TYLEX
CELLERA FARMA
paracetamol + codeína
Analgésico.
Apresentações.
Comprimidos de 500 mg de paracetamol e 7,5 mg de fosfato de codeína em embalagens com 12 comprimidos.
Comprimidos de 500 mg de paracetamol e 30 mg de fosfato de codeína em embalagens com 12, 24 e 36 comprimidos;
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 12 ANOS
Composição.
Cada comprimido de TYLEX® 7,5 mg contém: paracetamol 500 mg, fosfato de codeína 7,5 mg
Cada comprimido de TYLEX® 30 mg contém: paracetamol 500 mg, fosfato de codeína 30 mg
Excipientes: amido, bissulfito de sódio, celulose pó, docusato de sódio/benzoato de sódio e estearato de magnésio.
Informações técnicas.
1. INDICAÇÕES
TYLEX®7,5 mg é indicado para o alívio de dores de intensidade leve.
TYLEX® 30 mg é indicado para o alívio de dores de grau moderado a intenso, como nas decorrentes de traumatismo (entorses, luxações,
contusões, distensões, fraturas), pós-operatório, pós-extração dentária, neuralgia, lombalgia, dores de origem articular e condições similares.
2. RESULTADOS DE EFICÁCIA
Em um estudo clínico aberto, 50 atletas com traumatismos articulares e osteomusculares agudos, necessitando de analgesia, receberam 1 comprimido da associação paracetamol 500 mg + codeína 30mg, sendo permitido, a partir da 4ª hora, que tomassem 1 a 2 comprimidos com intervalo de 4 horas, até a 24ª hora. Setenta e oito por centro (78%) dos investigadores classificaram a eficácia do tratamento como excelente (46%) e boa (32%). A média da redução da dor (avaliada pela escala visual analógica) já a partir de 30 minutos foi de 54% (p < 0,001) quando comparada à dor inicial, e de 84% na 24ª hora de tratamento. Oitenta e oito por cento (88%) dos pacientes avaliaram a tolerabilidade do tratamento com a associação paracetamol 500mg + codeína 30mg como excelente (72%) e boa (16%).1
Em estudo duplo cego randomizado, 120 pacientes sofrendo de dor resultante de cirurgia odontológica foram tratados com dose única de paracetamol 1000mg, codeína 60mg, paracetamol 1000mg + codeína 60mg ou placebo. Uma análise fatorial demonstrou que 1000mg de paracetamol + 60mg de codeína promoveram efeito analgésico significativo (p < 0,05), avaliado através de diferentes medidas de eficácia. A incidência de eventos adversos não pareceu ser diferente entre os tratamentos, inclusive no tratamento com placebo.2
Referências bibliográficas:
1. Lasmar NP. Traumatismos articulares e osteomusculares agudos em atletas: analgesia com associação paracetamol-codeína. Farmacologia Clínica. 1988; 97(4): 277-82.
2. Bentley KC, Head T. The additive analgesic of acetaminophen 1000mg and codeine 60mg in dental pain. Clinical Pharmacology &Therapeutics. 1987; 42(6): 634-40.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
• codeína
A codeína é um analgésico opioide e antitussígeno. A codeína é um medicamento analgésico que age nos receptores m-opiáceos predominantemente através de seu metabólito ativo morfina, que é formado quase que exclusivamente pela enzima geneticamente polimórfica 2D6 do citocromo P450 (CYP2D6). A codeína também se liga fracamente aos receptores k, que mediam a analgesia, miose e sedação.
Os principais efeitos da codeína são no sistema nervoso central (SNC). A codeína é um agonista opiáceo, sua afinidade pelo receptor opiáceo é baixa. A codeína assemelha-se à morfina em possuir ações analgésicas, antitussígenas e antidiarreicas.
• paracetamol
O paracetamol é um analgésico não-salicilato, não-opiáceo de ação central. O paracetamol é um analgésico/antipirético clinicamente comprovado, e acredita-se que produz a analgesia pela elevação do limiar da dor e antipirese através da ação no centro hipotalâmico regulador do calor. Estudos de dose única (12,5 mg/kg) de paracetamol em crianças febris mostraram um início de redução da febre em 15 a 30 minutos.
Propriedades farmacocinéticas
Absorção
• codeína
A codeína é rapidamente e bem absorvida após administração oral de comprimidos e líquido, com uma biodisponibilidade de 50-80%. A codeína pode logo ser detectada no plasma, de 0,17 a 1 hora (h) após administração oral. A Tmáx de 30 mg e 60 mg de codeína ocorreu em 0,75 a 1 h e 0,61 a 1,3 h com Cmáx de 61 a 89,1 ng/mL e 122,8 a 214,2 ng/mL, respectivamente. A ASC para 30 mg e 60 mg de codeína é de 216 a 354,6 ng·h/·mL e 417 a 734 ng·h/·mL. A codeína pode ser administrada com ou sem alimentos.
Quando as doses de 30 mg de codeína e 1000 mg de paracetamol são administradas juntas, nenhuma interação farmacocinética medicamentosa entre a codeína e o paracetamol foi demonstrada.
• paracetamol
O paracetamol oral é rapidamente e quase que completamente absorvido a partir do trato gastrointestinal, principalmente no intestino delgado.
A absorção ocorre por transporte passivo. A taxa de absorção oral depende principalmente da taxa de esvaziamento gástrico.
A biodisponibilidade relativa varia de 85% a 99%. As concentrações plasmáticas máximas são normalmente atingidas cerca de 30 a 60 minutos após a administração oral.
Para indivíduos adultos, as concentrações plasmáticas máximas ocorrem em 1 hora após a ingestão, e varia de 14,8 a 17,6 mg/mL para uma dose única de 1000 mg. Concentrações plasmáticas máximas no estado de equilíbrio após doses de 1000 mg a cada 6 horas variam de 17,6 a 18,2 mg/mL. Os dados farmacocinéticos agrupados de cinco estudos patrocinados pela empresa de 59 crianças febris com idades de 6 meses a 11 anos, mostraram que a concentração média máxima de 12,08 ± 3,92 mg/mL foi obtida em 51 ± 39 min. (mediana, 35 min.) após uma dose de 12,5 mg/kg.
Embora as concentrações máximas de paracetamol sejam retardadas quando administradas com alimentos, a extensão da absorção não é afetada.
O paracetamol pode ser administrado independentemente dos horários das refeições.
Distribuição
• codeína
A codeína entra nos tecidos rapidamente e se concentra nos rins, pulmões, fígado e baço. A codeína é menos de 10% ligada a proteínas com um volume de distribuição (Vd) entre 3 a 4 L/kg.
• paracetamol
O paracetamol aparenta ser amplamente distribuído ao longo da maioria dos tecidos corporais, exceto gordura. Seu volume aparente de
distribuição é 0,7 a 1 L/kg em crianças e adultos. Uma proporção relativamente pequena (10% a 25%) de paracetamol se liga às proteínas
plasmáticas.
Metabolismo
• codeína
A codeína é metabolizada por O- e N-demetilação no fígado em morfina, norcodeína e outros metabólitos incluindo normorfina e hidrocodona.
Aproximadamente 50% sofre metabolismo pré-sistêmico no intestino e fígado.
O metabolismo para morfina é mediado pela isoenzima CYP2D6 do citocromo P450, que mostra polimorfismo genético. Uma proporção
significativa da população é de metabolizadores fracos ou rápidos de codeína devido a diferenças genéticas no metabolismo. Como consequência, eles apresentam efeitos analgésicos opioides ou eventos adversos imprevisíveis. A etnia é um fator na ocorrência de variabilidade de CYP2D6. Pacientes que são metabolizadores fracos (PMs) de CYP2D6, possuem uma deficiência ou são completamente desprovidos desta enzima e não irão obter efeito adequado. Aproximadamente 7 a 10% dos caucasianos, 0,5 a 1% dos chineses, japoneses e hispânicos, 1% dos árabes e 3% dos afro-americanos são metabolizadores fracos.
Metabolizadores ultrarrápidos convertem codeína em morfina mais rápido e completamente. Em metabolizadores ultrarrápidos (UMs), há um risco aumentado de desenvolver efeitos colaterais de toxicidade opioide mesmo em doses baixas. Sintomas gerais de toxicidade opioide incluem depressão do estado mental, hipoventilação, miose e hipoperistaltismo. A prevalência da presença deste genótipo de CYP2D6 varia e é estimada em 0,5 a 2% em asiáticos; 1 a 10% em caucasianos; 3 a 6,5% em afro-americanos; e 16 a 29% nos africanos do Norte, etíopes e árabes.
• paracetamol
O paracetamol é principalmente metabolizado no fígado e envolve três vias principais: conjugação com glicuronídeo; conjugação com sulfato; e oxidação através da via da enzima do citocromo P450. A via oxidativa forma um intermediário reativo que é detoxificado pela conjugação com glutationa para formar cisteína inerte e metabólitos do ácido mercaptúrico. A principal isoenzima do citocromo P450 envolvida in vivo parece ser a CYP2E1, embora CYP1A2 e CYP3A4 tenham sido consideradas as vias menores com base em dados microssômicos in vitro.
Subsequentemente, ambos CYP1A2 e CYP3A4 mostraram ter contribuição insignificante in vivo.
Em adultos, grande parte do paracetamol é conjugado com ácido glicurônico e, em uma extensão menor, com sulfato. Os metabólitos derivados do glicuronídeo, sulfato e glutationa carecem de atividade biológica. Em bebês prematuros, recém-nascidos e crianças jovens, o conjugado sulfato predomina. Em adultos com comprometimento hepático de diferentes gravidade e etiologia, vários estudos do metabolismo demonstraram que a biotransformação de paracetamol é semelhante àquele dos adultos saudáveis, mas de alguma forma mais lento. É importante ressaltar que a administração diária consecutiva de 4 g por dia induz à glicuronidação (uma via atóxica) em adultos saudáveis e com fígado comprometido, resultando no aumento da depuração total de paracetamol ao longo do tempo e acúmulo plasmático limitado.
Eliminação
A codeína e seus metabólitos ativos, como morfina, são excretados quase que totalmente pelos rins, principalmente como conjugados com ácido glicurônico. Apenas 3% a 16% da dose de codeína administrada, seja de maneira isolada ou com paracetamol, é excretada não metabolizada na urina. O T½ para 30 mg e 60 mg de codeína é 1,5 a 2,2 h e 2,1 a 4,5 h, respectivamente. Para codeína administrada com paracetamol, o T½ é semelhante ao da codeína isolada. No entanto, em um estudo de pacientes em hemodiálise, o T½ médio foi de 13 ± 3,3 h em comparação com indivíduos saudáveis no estudo com T½ de 4,5 ± 0,8 h. Pacientes com comprometimento renal devem ser dosados e titulados cuidadosamente devido ao possível acúmulo do medicamento e do metabólito.
A codeína possui uma depuração sistêmica relatada de 252 mL/min. e a sua depuração ao ser administrada com paracetamol é de 291 mL/min.
Embora nenhuma recomendação específica de administração esteja disponível para pacientes com disfunção hepática, doses menores e intervalos de dose prolongados devem ser considerados para se evitar acúmulo do medicamento.
• paracetamol
A meia-vida de eliminação do paracetamol é de cerca de 1 a 3,5 horas. É aproximadamente uma hora mais longa em recém-nascidos e em
pacientes cirróticos. O paracetamol é eliminado do organismo como conjugado de glicuronídeo (45-60%) e sulfato (25-35%), tióis (5-10%) como metabólitos de cisteína e mercapturato, e catecóis (3-6%) que são excretados na urina. A depuração renal de paracetamol não metabolizado é de cerca de 3,5% da dose.
Dados de segurança pré-clínicos
Resumo:
Os dados pré-clínicos não revelam riscos especiais para o ser humano, com base em estudos convencionais de toxicidade de dose única e repetida, genotoxicidade, carcinogenicidade e toxicidade para a reprodução e desenvolvimento.
Toxicologia geral
• codeína
Vários estudos de doses agudas e repetidas de codeína foram realizados em animais. Reduções discretas a moderadas no peso corporal com doses de 200 a 400 mg/kg de peso corporal/dia foram os únicos efeitos observados.
• paracetamol
Vários estudos de toxicidade aguda, subaguda e crônica em animais mostram que os efeitos tóxicos do paracetamol aparecem apenas com quantidades muito acima das doses terapêuticas.
Toxicologia genética
• codeína
Descobriu-se que a codeína era negativa em diversos estudos in vivo e in vitro e é considerada não genotóxica.
A codeína (até 10.000 mg/placa) e o fosfato de codeína (até 500 mg/placa) foram não mutagênicos no teste de Ames, com ou sem ativação
metabólica de S9. A codeína também foi negativa nos ensaios de genotoxicidade conduzidos em E. coli e células germinativas de Drosophila melanogaster. A codeína foi negativa para a indução de aberrações cromossômicas em células de ovário de hamster chinês (CHO) (até 3500 mg/ml na ausência de S9, ou até 10.000 mg/ml na presença de ativação metabólica de S9), mas não mostrou induzir um aumento significativo nas alterações de cromátides irmãs nas células CHO cultivadas na ausência e presença de ativação metabólica. A codeína foi negativa em um estudo de micronúcleo in vivo em camundongos quando administrada via intraperitoneal até 500 mg/kg/dia por cinco dias consecutivos. A codeína não mostrou evidência de ligação ao DNA in vitro com ou sem ativação metabólica de S9. Um relatório recente também concluiu que a codeína não é mutagênica no ensaio de micronúcleo em camundongos em uma dose oral de 26 mg/kg/dia em estudos agudos, bem como subagudos (7 dias).
• paracetamol
O paracetamol não mostrou qualquer evidência de atividade mutagênica em concentrações variando de 0,1 a 50 mg/placa quando testado para mutagenicidade no ensaio de salmonela (TA1535, TA1537, TA1538, TA100, TA97 e TA98) ou microssomo de mamíferos. O paracetamol não é mutagênico conforme demonstrado por resultados negativos no teste de Ames, mas se mostrou positivo como um clastógeno como demonstrado por resultados positivos no ensaio de aberrações cromossômicas.
Considerando estudos in vitro e in vivo, uma revisão abrangente e conclusiva, aceita pelo Comitê de Patentes de Produtos Médicos (CPMP) da União Europeia, relata que os efeitos genotóxicos do paracetamol aparecem apenas em doses induzindo toxicidade hepática e da medula óssea pronunciada e que o nível limiar para genotoxicidade não é alcançado nas doses recomendadas em bula.
Carcinogenicidade
• codeína
De acordo com estudos precursores de 2 anos conduzidos pelo Programa Nacional de Toxicologia (NTP), não há evidência de atividade carcinogênica da codeína em ratos e camundongos F344/N machos ou fêmeas expostos a 400 ppm (15 mg/kg/dia), 800 ppm (30 mg/kg/dia para machos e 40 mg/kg/dia para fêmeas) ou 1600 ppm (70 mg/kg/dia para machos e 80 mg/kg/dia para fêmeas), bem como camundongos B6C3F1 machos e fêmeas expostos a 750 ppm (100 mg/kg/dia), 1500 ppm (200 mg/kg/dia) ou 3000 ppm (300 mg/kg/dia). No entanto, feocromocitoma benigno da glândula adrenal e fibroadenoma/adenocarcinoma da glândula mamária foram observados em todos os grupos de dose de ratos machos e fêmeas, respectivamente. Em todos os grupos de dose de camundongos machos e fêmeas, foi observada hiperplasia de células foliculares da glândula tireoide. Os efeitos neoplásicos não foram observados nestes estudos conduzidos em ratos, bem como em camundongos.
• paracetamol
Baseado em vários estudos de longo prazo, o paracetamol não indica um potencial carcinogênico em doses não hepatotóxicas. Os resultados de carcinogenicidade de 2 anos do NTP em roedores mostraram que não há evidência de atividade carcinogênica de paracetamol em ratos F344/N machos (22, 109 e 222 mg/kg de paracetamol até 103 semanas). Não há evidência ambígua da atividade carcinogênica em ratos fêmeas (24, 118 e 240 de mg/kg de paracetamol até 103 semanas), com base no aumento de incidências de leucemia de células mononucleares. Não houve evidência de atividade carcinogênica em camundongos machos (79, 411 e 880 mg/kg de paracetamol até 103 semanas) e fêmeas (98, 534 e 987 mg/kg de paracetamol até 103 semanas). O nível sem observação de efeito adverso (NOAEL) para ratos machos mostrou ser 268 mg/kg. No entanto, o NOAEL para ratos fêmeas foi 118 mg/kg com base na incidência de leucemia de células mononucleares. Além disso, o NOAEL para camundongos machos e fêmeas foi de 880 e 987 mg/kg, respectivamente. Adicionalmente, os estudos precursores do NTP mostraram que paracetamol não é carcinogênico quando administrado em doses não hepatotóxicas de até 300 mg/kg/dia em ratos e de até 1000 mg/kg/dia em camundongos.
Teratogenicidade
• codeína
A codeína demonstrou não ser teratogênica em embriões de ratos e galinhas. Em hamsters e ratos, os efeitos teratogênicos foram observados após uma injeção subcutânea de alta dose no dia 8 de gestação.
Ratos que receberam até 120 mg/kg/dia por via oral nos dias da gestação (GD) de 6 a 15 e coelhos que receberam até 30 mg/kg/dia nos GD de 6 a 18 não mostraram efeitos teratogênicos. A codeína não foi teratogênica no embrião de galinha, mas mostrou ser teratogênica em roedores após injeção subcutânea. Em hamsters dourados não consanguíneos de Lakeview, uma injeção subcutânea única de fosfato de codeína (73 mg de codeína de base/kg) no dia 8 da gestação causou craniosquise em 6% de fetos de 12 dias. A administração de 110 mg/kg de fosfato de codeína em camundongos JBT/Jd no GD 9 causou dilatação hidrocefálica do quarto ventrículo cerebral em 15% dos fetos de 13 dias. Em camundongos albinos CF-1, injeção subcutânea de 100 mg/kg de sulfato de codeína no GD 8 e 9 produziu ossificação tardia de vários ossos em fetos de 18 dias.
• paracetamol
O paracetamol não demonstrou ser teratogênico em ratos ou camundongos. O paracetamol a 250 mg/kg/dia durante a organogênese não afetou a duração, o peso ou a incidência fetal de reabsorções, e não causou má formação ou fetotoxicidade em ratos. Nenhum efeito adverso no desenvolvimento do embrião a termo foi observado após o tratamento de camundongos fêmeas com 1430 mg/kg/dia de paracetamol no GD 8 ao 3. Nenhum efeito teratogênico do paracetamol foi observado nas doses de 100 e 250 mg/kg/dia administradas a camundongos entre o GD 6 e 13. O NOAEL para os efeitos embriotóxicos foi determinado como 250 mg/kg. Quando paracetamol foi administrado por gavagem a ratas prenhes a 150, 500 ou 1500 mg/kg/dia do primeiro dia da gravidez até o termo, não houve anormalidades morfológicas, mas lesões microscópicas dependentes da dose no fígado e rins maternos foram observadas. Um NOAEL de 125 mg/kg foi estabelecido para os achados de fígado e rins maternos.
Fertilidade
• codeína
As doses nas quais foram observadas toxicidade de desenvolvimento em animais foram várias vezes mais altas do que as doses recomendadas em humanos.
Em uma toxicidade reprodutiva e do desenvolvimento para codeína oral conduzida em hamsters sírios LGV (GD 5-15, até 150 mg/kg/dia), NOAELs para toxicidade materna e do desenvolvimento foram estabelecidos como 50 e 10 mg/kg/dia, respectivamente. Em um estudo semelhante conduzido em camundongos Swiss CD-1 (GD 6-15, até 300 mg/kg/dia), NOAELs para toxicidade materna e do desenvolvimento mostraram ser 150 e 75 mg/kg/dia, respectivamente. Quando a base de codeína foi administrada por via oral em ratos na dose de 120 mg/kg no momento da implantação, foi observada embriotoxicidade. As doses nas quais estas toxicidades do desenvolvimento foram observadas são geralmente várias vezes maiores do que as exposições humanas estimadas quando codeína é prescrita.
• Paracetamol
As doses nas quais foram encontradas toxicidade reprodutiva ou efeitos na fertilidade em animais foram muito maiores do que as doses recomendadas em humanos.
Em um estudo de toxicidade reprodutiva conduzido por NTP, os camundongos foram alimentados com uma dieta consistindo de paracetamol 0,25, 0,5, e 1,0% (357, 715 e 1430 mg/kg, respectivamente) na fase de criação contínua (consiste de uma exposição prematura de 7 dias, um período de coabitação de 98 dias e um período de segregação de 21 dias, que dura um total de 18 semanas) do estudo. A exposição contínua de camundongos a paracetamol 1% levou aos efeitos cumulativos na reprodução com crescimento tardio e esperma anormal em camundongos F1, e reduziu o peso no nascimento de crias F2, embora não tenha havido sinais de embrio ou teratogenicidade em doses menores. Um NOAEL de 715 mg/kg foi estabelecido para embriotoxicidade. Atrofia testicular e redução no peso dos testículos foram observadas em estudos de fertilidade de paracetamol (0,5, 0,7, 1,1, 1,4, 2,5, 3,0, 3,5 e 4,0 g/kg/dia durante 100 dias) em ratos. Não houve efeito na gestação ou prole quando paracetamol foi administrado em níveis de dose de 600 mg/kg/dia na dieta de ratos machos por 60 dias antes do acasalamento e em ratos fêmeas de 14 dias antes do acasalamento até o final da gestação.
4. CONTRAINDICAÇÕES
TYLEX® não deve ser administrado a pacientes que tenham previamente apresentado hipersensibilidade ao paracetamol, à codeína ou aos excipientes da formulação.
A codeína é contraindicada para dor em crianças abaixo de 12 anos.
A codeína é contraindicada para o tratamento da dor pós-operatória em crianças abaixo de 18 anos que foram submetidas à tonsilectomia e/ou adenoidectomia.
TYLEX® é contraindicado em metabolizadores ultrarrápidos de CYP2D6 que convertem a codeína no seu metabólito ativo completamente e mais rápido que outras pessoas. Esses indivíduos podem apresentar sinais de overdose/toxicidade incluindo sintomas tais como sonolência extrema, confusão ou respiração superficial, o que pode ser fatal.
Produtos que contêm codeína são contraindicados em mães que estejam amamentando.
5. ADVERTÊNCIAS E PRECAUÇÕES
• codeína
- A codeína não é recomendada para dor em adolescentes de 12 a 18 anos de idade.
- Risco de morte em metabolizadores ultrarrápidos de codeína: estes indivíduos convertem codeína em seu metabólito ativo, morfina, completamente e mais rápido que outras pessoas. Esta conversão rápida resulta em níveis séricos de morfina maiores do que os esperados.
Mesmo na posologia indicada, os indivíduos que são metabolizadores ultrarrápidos podem ter depressão respiratória fatal ou de risco à vida ou apresentar sinais de superdose (tais como sonolência extrema, confusão ou respiração superficial).
Depressão respiratória e morte ocorreram em crianças que receberam codeína no período pós-operatório após tonsilectomia e/ou adenoidectomia e apresentavam evidência de serem metabolizadores ultrarrápidas de codeína (ou seja, múltiplas cópias do gene para a isoenzima 2D6 do citocromo P450 ou concentrações altas de morfina). Crianças que são metabolizadores ultrarrápidos de codeína com apneia obstrutiva do sono quando tratadas com codeína para dor após tonsilectomia e/ou adenoidectomia podem ser particularmente sensíveis aos efeitos depressores respiratórios da codeína. A codeína é contraindicada em metabolizadores ultrarrápidos de CYP2D6.
- A codeína destina-se apenas para utilização a curto prazo. Não tome continuamente sem supervisão médica.
- A codeína é um agente opioide e apresenta o risco de uso indevido e abuso. Tolerância, dependência psicológica e/ou física pode ocorrer com o uso prolongado e/ou com altas doses de codeína.
- A codeína deve ser usada com cautela em pacientes com distúrbios convulsivos, lesões na cabeça e em condições na qual a pressão
intracraniana está elevada.
- A codeína deve ser usada com cautela em pacientes com função respiratória comprometida, como asma brônquica, edema pulmonar, doença obstrutiva das vias aéreas, depressão respiratória aguda, doença pulmonar grave, obesidade, apneia obstrutiva do sono ou distúrbios obstrutivos do intestino e em pacientes com risco de íleo paralítico. A terapia deve ser interrompida aos primeiros sinais de dor abdominal ou problemas respiratórios.
- A codeína deve ser utilizada com cautela em pacientes que fazem uso de medicações serotoninérgicas. Pergunte ao seu paciente se ele está utilizando medicamentos serotoninérgicos, antes de prescrever codeína.
Pacientes com comprometimento renal e hepático devem consultar um médico antes de usar TYLEX®.
- O uso deste medicamento deve ser descontinuado no primeiro sinal de toxicidade por codeína incluindo sintomas como confusão, respiração superficial e sonolência extrema os quais podem ser fatais.
- Oriente seu paciente a não ingerir bebidas alcoólicas, quando estiver usando TYLEX®. Pergunte ao seu paciente se ele está usando medicações benzodiazepínicas ou outros sedativos. O uso concomitante de opioides com benzodiazepínicos ou outros depressores do sistema nervoso central (SNC), incluindo o álcool, pode resultar em sedações profundas, depressão respiratória, coma e morte.
- A codeína pertence a uma classe de medicamentos chamados de opioides. Opioides foram associados às seguintes condições:
- Insuficiência adrenal, uma condição potencialmente fatal. A insuficiência adrenal pode apresentar sintomas e sinais inespecíficos, como náuseas, vômitos, anorexia, fadiga, fraqueza, tontura e pressão arterial baixa, que foram relatados com mais frequência após um mês de uso. Aconselhe o paciente a procurar atendimento médico se ele apresentar um conjunto desses sintomas.
- Deficiência de androgênio, que pode apresentar-se com sintomas e sinais inespecíficos, como baixa libido, impotência, disfunção erétil, amenorreia ou infertilidade. Aconselhe o paciente a procurar atendimento médico caso apresente algum desses sintomas.
- Síndrome serotoninérgica, uma condição rara, mas potencialmente fatal, resultante da administração concomitante de drogas
serotoninérgicas. Avise os pacientes sobre os sintomas da síndrome serotoninérgica para que procurem atendimento médico imediatamente se os sintomas se desenvolverem. Instrua os pacientes a informar seus médicos se eles estiverem tomando ou planejando tomar medicamentos serotoninérgicos.
- Distúrbios respiratórios relacionados ao sono, como síndromes da apneia do sono [incluindo apneia central do sono (ASC)] e
hipóxia (incluindo hipóxia relacionada ao sono). O uso de opioides aumenta o risco de ASC de maneira dose-dependente. Aconselhe os pacientes a informar seu médico se tiverem um histórico de distúrbios respiratórios relacionados ao sono ou se apresentarem sintomas desse distúrbio, por exemplo, se alguém perceber que eles param de respirar enquanto dormem.
- Hiperalgesia pode ocorrer com o uso de opioides, particularmente em altas doses. Um aumento inexplicado da dor ou níveis
aumentados de dor podem ocorrer com o aumento das doses de opioide. Se você estiver tomando algum opioide para dor, consulte um médico antes de utilizar este produto.
Atenção: pode causar dependência física ou psíquica.
• Paracetamol
- Advertência de superdose de paracetamol: administrar mais do que a dose recomendada (superdose) pode causar dano hepático. Em caso de superdose, oriente seu paciente a procurar auxílio médico imediatamente. Um cuidado médico rápido é fundamental para adultos, assim como para crianças, mesmo se você não perceber nenhum sinal ou sintoma.
- Advertência sobre álcool: avaliar com cautela a indicação de uso de paracetamol ou outros antipiréticos (produtos para adultos) em usuários crônicos de álcool.
- Pacientes com doença hepática devem consultar um médico antes de usar.
- Reações cutâneas sérias, como pustulose exantemática generalizada aguda, Síndrome de Stevens Johnson e necrólise epidérmica tóxica foram relatadas muito raramente em pacientes recebendo paracetamol. Pacientes devem ser informados sobre os sinais de reações cutâneas graves, e o uso do medicamento deve ser descontinuado no primeiro aparecimento de erupção cutânea ou qualquer outro sinal de hipersensibilidade.
- Não utilizar nenhum outro produto contendo paracetamol.
- Para produtos contendo um sulfito como excipiente: este produto contém um sulfito que pode causar reações do tipo alérgicas incluindo
sintomas anafiláticos e episódios asmáticos de risco à vida ou de menor gravidade em determinadas pessoas susceptíveis. A prevalência geral de sensibilidade ao sulfito na população geral é desconhecida e provavelmente baixa. A sensibilidade ao sulfito é observada mais frequentemente em pessoas asmáticas do que em não asmáticas.
- Se os sintomas persistirem ou piorarem, ou se ocorrerem novos sintomas, o paciente deve parar de usar e consultar um médico.
Oriente o seu paciente a não ultrapassar o limite máximo diário de paracetamol, a não consumir outro medicamento contendo paracetamol (devido ao risco de superdosagem) e a não consumir álcool durante o uso deste medicamento, pois essas ações aumentam o risco de dano hepático.
Gravidez (Categoria C) e lactação
Não há estudos adequados e bem controlados da combinação de codeína e paracetamol em gestantes ou lactantes. A combinação de codeína e paracetamol não deve ser utilizada durante a gravidez a menos que o potencial benefício do tratamento para a mãe supere os possíveis riscos ao feto em desenvolvimento. A combinação de codeína e paracetamol é contraindicada em mulheres que estejam amamentando.
Gravidez
• Codeína
Não há estudos adequados e bem controlados de codeína em mulheres grávidas ou que estejam amamentando.
A codeína é contraindicada para mulheres que amamentam.
A codeína atravessa a placenta. Recém-nascidos que foram expostos à codeína no útero podem desenvolver síndrome de abstinência (síndrome de abstinência neonatal) após o parto. Infarto cerebral foi relatado neste contexto.
• paracetamol
Não existem estudos adequados e bem controlados em mulheres grávidas ou que estejam amamentando para paracetamol.
Quando administrado à mãe em doses recomendadas, o paracetamol atravessa a placenta e alcança a circulação fetal em 30 minutos após a ingestão e é efetivamente metabolizado por conjugação com sulfato fetal. Quando tomado de acordo com as instruções, o paracetamol não afeta adversamente a mãe grávida ou o feto.
Lactação
• codeína
Em doses recomendadas, a codeína e seus metabólitos ativos estão presentes no leite materno em concentrações muito baixas.
Em mulheres com metabolismo normal de codeína (atividade normal de CYP2D6), a quantidade de codeína secretada no leite materno é baixa e dependente da dose. Apesar do uso comum dos produtos contendo codeína para tratar a dor pós-parto, relatos de eventos adversos em lactentes são raros. No entanto, algumas mulheres são metabolizadoras ultrarrápidas de codeína. Estas mulheres atingem níveis séricos maiores do que os esperados do metabólito ativo da codeína, morfina, levando a níveis maiores do que os esperados de morfina no leite materno e altos níveis séricos de morfina potencialmente perigosos nos bebês amamentados. Ocorreram mortes em lactentes que foram expostos a altos níveis de morfina no leite materno, pois suas mães eram metabolizadoras ultrarrápidas de codeína. Portanto, o uso materno de codeína pode potencialmente levar a reações adversas graves, incluindo morte, em lactentes. A codeína é contraindicada em mulheres amamentando.
• paracetamol
O paracetamol é excretado no leite materno em concentrações baixas (0,1% a 1,85% da dose materna ingerida). A ingestão materna de paracetamol em doses recomendadas não apresenta risco para o lactente.
A combinação de codeína e paracetamol é contraindicada durante a amamentação e não deve ser usada durante a gravidez a menos que o benefício potencial do tratamento para a mãe supere os possíveis riscos para o feto em desenvolvimento. Pergunte à sua paciente se ela está gestante antes de prescrever a medicação.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Oriente seu paciente a não dirigir veículos ou operar máquinas durante todo o tratamento, pois sua habilidade e capacidade de reação podem estar prejudicadas.
O uso deste medicamento pode causar tontura, desmaios ou perda da consciência, expondo o paciente a quedas ou acidentes.
6. INTERAÇÕES MEDICAMENTOSAS
Depressores do SNC
O uso concomitante com depressores do sistema nervoso central (SNC) (por exemplo, barbitúricos, hidrato de cloral, benzodiazepínicos, fenotiazinas, álcool e relaxantes musculares de ação central) pode causar depressão aditiva no SNC e depressão respiratória.
Analgésicos opioides
Uso concomitante com outros agonistas do receptor opioide pode causar depressão aditiva no SNC, depressão respiratória e efeitos hipotensores.
Inibidores de CYP2D6
Acredita-se que a analgesia da codeína seja dependente da isoenzima CYP2D6 do citocromo P450 catalisada pela o-demetilação para formar o metabólito ativo morfina, embora outros mecanismos tenham sido citados. Interações com quinidina, metadona e paroxetina (inibidores de CYP2D6) levando à diminuição de concentrações plasmáticas de morfina foram descritas, o que pode ter potencial para diminuir a analgesia da codeína.
Medicamentos serotonérgicos
O uso concomitante de opioides com outras drogas que afetam o sistema neurotransmissor serotoninérgico, como inibidores seletivos de recaptação de serotonina (ISRSs), inibidores seletivos de recaptação de serotonina e noradrenalina (ISRSNs), antidepressivos tricíclicos (ADTs), triptanos, antagonistas dos receptores 5-HT3, drogas que afetam o sistema neurotransmissor da serotonina (por exemplo, mirtazapina, trazodona, tramadol) e inibidores da monoaminoxidase (MAO) (usados para tratar transtornos psiquiátricos e outros, como linezolida e azul de metileno intravenoso) podem resultar na síndrome da serotonina.
Compostos semelhantes à varfarina
Para a maioria dos pacientes, o uso ocasional de paracetamol geralmente possui pequeno ou nenhum efeito no índice de normatização internacional (INR) em pacientes recebendo tratamento crônico com varfarina; no entanto, há controvérsia em relação à possibilidade de o paracetamol potencializar os efeitos anticoagulantes da varfarina e outros derivados cumarínicos. Os pacientes devem ser instruídos a perguntarem ao médico ou farmacêutico se eles estão utilizando a varfarina, medicamento que afina o sangue, ou outros derivados cumarínicos antes de utilizar este medicamento.
Flucloxacilina
Acidose metabólica de alto gap aniônico devido ao ácido piroglutâmico (5-oxoprolinemia) tem sido relatada com o uso concomitante de doses terapêuticas de paracetamol e flucloxacilina. Pacientes relatados como de maior risco são mulheres idosas com doenças subjacentes, por exemplo, sepse, anormalidades da função renal e desnutrição. A maioria dos pacientes melhoram após interromper a utilização de um ou de ambos os medicamentos. Os pacientes devem ser instruídos a perguntarem ao seu médico se estão utilizando o antibiótico flucloxacilina antes de utilizar este medicamento.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (entre 15°C e 30°C). Proteger da luz e umidade.
Este medicamento possui prazo de validade de 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico:
TYLEX® 7,5 mg: Comprimidos brancos ou levemente cinzentos, faces planas, redondos e chanfrados, com vinco diametral em uma das faces.
TYLEX® 30 mg: Comprimidos brancos ou levemente cinzentos, faces planas, redondos e chanfrados, com vinco diametral em uma das faces.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
A dose deve ser ajustada de acordo com a intensidade da dor e a resposta do paciente. De modo geral, de acordo com o processo doloroso, recomenda-se:
TYLEX® 7,5 mg = 1 comprimido a cada 4 horas.
TYLEX® 30 mg = 1 comprimido a cada 4 horas.
Em adultos, nas dores de grau mais intenso (como por exemplo, as decorrentes de determinados pós-operatórios, traumatismos graves,
neoplasias) recomendam-se 2 comprimidos a cada 6 horas, não ultrapassando o máximo de 8 comprimidos de TYLEX® 7,5 mg ou TYLEX® 30 mg em um período de 24 horas.
A dose diária máxima para adultos é de:
- fosfato de codeína: 240 mg a cada 24 horas.
- paracetamol: 4000 mg a cada 24 horas.
Este medicamento não deve ser partido ou mastigado.
9. REAÇÕES ADVERSAS
Dados de estudos clínicos
A segurança de codeína e paracetamol a partir de dados de estudos clínicos é baseada em dados de 27 estudos clínicos randomizados, controlados por placebo, de dose única ou doses múltiplas, para o tratamento da dor secundária à cirurgia dentária, cirurgia geral ou artrite reumatoide.
A tabela a seguir inclui eventos adversos que ocorreram quando mais de um evento foi relatado, e a incidência foi maior do que o placebo e em ≥ 1% dos pacientes. Um traço (-) representa uma incidência de < 1%.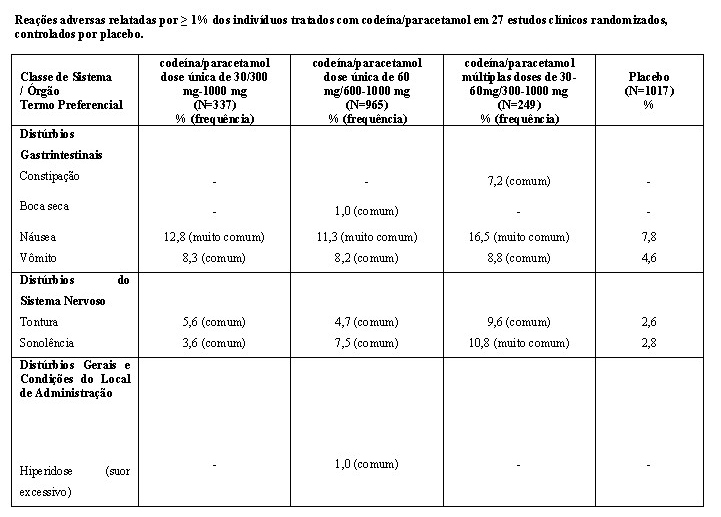
Dados pós-comercialização
Muito comum ≥ 1/10
Comum ≥ 1/100 e < 1/10
Incomum ≥ 1/1.000 e < 1/100
Rara ≥ 1/10.000 e < 1/1.000
Muito rara < 1/10.000
Desconhecida (não pode ser estimada a partir dos dados disponíveis)
Reações adversas ao medicamento (ADRs) identificadas durante a experiência pós-comercialização com codeína e paracetamol estão incluídas na tabela a seguir. As frequências são fornecidas de acordo com a seguinte convenção:
A codeína é um agente opioide. Opioides têm sido associados com as seguintes reações adversas:
• Sedação
• Vertigem
• Broncoespasmo
• Distúrbio gastrointestinal, como dispepsia, náusea, vômito, constipação
• Humor eufórico
• Dependência de drogas pode se desenvolver após o uso prolongado de altas doses
Em casos de eventos adversos, notifique pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
• codeína
Riscos de superdose por codeína incluem parada cardiorrespiratória, edema cerebral, coma, estado confusional, convulsão, hipotensão, hipóxia, íleo paralítico, miose, insuficiência renal, depressão respiratória e insuficiência respiratória, letargia e vômito.
Em particular, agitação e/ou convulsões podem ocorrer em crianças jovens após superdose.
• paracetamol
Transtornos Hepatobiliares
Se um produto contendo paracetamol de liberação prolongada estiver envolvido ou se a formulação exata não for conhecida, recomenda-se obter um nível adicional de paracetamol plasmático de 4 a 6 horas após o nível inicial de paracetamol, pois esses níveis continuarão aumentando com os produtos de liberação prolongada e podem alterar as decisões de tratamento.
Em adultos e adolescentes (≥ 12 anos de idade), pode ocorrer toxicidade hepática após ingestão de mais de 7,5 a 10 gramas durante um período de 8 horas ou menos. Fatalidades são pouco frequentes (menos de 3-4% dos casos não tratados) e foram raramente relatadas com superdoses de menos de 15 gramas. Em crianças ( < 12 anos de idade), uma superdose aguda de menos de 150 mg/kg não foi associada com toxicidade hepática.
Os sintomas iniciais após uma superdose potencialmente hepatotóxica podem incluir

