x/dia, respectivamente.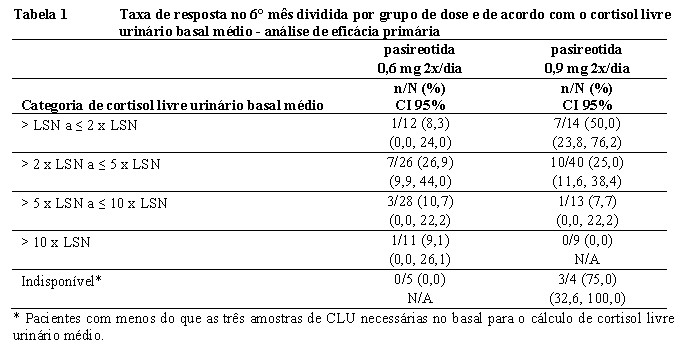
Uma análise de eficácia de apoio foi conduzida na qual os pacientes foram classificados novamente em 3 categorias de resposta independentemente da elevação da titulação no 3° mês: controlados (cortisol livre urinário ≤1,0 x LSN), parcialmente controlados (cortisol livre urinário > 1,0 x LSN mas com redução de cortisol livre urinário ≥ 50% em comparação com os valores basais) ou não controlados (todos os outros pacientes). As taxas de resposta para aqueles considerados controlados e parcialmente controlados no 6°mês constituíram 34% e 41% (0,6 mg 2x/dia e 0,9 mg 2x/dia, respectivamente) de pacientes randomizados (Tabela 2). Os pacientes que não evidenciavam controle tanto no 1° mês quanto no 2° mês tinham probabilidade de 90% de permanecer sem controle no 6° mês ou no 12° mês.
Em ambos os grupos de dose, o Upelior® resultou em uma redução rápida e robusta do cortisol livre urinário médio após o 1° mês de tratamento o qual foi mantido com o tempo (Figura 1). Reduções e elevações da dose pareceram ter efeito mínimo sobre a resposta do cortisol livre urinário, ainda que alguns pacientes tenham experimentado maior redução dos níveis de cortisol livre urinário com a titulação da dose.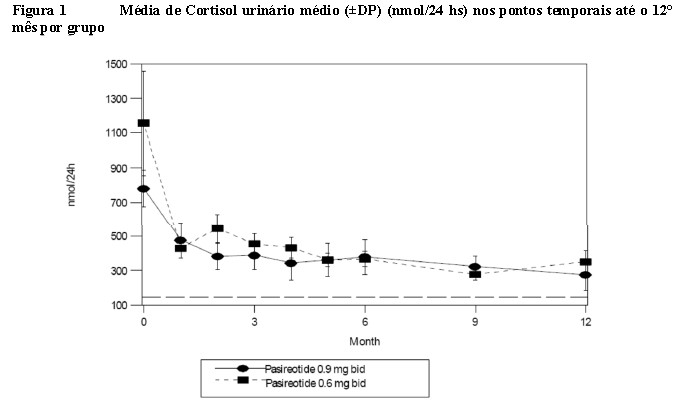
Observação: os pacientes foram randomizados para o Upelior® 0,6 mg ou 0,9 mg na linha de base. Pelo menos 3 amostras de cortisol livre urinário de 24 horas contribuíram para os resultados da média dos pacientes nos meses 0 (referência basal), 3, 6 e 12. Pelo menos 2 amostras de cortisol livre urinário de 24 horas contribuíram para os resultados médios nos outros pontos temporais. A linha de referência é o limite superior da normalidade para o cortisol livre urinário, que é de 145 nmol/24 h. (+/- erros-padrão são exibidos).
Reduções robustas também foram demonstradas pelo percentual geral de alteração nos níveis de cortisol livre urinário médios e medianos nos 6° mês e no 12° mês em comparação com os valores basais (Tabela 3). Reduções do cortisol sérico médio e dos níveis de ACTH plasmático também foram observadas em cada ponto temporal para cada grupo.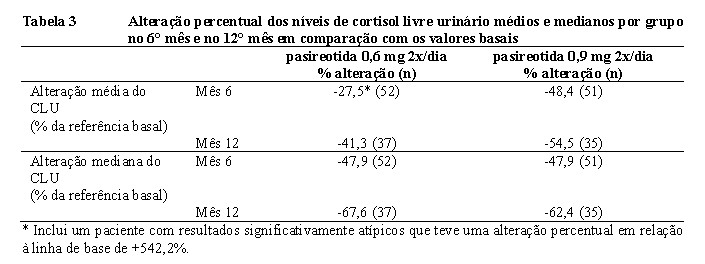
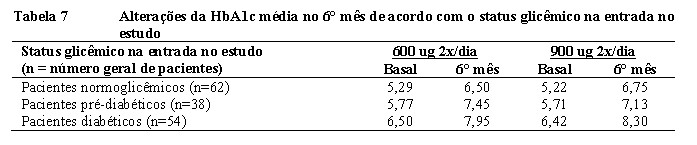
No geral, no mês 6, houve mudanças favoráveis em praticamente todos os sinais e sintomas estudados associados à Doença de Cushing (isto é, pressão sanguínea sistólica sentado, pressão sanguínea diastólica sentado, IMC, circunferência da cintura, colesterol total, escore da depressão de Beck, escore do hirsutismo de Ferriman-Galway e % da composição corporal-gordura localizada) em ambos os grupos. As reduções gerais desses parâmetros tendem a ser maiores nos pacientes que tiveram normalização dos níveis de cortisol livre urinário. Tendências similares foram observadas no 12° mês, com redução dos triglicerídeos séricos também nesse ponto temporal. Nenhuma alteração da densidade mineral óssea foi observada. O rubor facial melhorou em 36,7% (18/49) e 59,6% (28/47) dos pacientes tratados com 0,6 e 0,9 mg 2x/dia, respectivamente. Mais de um terço dos pacientes de ambos os grupos de tratamento também demonstrou melhoria do acúmulo de gordura supraclavicular e dorsal. Efeitos similares foram registrados na consulta do 12° mês. Os escores médios e globais médios no Cushing QoL foram similares para os dois grupos na primeira consulta. Na consulta do 3° mês, os pacientes de ambos os grupos relataram elevações dos escores, indicando melhora da QVRS relatada pelo paciente. No 6° mês, as melhorias medianas em relação à primeira consulta foram de 13,2% e 30% nos grupos de 0,6 e 0,9 mg 2x/dia, respectivamente. No 12° mês, as melhorias medianas em relação a primeira consulta foram de 26% e 20,6% nos grupos de 0,6 e 0,9 mg 2x/dia, respectivamente.
Referências bibliográficas
1. 2.5 Clinical Overview - Cushing's Disease. Novartis Pharma AG. Aug 2010.
2. Study CSOM230B2305 - A randomised, double-blind study to assess the safety and efficacy of different dose levels of Pasireotide (SOM230) s.c. over a 6 month treatment period in patients with de novo, persistent or recurrent Cushing's disease.
3. 2.7.3 Summary of Clinical Efficacy - Cushing's disease. Novartis Pharma AG. Aug 2010.
4. [Webb SM, Badia X, Barahona MJ et al (2008)] Evaluation of health-related quality of life in patients with Cushing's syndrome with a new questionnaire. Eur J Endocrinol; 158(5):623-30.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico
Somatostatina e análogos, H01CB05.
Mecanismo de ação
A pasireotida é um novo ciclohexapeptídeo injetável análogo da somatostatina. Assim como os hormônios peptídicos naturais, a somatostatina-14 e a somatostatina-28 (também conhecidos como Fator de Inibição da Liberação da Somatotropina [SRIF]) e outros análogos da somatostatina, a pasireotida exerce sua atividade farmacológica via ligação aos receptores da somatostatina (SSTR). Cinco subtipos de receptores da somatostatina humana são conhecidos: SSTR 1, 2, 3, 4 e 5. Esses subtipos de receptores são expressos em diferentes tecidos sob condições fisiológicas normais. Os análogos da somatostatina se ligam com potências diferentes a receptores SSTR (Tabela 4). A pasireotida se liga com alta afinidade a quatro dos cinco SSTRs.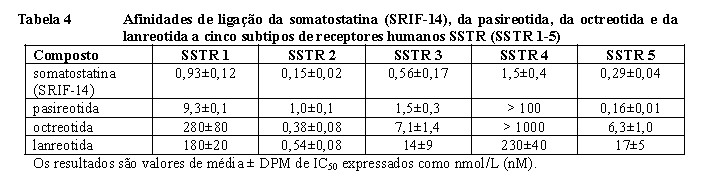
Farmacodinâmica
Os receptores da somatostatina são expressos em muitos tecidos, especialmente em tumores neuroendócrinos nos quais hormônios são secretados em excesso, incluindo o hormônio adrenocorticotrófico (ACTH) na Doença de Cushing. Devido ao seu amplo perfil de ligação aos receptores da somatostatina, a pasireotida tem o potencial de tratar doenças caracterizadas pela expressão desses receptores nos tecidos-alvo.
Estudos in vitro mostraram que células tumorais corticotróficas de pacientes com Doença de Cushing apresentam altos níveis de SSTR 5, enquanto os outros subtipos de receptores ou não são expressos ou são expressos em um nível mais baixo. A pasireotida se liga aos receptores SSTR dos corticotrofos de adenomas produtores de ACTH e os ativa, resultando em inibição da secreção de ACTH. A alta afinidade da pasireotida para quatro dos cinco SSTR, especialmente ao SSTR 5 (vide Tabela 4), proporciona a base para que o tratamento com a pasireotida seja eficaz para pacientes com Doença de Cushing.
Metabolismo da glicose
Em um estudo duplo-cego randomizado realizado em voluntários sadios para estudar o mecanismo do desenvolvimento da hiperglicemia após administração da pasireotida (Upelior® s.c) em doses de 600 e 900 microgramas 2x/dia observou-se reduções significativas na secreção de insulina, bem como das incretinas (ou seja, pepetídeo semelhante ao glucagon do tipo 1 [GLP-1] e polipeptídeos insulinotrópicos dependentes da glicose [GIP]). A pasireotida não afetou a sensibilidade à insulina. Em outro estudo randomizado realizado em voluntários sadios, os efeitos da pasireotida na glicose sanguínea foram investigados através da comparação entre as administrações de Upelior® s.c. 600 microgramas 2x/dia em monoterapia e com a coadministração de um medicamento anti-hiperglicêmico (metformina, nateglinida, vildagliptina ou liraglutida, respectivamente. A insulina não foi estudada) ao longo de um período de 7 dias. Nesse estudo, a terapia com incretinomiméticos (agonistas GLP-1 e inibidores DDP-IV) foi mais eficaz no tratamento da hiperglicemia associada a pasireotida em voluntários sadios.
Eletrofisiologia cardíaca
O efeito da pasireotida (administrada como Upelior® s.c.) sobre o intervalo QT foi avaliado em dois estudos desenhados para avaliação do QT, aberto, controlados e cruzados. No primeiro estudo que investigou uma dose de 1950 mg administrada 2x/dia, a alteração média máxima de QTcF com subtração do placebo com relação à referência basal (DDQTcI) foi de 17,5 ms (IC 90%: 15,53; 19,38). No segundo estudo, que investigou doses de 600 mg e 1950 mg 2x/dia, as alterações médias máximas do QTcF com subtração do placebo com relação às referências basais (DDQTcI) foram de 13,19 ms (IC 90%: 11,38; 15,01) e 16,12 ms (IC 90%: 14,30; 17,95 ms), respectivamente. Em ambos os estudos a alteração média máxima com subtração do placebo com relação à referência basal, ocorreu após 2 horas da dose. Ambas as doses de Upelior® reduziram a frequência cardíaca com uma diferença máxima em relação ao placebo observada em 1 hora para a dose de 600 mg 2x/dia (-10,39 bpm) e a 0,5 hora para a dose de 1950 mg 2x/dia (-14,91 bpm). Nenhum episódio de torsade de pointes foi observado (vide "Advertências e precauções").
O aumento do intervalo QT com a administração de pasireotida não é mediado por um efeito sobre o canal de potássio hERG. Restituição cardíaca, a capacidade do coração para se recuperar a partir de cada batida anterior, foi medida em contínuos ECGs de 24 horas para determinar o efeito da pasireotida na vulnerabilidade arrítmica. A pasireotida melhorou significativamente todos os parâmetros de restituição, na presença de prolongamento do intervalo QT, indicando que o prolongamento do intervalo QT mediado pela pasireotida pode não estar associado a um aumento do risco de pró-arritmia. Além disso, a análise morfológica quantitativa da onda T não mostrou alterações indicativas de heterogeneidade espacial que comprometesse a repolarização cardíaca durante o tratamento com a pasireotida.
Farmacocinética
Em voluntários sadios, a pasireotida demonstra farmacocinética aproximadamente linear para um intervalo que variou de 0,0025 a 1,5 mg em dose única. Em pacientes com Doença de Cushing, a pasireotida demonstra uma relação dose-exposição linear em um intervalo de dose de 0,3 a 1,2 mg com doses 2x/dia.
- Absorção
Em voluntários sadios, a pasireotida s.c. é rapidamente absorvida e o pico de concentração plasmática é alcançado em um Tmax de 0,25-0,5 horas. A Cmax e ASC são aproximadamente proporcionais à dose após a administração tanto de uma dose única quanto de doses múltiplas. Nenhum estudo foi conduzido para avaliar a biodisponibilidade da pasireotida em humanos. Com base nos dados de biodisponibilidade absoluta de estudos pré-clínicos com ratos e macacos, prevê-se que a biodisponibilidade absoluta da pasireotida s.c. seja completa em humanos. É improvável que haja efeito de alimentos, uma vez que o Upelior® é administrado pela via parenteral.
- Distribuição
Em voluntários sadios, a pasireotida é amplamente distribuída com um aparente volume de distribuição amplo (Vz/F > 100 L). A distribuição entre o sangue e o plasma é independente da concentração e mostra que a pasireotida se localiza principalmente no plasma (91%). A ligação às proteínas plasmáticas é moderada (88%) e independente da concentração. A pasireotida tem permeabilidade passiva baixa e tem probabilidade de ser um substrato de P-gp, mas esperase que o impacto da P-gp sobre o ADME (absorção, distribuição, metabolismo, excreção) da pasireotida seja baixo. Em doses terapêuticas, a pasireotida não é um substrato da BCRP (proteína de resistência ao câncer de mama), do OCT1 (transportador 1 de cátions orgânicos) nem dos OATP (polipeptídeos transportadores de ânions orgânicos) 1B1, 1B3 ou 2B1.
- Biotransformação/metabolismo
A pasireotida demonstrou alta estabilidade metabólica em microssomos do fígado e do rim humanos. Em voluntários sadios, a pasireotida em sua forma inalterada é a forma predominantemente encontrada no plasma, na urina e nas fezes.
- Eliminação
A pasireotida s.c. é eliminada principalmente pela via hepática (excreção biliar) com uma pequena contribuição da via renal. No estudo do ADME humano com pasireotida s.c. administrada em dose única de 600 microgramas, 55,9 ± 6,63% da dose de radioatividade foi recuperada após os primeiros 10 dias após a administração, incluindo 48,3 ± 8,16% da radioatividade nas fezes e 7,63 ± 2,03% na urina. A eliminação (CL/F) da pasireotida em voluntários sadios e em pacientes com Doença de Cushing é de ~7,6 litros/h e ~3,8 litros/h, respectivamente.
- Farmacocinética no estado de equilíbrio
Após múltiplas doses s.c., a pasireotida demonstra farmacocinética linear e independente do tempo no intervalo de dose de 0,05 a 0,6 mg 1x/dia em voluntários sadios, e 0,3 mg a 1,2 mg 2x/dia em pacientes com Doença de Cushing. Com base na taxa de acúmulo ASC, a meia-vida efetiva (t1/2,eff) em voluntários sadios foi de aproximadamente 12 horas (em média entre 10 e 13 horas para as doses de 0,05, 0,2 e 0,6 mg 1x/dia) (vide "Posologia e modo de usar").
Populações especiais
- Pacientes geriátricos (65 anos ou mais)
Descobriu-se que a idade é uma covariável na análise farmacocinética da população de pacientes com Doença de Cushing. Redução da eliminação corporal total e elevação das exposições da farmacocinética foram vistas com o aumento da idade. Em estudos na faixa etária de 18 a 73 anos, a área sob a curva no estado de equilíbrio para um intervalo de dosagem de 12 horas (ASCss) varia de 86% a 110% da área para um paciente típico de 41 anos. Esta variação é moderada e considerada de menor significância considerando a ampla faixa de idade na qual o efeito foi observado. Os dados referentes a pacientes com Doença de Cushing com mais de 65 anos de idade são limitados, mas não sugerem nenhuma diferença clinicamente significativa na segurança e eficácia em relação a pacientes mais jovens.
- Pacientes pediátricos
Nenhum estudo foi realizado com pacientes pediátricos.
- Pacientes com insuficiência renal
A via renal contribui pouco para a eliminação da pasireotida em humanos. Em um estudo clínico com administração de dose única de 900mg de pasireotida como Upelior® em pacientes com função renal comprometida, insuficiência renal leve, moderada, grave ou em estágio terminal, não teve impacto significativo na farmacocinética de pasireotida.
- Pacientes com insuficiência hepática
Em um estudo clínico com administração de dose única de 600mg de pasireotida administrada como Upelior® em pacientes com função hepática comprometida (Child-Pugh A, B e C), pacientes com comprometimento hepático moderado e grave (Child-Pugh B e C) apresentaram exposições significativamente mais elevadas do que pacientes com função hepática normal. Com a correção de efeito de covariáveis (idade, IMC e albumina), a ASCinf foi elevada em 60% e 79%, a Cmax se elevou em 67% e 69% e a CL/F se reduziu em 37% e 44%, respectivamente, nos grupos de comprometimento moderado e grave em relação ao grupo de controle.
- Demografia
Análise populacional da farmacocinética da pasireotida (administrada como Upelior® s.c.) sugerem que a raça e o gênero não influenciam os parâmetros farmacocinéticos. Descobriu-se que o peso corporal magro, que subtrai a gordura do peso corporal total estimado, é uma covariável na análise farmacocinética da população dos pacientes com Doença de Cushing. Na faixa de peso corporal magro estudada de 33 a 83 kg, prevê-se que a ASCs varia de 67% a 134% da do paciente típico de 49 kg (a faixa de peso corporal total correspondente foi de 43,0 a 175 kg, com uma média de 77,4 kg). Esta variação é considerada moderada e de menor significado clínico.
Dados de segurança pré-clínicos
Os estudos de segurança pré-clínicos incluem farmacologia de segurança, toxicidade com doses repetidas, genotoxicidade e potencial carcinogênico, toxicidade para a reprodução e desenvolvimento. A maioria dos efeitos vistos nos estudos da toxicidade com doses repetidas foram reversíveis e atribuíveis à farmacologia da pasireotida. Efeitos em estudos pré-clínicos foram observados nas exposições consideradas similares a, ou além da exposição humana máxima. Nos estudos da farmacologia de segurança, a pasireotida não teve efeitos adversos sobre a função respiratória ou cardiovascular. Reduções da atividade geral e comportamental foram observadas em camundongos na dose de 12 mg/kg, equivalente a aproximadamente 32 vezes a dose terapêutica máxima recomendada a humanos com base na superfície corporal. A pasireotida não foi genotóxica em uma bateria de ensaios in vitro (teste de mutação de Ames em Salmonella e E coli. e teste de mutação em linfócitos periféricos humanos). A pasireotida não foi genotóxica em um teste com medula óssea de ratos in vivo com doses de até 50 mg/kg, aproximadamente 250 vezes a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2. Estudos da carcinogenicidade conduzidos em ratos e camundongos transgênicos não identificaram nenhum potencial carcinogênico. Em estudos de desenvolvimento embrio-fetal com ratos e coelhos, a pasireotida não foi teratogênica em doses maternalmente tóxicas (respectivamente 10 e 5 mg/kg/dia), levando a exposições (ASC 0 a 24 horas) respectivamente 145 e 40 vezes maiores do que a dose terapêutica máxima recomendada a humanos. A uma razão de 10 mg/kg/dia em ratos, a frequência de reabsorções precoces/totais e membros com má rotação foi elevada. A uma razão de 5 mg/kg/dia em coelhos, foram observados mais abortos, fetos com pesos menores e variações esqueléticas. Fetos com peso reduzido e subsequente retardo da ossificação foram observados com 1 mg/kg/dia (6,5 vezes a dose terapêutica máxima recomendada a humanos). A pasireotida não apresentou efeito sobre o parto em ratos que receberam até 10 mg/kg/dia (52 vezes a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2). Os dados toxicológicos disponíveis em animais mostraram a excreção da pasireotida no leite. O retardo do crescimento fisiológico, atribuído à inibição do GH, foi observado com 2 mg/kg/dia (10 vezes a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2) durante um estudo pré e pós-natal em ratos. Após o desmame, os ganhos de peso corporal nos filhotes de ratos expostos a pasireotida foram comparáveis aos dos controles, mostrando reversibilidade. A pasireotida não afetou a fertilidade em ratos do sexo masculino em doses de até 10 mg/kg/dia (uma dose 52 vezes maior do que a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2). Nas ratas, como esperado pela farmacologia da pasireotida, a fertilidade foi reduzida em doses diárias de 0,1 mg/kg/dia (0,6 vezes a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2), conforme demonstrado por números reduzidos de corpos lúteos e locais de implantação. Ciclos anormais ou aciclicidade foram observados com 1 mg/kg/dia (administração 5 vezes maior do que a dose terapêutica máxima recomendada a humanos com base na superfície corporal, mg/m2).
4. CONTRAINDICAÇÕES
Insuficiência hepática grave (Child Pugh C). Hipersensibilidade a pasireotida ou a qualquer outro componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipocortisolismo
O tratamento com Upelior® leva a uma rápida supressão da secreção de ACTH (hormônio adrenocorticotrófico) em pacientes com Doença de Cushing. Como qualquer outra terapia bem-sucedida direcionada à hipófise, a rápida e completa ou quase completa supressão do ACTH pode levar a uma redução dos níveis de cortisol circulante e potencialmente a hipocortisolismo/hipoadrenalismo temporário. Casos de hipocortisolismo foram relatados no estudo de Fase III em pacientes com Doença de Cushing (vide "Reações adversas"), geralmente nos primeiros dois meses de tratamento. Exceto por um caso no qual o tratamento foi descontinuado, todos os outros casos foram tratáveis mediante redução da dose de Upelior® e/ou adição de terapia com glicocorticoides em baixa dosagem e por curto período. Portanto, é necessário monitorar e instruir os pacientes sobre os sinais e sintomas associados ao hipocortisolismo (p. ex. fraqueza, fadiga, anorexia, náusea, vômitos, hipotensão, hiponatremia ou hipoglicemia). Em caso de hipocortisolismo comprovado, terapia de substituição temporária exógena de esteroides (glicocorticoide) e/ou redução da dose ou interrupção do tratamento com Upelior® pode ser necessária.
Metabolismo da glicose
Alterações dos níveis plasmáticos de glicose foram observadas em voluntários sadios e pacientes tratados com pasireotida. Hiperglicemia e, menos frequentemente, hipoglicemia, foram observadas em participantes dos estudos clínicos com pasireotida (vide "Reações adversas"). O desenvolvimento de hiperglicemia parece estar relacionado a uma redução da secreção de insulina (particularmente no período pós-administração), assim como dos hormônios incretina (p. ex., Peptídeo 1 semelhante ao Glucagon [GLP-1] e Polipeptídeo Insulinotrópico Glicose-dependente [GIP]. O grau de hiperglicemia parece ser mais elevado em pacientes com quadro clínico de pré-diabetes ou diabetes mellitus estabelecido. O início do tratamento com medicamentos antidiabéticos foi associado à diminuição na HbA1c < 7% e GPJ < 130 mg/dL em 43% e 72% dos pacientes com Doença de Cushing, respectivamente. Reduções da dose ou interrupção do tratamento com pasireotida devido à hiperglicemia foram infrequentes. O status glicêmico (glicemia de jejum/hemoglobina A1c [GPJ/HbA1c]) deve ser avaliado antes do início do tratamento com pasireotida. O monitoramento de GPJ/HbA1c durante o tratamento deve seguir as diretrizes estabelecidas. O automonitoramento da glicose sanguínea e/ou avaliações da GPJ devem ser realizadas semanalmente nos primeiros dois ou três meses e periodicamente a partir daí, como clinicamente apropriado, assim como ao longo das primeiras 2 a 4 semanas após qualquer aumento da dose. Após a interrupção do tratamento, monitoramento glicêmico (p. ex., GPJ ou HbA1c) deve ser feito de acordo com a prática clínica. Caso se desenvolva hiperglicemia em um paciente tratado com Upelior®, recomenda-se que se inicie ou ajuste o tratamento antidiabético seguindo-se as diretrizes de tratamento estabelecidas para o trato da hiperglicemia. Se a hiperglicemia não controlada persistir apesar do tratamento médico apropriado, a dose de Upelior® deverá ser reduzida ou o tratamento descontinuado. Houve casos pós-comercialização de cetoacidose com Upelior® em pacientes com e sem histórico de diabetes. Em alguns casos, fatores predisponentes à cetoacidose, como doença aguda, infecção, distúrbios pancreáticos (p. ex.: malignidade pancreática ou cirurgia pancreática) e abuso de álcool estavam presentes. Pacientes que apresentam sinais e sintomas consistentes com acidose metabólica grave devem ser avaliados quanto à cetoacidose, independentemente do histórico de diabetes.
Em pacientes com controle glicêmico inadequado (conforme definido pelos valores de HbA1c > 8% enquanto recebe terapia antidiabética), o controle e o monitoramento da diabetes devem ser intensificados antes do início e durante o tratamento com a pasireotida.
Eventos cardiovasculares relacionados
Bradicardia foi relatada com o uso de pasireotida (vide "Reações adversas"). Pacientes com doença cardíaca e/ou fatores de risco para bradicardia, tais como: histórico de bradicardia clinicamente significativa ou infarto agudo do miocárdio, bloqueio cardíaco de alto grau, insuficiência cardíaca congestiva (NYHA Classe III ou IV), angina instável, taquicardia ventricular prolongada, fibrilação ventricular, devem ser monitorados cuidadosamente. Ajustes da dose de fármacos tais como betabloqueadores, bloqueadores dos canais de cálcio ou agentes para controle do equilíbrio eletrolítico podem ser necessários. Foi demonstrado que a pasireotida administrada como Upelior® s.c. prolongou o intervalo QT em indivíduos sadios com base em dois estudos (vide "Características farmacológicas"). Análises adicionais dos dados QT ao longo do estudo, incluindo análise da restituição cardíaca do batimento quantitativo no ECG, mostraram que a pasireotida não altera a repolarização cardíaca, da mesma maneira como os medicamentos conhecidos por prolongarem o intervalo QT, que estão associados com pró-arritmia (vide "Características farmacológicas - eletrofisiologia cardíaca"). Em estudos clínicos com pacientes com Doença de Cushing, QTcF > 500 msec foi observado em dois de 201 pacientes. Esses episódios foram esporádicos e de ocorrência única, sem observação de qualquer consequência clínica. Episódios de torsades de pointes não foram observados em nenhum estudo clínico com pasireotida. A pasireotida deve ser usada com cautela em pacientes com risco significativo de desenvolver prolongamento de QT, incluindo: ·
- síndrome congênita do prolongamento de QT; ·
- doença cardíaca não controlada ou significativa, incluindo infarto do miocárdio recente, insuficiência cardíaca congestiva, angina instável ou bradicardia clinicamente significativa; ·
- administração de medicamentos antiarrítmicos ou outras substâncias que tem ação conhecida de prolongamento de QT (vide "Interações medicamentosas");
- hipocalemia e/ou hipomagnesemia.
Um ECG basal é recomendado antes do início da terapia com o Upelior® e o monitoramento do efeito sobre o intervalo QTc é aconselhável quando se inicia o tratamento com Upelior®, quando clinicamente indicado. Hipocalemia ou hipomagnesemia devem ser corrigidas antes da administração do Upelior® e os eletrólitos devem ser monitorados periodicamente durante a terapia.
Exames do fígado
Elevações leves e temporárias de aminotransferases são comumente observadas em indivíduos sadios e pacientes tratados com pasireotida. Alguns casos de elevações concomitantes de ALT (alanina aminotransferase) maior que 3 x LSN e bilirrubina superior a 2x LSN também foram observadas (vide "Reações adversas"). O monitoramento da função hepática é recomendado antes do tratamento com o Upelior® e após a primeira ou segunda semanas, e então mensalmente nos primeiros 3 meses de tratamento. Após esse período, a função hepática deverá ser monitorada conforme clinicamente indicada. Pacientes que desenvolvem níveis de transaminase elevados devem ser monitorados com uma segunda avaliação da função hepática para confirmação do resultado. Se o resultado for confirmado, o paciente deverá ser acompanhado com monitoramento frequente da função hepática até que os valores retornem aos níveis prétratamento. A terapia com pasireotida deve ser interrompida se o paciente desenvolver icterícia ou outros sinais sugestivos de comprometimento hepático clinicamente significativo, em caso de aumento progressivo da AST (aspartato aminotransferase) ou ALT de 5x LSN ou mais, ou ALT ou AST com elevações superiores a 3x LSN ocorrerem concomitantemente com elevações da bilirrubina maiores que 2x LSN. Após a interrupção do tratamento com pasireotida, os pacientes devem ser monitorados até o desfecho. O tratamento não deve ser reiniciado se suspeitar-se que as anormalidades da função hepática estão relacionadas ao Upelior®.
Vesícula biliar e eventos relacionados
A colelitíase (cálculos biliares) é uma reação adversa conhecida associada com o uso prolongado de análogos da somatostatina e foi frequentemente relatada em estudos clínicos com a pasireotida (vide "Reações adversas"). Houve casos pós-comercialização de colangite em pacientes que utilizavam Upelior®, que namaioria dos casos foram relatados como uma complicação de cálculos biliares. É recomendado, portanto, o exame por ultrassom da vesícula biliar antes, e a intervalos de 6 a 12 meses durante a terapia com Upelior®. A presença de cálculos biliares em pacientes tratados com Upelior® é em sua grande maioria assintomática; cálculos sintomáticos devem ser tratados de acordo com a prática clínica.
Hormônios hipofisários
A deficiência de hormônios secretados pela hipófise é comum após a cirurgia transfenoidal e ainda mais frequentemente observada após a radioterapia hipofisária. Pacientes com Doença de Cushing com doença persistente ou recorrente podem, portanto, apresentar deficiência de um ou mais hormônios hipofisários. A atividade farmacológica da pasireotida mimetiza a da somatostatina, a inibição de hormônios hipofisários, diferente do ACTH, não pode ser descartada. Portanto, o monitoramento do funcionamento da hipófise (p. ex., TSH/T4 livre, GH/IGF-1) antes do início da terapia com Upelior® e periodicamente durante o tratamento, deve ser conduzido conforme clinicamente apropriado.
Interação medicamento-medicamento
A pasireotida pode reduzir a biodisponibilidade relativa da ciclosporina (vide "Interações medicamentosas"). A administração concomitante de Upelior® e ciclosporina pode exigir ajuste da dose da ciclosporina para a manutenção dos níveis terapêuticos do fármaco.
Gravidez e amamentação
-Mulheres em idade fértil e medidas contraceptivas
Estudos em animais demonstraram que pasireotida é prejudicial ao feto em desenvolvimento. Mulheres com potencial reprodutivo são recomendadas a usar método contraceptivo eficaz durante o tratamento com pasireotida e, também, devem ser avisadas que o tratamento com pasireotida pode levar a uma melhora na fertilidade.
- Gravidez
Não existem estudos adequados e bem controlados com mulheres grávidas. Estudos em animais demonstraram toxicidade reprodutiva (vide "Dados de segurança pré-clínicos"). O risco potencial para humanos é desconhecido. O Upelior® deve ser prescrito para mulheres grávidas apenas se os benefícios superarem os riscos potenciais para o feto.
Este medicamento pertence à categoria C de risco na gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
- Parto
Não existem dados disponíveis em humanos. Estudos com ratos não demonstraram efeitos sobre o parto (vide "Dados de segurança pré-clínicos").
- Amamentação
Não se sabe se a pasireotida é excretada no leite humano. Dados disponíveis referentes a ratos com pasireotida via s.c. demonstraram excreção da pasireotida no leite (vide "Dados de segurança pré-clínicos"). Como não podem ser excluídos riscos para lactantes, o Upelior® não deve ser usado por mulheres que estejam amamentando.
- Fertilidade
Estudos com ratos com pasireotida via S.C. demonstraram efeitos sobre os parâmetros de reprodução em fêmeas (vide "Dados de segurança pré-clínicos"). A relevância clínica desses efeitos em humanos é desconhecida. Os benefícios terapêuticos de uma redução ou normalização dos níveis séricos de cortisol em pacientes femininos com doença de Cushing tratados com pasireotida pode conduzir a uma melhora na fertilidade.
6. INTERAÇÕES MEDICAMENTOSAS
A pasireotida tem uma ligação moderada às proteínas e é altamente estável metabolicamente. A pasireotida parece ser um substrato da transportadora de efluxo P-gp (glicoproteína P), mas não é inibidor ou indutor da Pgp. Além disso, em níveis de dosagem terapêutica, não se espera que a pasireotida seja:
- um substrato, inibidor ou indutor da CYP450 (citocromo P450);
- um substrato da transportadora de efluxo BCRP (proteína de resistência ao câncer de mama) nem dos transportadores de influxo OCT1 (transportador 1 de cátions orgânicos) e OATP (polipeptídeo transportador de ânions orgânicos) 1B1, 1B3 ou 2B1;
- um inibidor da UGT1A1 (uridina difosfato glucuronosiltransferase 1A1, transportador de influxo OAT1 ou OAT3, OATP 1B1 ou 1B3 e OCT1 ou OCT2, transportador de efluxo P-gp, BCRP, MRP2 (proteína da resistência multi-droga 2) ou BSEP (bomba de transporte de sais biliares).
Com base em todos esses dados in vitro, o potencial para ligação às proteínas, metabolismo e/ou transporte mediado DDI é baixo entre a pasireotida e comedicamentos in vivo.
Interações farmacocinéticas antecipadas resultantes em efeitos sobre a pasireotida
A influência de um inibidor da P-gp na farmacocinética da pasireotida subcutânea foi testada em um estudo de interação medicamento-medicamento com coadministração de verapamil em voluntários sadios. Nenhuma alteração na taxa ou na extensão da disponibilidade da pasireotida foi observada.
Interações antecipadas resultantes em efeitos sobre outros fármacos
Dados limitados publicados sugerem que análogos da somatostatina podem ter um efeito indireto sobre a diminuição do clearance (depuração) metabólico em compostos metabolizados por enzimas CYP450, através da supressão do hormônio do crescimento. A possibilidade de que a pasireotida exerça tal efeito indireto não pode ser excluída com base nos dados disponíveis. Deve-se ter cautela ao administrar-se a pasireotida concomitantemente com fármacos de baixo índice terapêutico e que sejam metabolizados principalmente pela CYP3A4 (p. ex., quinidina, terfenadina).
Em cães, descobriu-se que a pasireotida reduz o nível sanguíneo de ciclosporina ao reduzir sua absorção intestinal. Não se sabe se tal interação ocorre nos humanos. Portanto pode ser necessário ajustar a dose de ciclosporina durante a coadministração de pasireotida e ciclosporina (vide "Advertências e precauções"). Dados limitados referentes a outros análogos da somatostatina sugerem que a coadministração com bromocriptina pode aumentar a disponibilidade de bromocriptina. Os dados disponíveis não podem excluir a possibilidade da pasireotida exercer tal efeito.
Interações farmacodinâmicas antecipadas
Medicamentos que prolongam o intervalo QT
Upelior® deve ser utilizado com cautela em pacientes que estão concomitantemente tomando medicamentos que prolongam o intervalo QT, como antiarrítmicos classe I (ex. quinidina, procainamida, disopiramida), antiarrítmicos de classe III (ex. amiodarona, dronedarona, sotalol, dofetilida, ibutilida), certos antibacterianos (eritromicina intravenosa, pentamidina injetável, claritromicina, moxifloxacina), certos antipsicóticos (ex. clorpromazina, tiorizadina, flufenazina, pimozida, haloperidol, tiaprida, amisulprida, sertindol, metadona), certos anti-histamínicos (ex. terfenadina, astemizol, mizolastina), antimaláricos (ex. cloroquina, halofantrina, lumefantrina), certos antifúngicos (cetoconazol, exceto em shampoo).
É necessário cautel