TREZETE
ACHÉ
rosuvastatina + ezetimiba
Hipocolesterolemiante.
Apresentações.
Comprimidos revestidos 10 mg + 10 mg: embalagens com 10 e 30 comprimidos.
Comprimidos revestidos 20 mg + 10 mg: embalagens com 10 e 30 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de TREZETE 10 mg + 10 mg contém: rosuvastatina cálcica (equivalente a 10 mg de rosuvastatina) 10,4 mg; ezetimiba 10 mg. Excipientes: celulose microcristalina, crospovidona, dióxido de silício, estearato de magnésio, lactose monoidratada, laurilsulfato de sódio, povidona, amarelo crepúsculo, amarelo de quinolina, dióxido de titânio, álcool polivinílico, macrogol e talco.
Cada comprimido revestido de TREZETE 20 mg + 10 mg contém: rosuvastatina cálcica (equivalente a 20 mg de rosuvastatina) 20,8 mg ezetimiba 10 mg. Excipientes: celulose microcristalina, crospovidona, dióxido de silício, estearato de magnésio, lactose monoidratada, laurilsulfato de sódio, povidona, óxido de ferro amarelo, dióxido de titânio, álcool polivinílico, macrogol e talco.
Informações técnicas.
1. INDICAÇÕES
TREZETE deve ser usado como terapia adjuvante à dieta em pacientes considerados como de alto ou muito alto risco cardiovascular, quando a resposta à dieta e aos exercícios é inadequada em pacientes adultos com hipercolesterolemia primária (familiar heterozigótica ou não familiar) ou com dislipidemia mista.
Em pacientes adultos com hipercolesterolemia TREZETE é indicado para:
redução do LDL-colesterol; colesterol total e triglicérides elevados; diminuição de ApoB; não HDL-C; das razões LDL-C/HDL-C; não HDL-C/HDL-C; ApoB/Apo A-I; C-total/HDL-C e aumento de HDL-C.
2. RESULTADOS DE EFICÁCIA
O Estudo LANCE (Estudo clínico fase III randomizado, aberto, multicêntrico, comparativo de rosuvastatina + ezetimiba versus sinvastatina + ezetimiba, para avaliação da eficácia e segurança em pacientes de alto risco com hipercolesterolemia primária ou dislipidemia mista) teve como objetivo primário avaliar a eficácia da associação rosuvastatina + ezetimiba em comparação com a combinação sinvastatina + ezetimiba no controle dos níveis de colesterol LDL em relação ao basal, em participantes de alto risco cardiovascular, portadores de hipercolesterolemia primária ou dislipidemia mista.
Os objetivos secundários do estudo foram avaliar a variação dos níveis de HDL, triglicérides, PCR e Apolipoproteína B após nove semanas de tratamento em relação aos níveis basais.
Os desfechos de eficácia considerados para a combinação de rosuvastatina + ezetimiba foram a taxa de participantes que atingiram níveis de LDL abaixo de 100mg/dL após nove semanas de tratamento e a eficácia comparativa em relação aos níveis basais para atingir níveis de LDL-c < 70 mg/dL, Colesterol não-HDL < 100 mg/dL, bem como avaliar os efeitos do tratamento sobre os níveis de HDL-C, triglicérides, Apolipoproteína B e Proteína C reativa.
O estudo compreendeu um período de quinze semanas, sendo dividido em período de triagem (uma semana), período de uniformização (no qual todos os participantes da pesquisa utilizaram monoterapia com sinvastatina 20 mg por um período de cinco semanas) e período de tratamento (no qual os participantes da pesquisa que apresentavam níveis de LDL iguais ou acima de 100mg/dL foram randomizados para um dos tratamentos em estudo (rosuvastatina 10 mg + ezetimiba 10 mg ou sinvastatina 20 mg + ezetimiba 10 mg). O tratamento com as dosagens iniciais das combinações teve duração de cinco semanas e, após quatro semanas de tratamento, os participantes que apresentavam níveis abaixo de 100mg/dL, tiveram a dosagem da medicação mantida por mais quatro semanas e os que apresentaram níveis iguais ou acima de 100 mg/dL passaram a receber as dosagens de rosuvastatina 20 mg + ezetimiba 10 mg ou sinvastatina 40 mg + ezetimiba 10 mg por um período de mais quatro semanas.
Dos 129 participantes randomizados, 66 (51,2%) receberam rosuvastatina + ezetimiba e 63 (48,8%) receberam sinvastatina + ezetimiba. No total, 117 pacientes (90,7%) concluíram as nove semanas de tratamento, sendo 59 (89,4%) e 58 (92,1%) em cada grupo, respectivamente. Todos os participantes da pesquisa randomizados foram incluídos nas populações de segurança e intenção de tratar.
Os valores médios de CT e LDL-C antes do início do período de uniformização com sinvastatina eram de 247,88 (± 31,95) mg/dL e 162,29 (± 27,12) mg/dL.
Os valores basais para o início do período de tratamento com as combinações foram considerados a partir do final do período de uniformização com o uso de sinvastatina, sendo a média dos valores de LDL-C 124,79 (± 19,97) mg/dL para o grupo randomizado para rosuvastatina + ezetimiba (R/E) e 121,27 (± 17,03) mg/dL para o grupo randomizado para sinvastatina + ezetimiba (S/E).
O tratamento com as combinações fixas teve a duração de 9 semanas, sendo que nas primeiras 4 semanas foram utilizadas as dosagens de rosuvastatina 10 mg/ezetimiba 10 mg ou sinvastatina 20 mg/ezetimiba 10 mg administradas uma vez ao dia. Na análise descritiva, após 4 semanas de uso das combinações, o valor médio de LDL-C no grupo R/E foi 75,03 (± 20,91) mg/dL e 87,98 (± 23,44) mg/dL, no grupo S/E. Aumento de dosagem em função da não obtenção de meta de LDL < 100 mg/ml foi necessário em 6 pacientes no grupo R/E e 19 pacientes no grupo S/E, utilizando-se rosuvastatina 20 mg + ezetimiba 10 mg e sinvastatina 40 mg + ezetimiba 10 mg por mais 4 semanas, sendo a média de LDL-C na semana 9 do estudo de 76,25 (± 23,79) mg/dL e 89,34 (± 23,36) mg/dL, respectivamente para os grupos.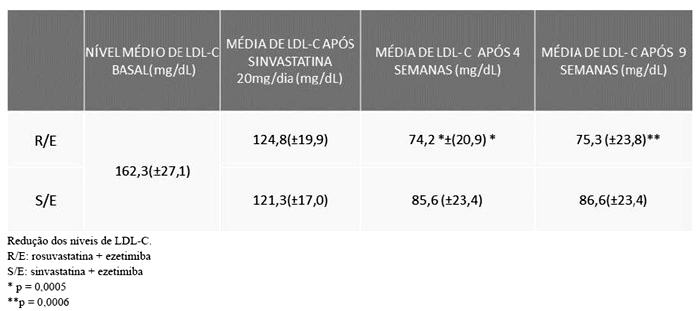
A variação absoluta dos níveis de LDL-C do período de uniformização com sinvastatina (semana -3) até o final do estudo com o uso das combinações foi de 86,78 (± 38,66) mg/dL e 72,54 (± 34,50) mg/dL para os grupos R/E e S/E respectivamente.
Na avaliação dos percentuais médios absolutos totais de redução de LDL-C, as variações totais foram de -51,3 % e -43,3%, respectivamente para as combinações R/E e S/E.
A estimativa de mínimos quadrados da variação do valor final de LDL-C em relação ao basal (LDLfinal/LDLbasal) mostrou que a combinação R/E proporcionou uma redução nos níveis de LDL-C, em média, 15% (IC95%: 6,1%; 22,2%) maior em relação à combinação S/E e uma redução média dos níveis de LDL-C de -39,5% (IC 95%: -44%; -35%) para o grupo R/E e -29,1% (IC95%: -34%; -24%) para o grupo S/E, sendo a diferença entre os grupos de -10,3% (IC95%: (-17%; -4,0%).
A média ajustada de LDL-C ao longo das semanas de tratamento mostrou um valor 13% menor na semana 4 do estudo para o grupo R/E em relação ao grupo S/E (p = 0,0005), com resultados semelhantes sendo observados na semana 9 (p = 0,0006).
A meta de LDL-C < 100 mg/dL foi atingida por 84,8% dos pacientes no grupo R/E em comparação a 68,2% no grupo sinvastatina/ezetimiba após 4 semanas (p = 0,0257) e em 81,2% versus 73,0%, respectivamente, após 9 semanas (p = 0,23). A obtenção da meta de LDL-C < 70 mg/dL foi obtida em proporção significativamente superior para o grupo R/E tanto na semana 4 (45,4% versus 15,9%, p = 0,003) como na semana 9 (40,9% versus 15,9%, p = 0,0017)
Na semana 4, observou-se que o nível médio de Apolipoproteína B para o grupo R/E foi 13% menor em relação ao nível do grupo S/E (p = 0,0005) e o nível médio de colesterol não HDL-C foi 12% menor, respectivamente para os grupos.
O estudo EXPLORER investigou a eficácia e segurança da rosuvastatina 40 mg isoladamente ou em combinação com ezetimiba 10 mg em pacientes com alto risco de doença cardíaca coronariana. Quatrocentos e sessenta e nove pacientes foram aleatoriamente designados para tratamento com rosuvastatina isoladamente ou em combinação com ezetimiba por 6 semanas. O desfecho primário foi o percentual de pacientes que atingiram a meta de colesterol LDL ( < 100 mg/dL) do Painel do Grupo americano de Tratamento Adulto (ATP III) na semana 6. Os desfechos secundários incluíram a porcentagem de pacientes que atingiram a meta ATPIII de lipoproteína não-HDL (colesterol não HDL < 130 mg/dL e colesterol LDL 100 mg/dl quando o valor basal de triglicèrides fosse ≥ 200 mg/dL); as metas Europeias 2003 de colesterol LDL ( < 2,5 ou 3,0 mmol/L [ < 100 ou 115 mg/dL], dependendo da categoria de risco); as metas Europeias 2003 combinadas para colesterol LDL e colesterol total (CT) (colesterol LDL < 2,5 ou 3,0 mmol/L [ < 100 ou 115 mg/dL] e CT < 4,5 ou 5,0 mmol/L [ < 175 ou 190 mg/dL], dependendo da categoria de risco); e alteração percentual desde a linha de base no colesterol LDL, colesterol HDL, CT, TG, colesterol não-HDL, proporções lipídicas (colesterol LDL/HDL, CT/colesterol HDL e colesterol não-HDL/HDL), Apolipoproteína A-I, Apolipoproteína B, proporção de Apolipoproteína B/Apolipoproteína A-I e alterações na proteína C reativa de alta sensibilidade (PCR-as), e parâmetros de segurança e tolerabilidade na semana 6.
Como resultado, significativamente mais pacientes recebendo rosuvastatina/ezetimiba do que a rosuvastatina isolada atingiram a meta ATPIII de colesterol LDL ( < 100 mg/dL, 94,0% vs 79,1%, p < 0,001) e a meta de colesterol LDL ( < 70 mg/dL) para pacientes de muito alto risco (79,6% vs 35,0%, p < 0,001) na semana 6. A combinação de rosuvastatina/ezetimiba reduziu o colesterol LDL significativamente mais do que a rosuvastatina (-69,8% vs. -57,1%, p < 0,001). As metas ATP III para colesterol não-HDL e LDL para pacientes com triglicérides (TG) ≥ 200 na linha de base (88 pacientes [37.4%] no grupo da terapia combinada; 80 pacientes [34.8%] no grupo da monoterapia) foram alcançadas em significativamente maior porcentagem de pacientes no grupo da terapia combinada que no da monoterapia (p < 0,001).
Adicionalmente, na semana 6, significativamente mais pacientes de muito alto risco (196 no grupo da terapia combinada e 197 no grupo da monoterapia) atingiram a meta opcional de colesterol LDL de < 70 mg/dL (79,6 % vs. 35,0%, p < 0,001). Significativamente mais pacientes alcançaram ambas as metas atualizadas (colesterol não-HDL < 100 mg/dL e LDL < 70 mg/dL, ou não-HDL < 130 mg/dl e LDL < 100 mg/dL, dependendo da categoria de risco, para pacientes com TG ≥ 200 mg/dL na linha de base) com a terapia combinada do que com a monoterapia (79,5% vs. 27,5%, p < 0,001).
As análises de acordo com as metas Europeias 2003 de colesterol LDL (colesterol LDL < 2,5 ou 3,0 mmol/L [ < 100 ou 115 mg/dL] dependendo da categoria de risco) e as metas combinadas de colesterol LDL e TC (colesterol LDL < 2,5 ou 3,0 mmol/L [ < 100 ou 115 mg/dl] e TC < 4,5 ou 5,0 mmol/L [ < 175 ou 190 mg/dL], respectivamente, dependendo da categoria de risco) indicaram que uma porcentagem significativamente maior de pacientes no grupo de terapia combinada atingiu a meta em comparação com aqueles no grupo de monoterapia na semana 6 (93,6% vs 74,3% de colesterol LDL e TC 90,6% vs 68,3%, p < 0,001 para ambos).
Às 6 semanas, o colesterol LDL diminuiu para 56,9 e 81,5 mg/dL nos grupos de combinação e monoterapia, respectivamente. Reduções percentuais significativamente maiores nos níveis de colesterol LDL foram alcançadas com a terapia combinada do que com a monoterapia (percentagem média de redução -69,8% vs -57,1%, p < 0,001). Significativamente (p < 0,001) maiores diminuições em CT, colesterol não-HDL e TGs foram observadas também na semana 6 no grupo de terapia combinada em comparação com o grupo de monoterapia. Ambos os tratamentos aumentaram o colesterol HDL de forma semelhante na semana 6. As taxas de colesterol LDL/HDL, colesterol CT/HDL e colesterol não-HDL/HDL diminuíram significativamente mais em pacientes que receberam terapia combinada do que naqueles recebendo monoterapia (todos p < 0,001). O valor médio da semana 6 para a relação colesterol LDL/HDL foi 1,1. Diminuições significativas da Apolipoproteína B e da proporção Apolipoproteína B/Apolipoproteína A-I foram observadas no grupo de terapia combinada em comparação com o grupo de monoterapia (p < 0,001 para ambos). A Apolipoproteína A-I aumentou 3,2% e 1,6% nos grupos de terapia combinada e monoterapia, respectivamente (p < 0,202).
Após 6 semanas de tratamento, a redução percentual mediana em hs-CRP foi significativamente maior com a terapia combinada do que com a monoterapia (-46,4% vs -28,6%, p < 0,001). Dos 183 pacientes com hs-CRP mediana > 3 mg/L na linha de base (90 no grupo de terapia combinada, 93 no grupo da monoterapia), uma proporção mais elevada dos pacientes tratados com a terapia combinada do que com monoterapia alcançaram hs-CRP < 3 mg/L (56,7% vs 37,6%) e < 1 mg/L (14,4% vs 6,5%). Ambos os tratamentos foram bem tolerados. A rosuvastatina 40 mg foi eficaz na melhora do perfil lipídico aterogênico nessa população de alto risco. A combinação de rosuvastatina com ezetimiba melhorou a redução do colesterol LDL e permitiu que mais pacientes atingissem as metas de colesterol LDL. Concluiu-se que a rosuvastatina associada à ezetimiba pode melhorar o manejo de pacientes de alto risco que não conseguem atingir a meta em monoterapia com estatina na dose máxima.
O estudo GRAVITY comparou eficácia, segurança e efeito sobre biomarcadores de rosuvastatina 10 ou 20 mg associada à ezetimiba 10 mg (RSV10/EZE10 e RSV20/EZE10) com sinvastatina 40 mg ou 80 mg associada à EZE10 (SIM40/EZE10 e SIM80/EZE10) em pacientes com doença arterial coronariana (DAC) ou risco equivalente a DAC. Após interrupção da terapia hipolipemiante vigente seguida de uma fase de adaptação dietética do estudo, os pacientes (n = 833) elegíveis para inclusão na fase de tratamento randomizado, cujos requisitos foram LDL-C em jejum de 130 a < 220 mg/dL (3,4 a < 5,7 mmol/L) e triglicerídeos em jejum (TG) < 400 mg/dL (4,5 mmol/L), receberam aleatoriamente um dos quatro tratamentos monoterápicos por 6 semanas, rosuvastatina 10 ou 20 mg, sinvastatina 40 ou 80 mg (1:1:1:1). Em seguida, ezetimiba 10 mg foi adicionada ao tratamento com estatina na mesma dose (RSV10/EZE10, RSV20/EZE10, SIM40/EZE10 ou SIM80/EZE10) por mais 6 semanas. O desfecho primário foi a alteração do LDL-C, após 12 semanas. Além disso, alterações em biomarcadores relacionados à síntese (lanosterol), metabolismo (colesterol livre) e absorção (b-sitosterol) de colesterol, síntese de ácidos biliares (7-a-hidroxi-4-colesteno-3-ona [C4]) e aterosclerose (concentração e atividade de 7-cetocolesterol e fosfolipase A2 associada a lipoproteína [Lp-PLA2]).
Como resultado, RSV20/EZE10 reduziu o LDL-C a partir da linha de base em 63,5% vs. reduções de 55,2-57,4% alcançadas com SIM40/EZE10 ou SIM80/EZE10, respectivamente (p < 0,001). As reduções de LDL-C com ROS10/EZE10 (59,7%) foram significativamente maiores (p = 0,002) vs. SIM40/E10. Quando comparada à estatina em monoterapia, novas reduções de LDL-C de ~ 10% -14% foram obtidas com a adição de ezetimiba.
Uma proporção significativamente maior (p ≤ 0,007) dos pacientes atingiu a meta de LDL < 100 mg/dL e < 70 mg/dL com RSV20/EZE10 (95,6% e 77,0%, respectivamente) do que com qualquer dose de sinvastatina com ezetimiba (87,4-88,6% e 55,3-67,7%, respectivamente). A proporção de pacientes que atingiram o objetivo de LDL-C < 100 mg/dL foi significativamente maior com RSV10/EZE10 (93,3%) vs. SIM40/EZE10 (p = 0,03).
Com a terapia combinada, os níveis de HDL-c aumentaram em relação aos valores basais em 3,9% e 7,5% entre os grupos, com aumentos significativos (p < 0,05) observados com RSV20/EZE10 vs. qualquer uma das doses de sinvastatina com ezetimiba. ROS20/EZE10 resultou em significativamente (p < 0,001) maiores reduções nos níveis de CT, TG, colesterol não-HDL e ApoB e nas razões CT/HDL, LDL/HDL, não-HDL/HDL e ApoB/ApoA-I vs. qualquer uma das doses de sinvastatina com ezetimiba. Reduções nesses parâmetros também foram significativamente (p < 0,05) maiores com RSV10/EZE10 vs. SIM40/EZE10. Todos os tratamentos resultaram em reduções médias do nível de hsCRP a partir da linha de base. Não houve diferenças estatisticamente significativas entre os grupos nas alterações da ApoA-1 ou hsCRP. A análise opcional de biomarcadores incluiu 542 pacientes. Os níveis de biomarcadores basais foram semelhantes entre os grupos. Significativas (p < 0,05) reduções a partir da linha de base ao fim de 6 semanas em monoterapia foram observados em todos os grupos para lanosterol, 7-cetocolesterol, colesterol livre e concentração e atividade de Lp-PLA2 e C4 com ambas as doses de rosuvastatina; as alterações a partir da linha de base para monoterapia em b-sitosterol não foram significativas para qualquer grupo de tratamento.
As reduções médias da linha de base com a terapia combinada foram numericamente maiores para a maioria dos biomarcadores ao final das monoterapias, exceto para C4 e lanosterol. Reduções do fim da monoterapia ao fim da terapia combinada foram significativas (p ≤ 0,002) para todos os grupos de tratamento para 7-cetocolesterol (exceto para RSV20/EZE10), b-sitosterol, colesterol livre e atividade e concentração de Lp-PLA2. Não houve reduções significativas do final da monoterapia para o final da terapia combinada para C4 e lanosterol. Todos os tratamentos foram bem tolerados.
Concluiu-se que a coadministração de rosuvastatina 10 ou 20 mg e ezetimiba 10 mg alcançou melhora significativa nos perfis lipídicos de pacientes de alto risco, quando comparado com sinvastatina 40 ou 80 mg associada à ezetimiba 10 mg.
Referências:
BALLANTYNE, Christie M. et al. Efficacy and safety of rosuvastatin 40 mg alone or in combination with ezetimibe in patients at high risk of cardiovascular disease (results from the EXPLORER study). The American journal of cardiology, v. 99, n. 5, p. 673-680, 2007.
BALLANTYNE, Christie M. et al. Efficacy, safety and effect on biomarkers related to cholesterol and lipoprotein metabolism of rosuvastatin 10 or 20 mg plus ezetimibe 10 mg vs. simvastatin 40 or 80 mg plus ezetimibe 10 mg in high-risk patients: results of the GRAVITY randomized study. Atherosclerosis, v. 232, n. 1, p. 86-93, 2014.
Clinical study report NCT01420549: Efficacy and Safety of FDC in High Risk Patients With Primary Hypercholesterolemia or Mixed Dyslipidemia (LANCE).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
a) rosuvastatina
Mecanismo de ação: A rosuvastatina é um seletivo e potente inibidor competitivo da HMG-CoA redutase, a enzima que limita a taxa de conversão da 3-hidroxi-3-metilglutaril coenzima A para mevalonato, um precursor do colesterol. Os triglicérides (TG) e o colesterol são incorporados no fígado à Apolipoproteína B (ApoB), e liberados no plasma como lipoproteína de densidade muito baixa (VLDL), para serem distribuídos nos tecidos periféricos. As partículas VLDL são ricas em triglicérides. A lipoproteína de baixa densidade (LDL), rica em colesterol, é formada a partir de VLDL e captada principalmente através do receptor de LDL de alta afinidade no fígado.
A rosuvastatina exerce seus efeitos modificadores sobre os lipídios de duas maneiras: ela aumenta o número de receptores LDL hepáticos na superfície celular, aumentando a captação e o catabolismo do LDL, e inibe a síntese hepática de VLDL, reduzindo, assim, o número total de partículas de VLDL e LDL.
A lipoproteína de alta densidade (HDL) que contém ApoA-I é envolvida, entre outros, no transporte do colesterol dos tecidos de volta para o fígado (transporte reverso de colesterol).
O envolvimento do LDL-C na aterogênese está bem documentado. Estudos epidemiológicos estabeleceram que LDL-C e TG altos e HDLC e ApoA-I baixos foram associados a um maior risco de doença cardiovascular. Estudos de intervenção mostraram os benefícios da redução de LDL-C e TG ou do aumento do HDL-C sobre as taxas de mortalidade e de eventos cardiovasculares (CV). Dados mais recentes associaram os efeitos benéficos dos inibidores da HMG-CoA redutase à diminuição do não-HDL (por ex.: todo colesterol circulante que não está em HDL) e da ApoB ou à redução da razão ApoB/ApoA-I.
b) ezetimiba
Mecanismo de ação:
A ezetimiba pertence a uma nova classe de compostos hipolipemiantes que inibem de forma seletiva a absorção intestinal de colesterol e de fitosteróis relacionados. A ezetimiba é ativo e potente por via oral e apresenta mecanismo de ação exclusivo, que difere de outras classes de compostos redutores do colesterol (por exemplo, estatinas, sequestrantes de ácidos biliares [resinas], derivados do ácido fíbrico e fitosteróis). A meta molecular da ezetimiba é o transportador de esterol, Niemann-Pick C1-Like 1 (NPC1L1), responsável pela captação intestinal de colesterol e de fitosteróis. A ezetimiba localiza-se na borda em escova dos enterócitos do intestino delgado, onde inibe a absorção do colesterol, promovendo redução do aporte de colesterol do intestino para o fígado. Isto leva à redução do estoque de colesterol hepático e ao aumento da depuração do colesterol sanguíneo. A ezetimiba não aumenta a excreção de ácido biliar (como os sequestrantes de ácidos biliares) e não inibe a síntese hepática de colesterol (como as estatinas).
Propriedades Farmacocinéticas
a) rosuvastatina
Absorção: A rosuvastatina é administrada por via oral na forma ativa, com picos de níveis plasmáticos ocorrendo 5 horas após a administração. A absorção aumenta linearmente com a faixa de dose. A meia-vida é de 19 horas e não aumenta com a elevação da dose.
Distribuição: A biodisponibilidade absoluta é de 20%. Há um acúmulo mínimo com dose única diária repetida. Aproximadamente 90% da rosuvastatina liga-se às proteínas plasmáticas, principalmente à albumina.
Metabolismo: A rosuvastatina sofre metabolismo de primeira passagem no fígado, que é o local primário da síntese de colesterol e da depuração de LDL-C. Mais de 90% da atividade inibitória para a HMG-CoA redutase circulante é atribuída ao princípio ativo.
Excreção: A rosuvastatina sofre metabolismo limitado (aproximadamente 10%), principalmente para a forma N-desmetila, e 90% são eliminados como droga inalterada nas fezes, sendo o restante excretado na urina.
Populações especiais
Idade e sexo: não houve efeito clinicamente relevante associado à idade ou sexo na farmacocinética da rosuvastatina em adultos. A farmacocinética da rosuvastatina em crianças e adolescentes com hipercolesterolemia familiar heterozigótica foi similar à de voluntários adultos.
Raça: estudos farmacocinéticos mostram uma elevação de aproximadamente duas vezes na mediana da área sob a curva (AUC) em descendentes asiáticos comparados com caucasianos. Uma análise da farmacocinética da população não revelou diferenças clinicamente relevantes na farmacocinética entre caucasianos, hispânicos e negros ou grupos de afrocaribenhos.
Insuficiência renal: em um estudo realizado em indivíduos com graus variáveis de insuficiência renal, a doença renal de leve a moderada apresentou pouca influência nas concentrações plasmáticas da rosuvastatina. Entretanto, indivíduos com insuficiência grave (depuração de creatinina < 30 mL/min) apresentaram um aumento de 3 vezes na concentração plasmática em comparação com voluntários sadios.
Insuficiência hepática: em um estudo realizado em indivíduos com graus variáveis de insuficiência hepática, não houve evidência de aumento da exposição à rosuvastatina, exceto em 2 indivíduos com doença hepática mais grave (graus 8 e 9 de Child-Pugh). Nestes indivíduos, a exposição sistêmica foi aumentada em no mínimo 2 vezes em comparação aos indivíduos com grau menor de Child-Pugh.
Polimorfismos genéticos: a disponibilidade dos inibidores da HMG-CoA redutase, incluindo a rosuvastatina, envolve OATP1B1 e as proteínas transportadoras BCRP.
Em pacientes com polimorfismos genéticos em SLCO1B1 (OATP1B1) e/ou ABCG2 (BCRP) existe um risco de maior exposição à rosuvastatina. Polimorfismos individuais de SLCO1B1 c.521CC e ABCG2 c.421AA estão associados com uma exposição (AUC) à rosuvastatina aproximadamente 1,6 ou 2,4 vezes maior, respectivamente, em comparação com os genótipos SLCO1B1 c.521TT ou ABCG2 c.421CC.
b) ezetimiba
Absorção: após administração oral, a ezetimiba é rapidamente absorvida e extensivamente conjugada a um glicuronídeo fenólico farmacologicamente ativo (glicuronídeo da ezetimiba), cujas concentrações plasmáticas máximas (Cmáx) médias ocorrem em 1 a 2 horas. Já para a ezetimiba, estas concentrações são atingidas em 4 a 12 horas. A biodisponibilidade absoluta da ezetimiba não pode ser determinada já que o composto é praticamente insolúvel em meios aquosos próprios para injeção. A administração concomitante de alimentos (com teores de gorduras baixos ou altos) não exerceu qualquer efeito sobre a biodisponibilidade oral da ezetimiba.
Distribuição: a ezetimiba e o glicuronídeo da ezetimiba estão 99,7% e 88% a 92% ligadas às proteínas plasmáticas de seres humanos, respectivamente.
Metabolismo: a ezetimiba é metabolizada principalmente no intestino delgado e no fígado, por meio da conjugação do glicuronídeo (uma reação de fase II) e excreção biliar subsequente. Observou-se metabolismo oxidativo mínimo (uma reação de fase I) em todas as espécies avaliadas. A ezetimiba e o glicuronídeo da ezetimiba são os principais derivados do fármaco detectados no plasma, constituindo aproximadamente 10% a 20% e 80% a 90% do total, respectivamente. Tanto a ezetimiba quanto o glicuronídeo da ezetimiba são eliminados lentamente do plasma, com evidência de recirculação entero-hepática significativa. A meia-vida da ezetimiba e do glicuronídeo da ezetimiba é de aproximadamente 22 horas.
Eliminação: após administração oral de 20 mg de (14C) ezetimiba a seres humanos, a ezetimiba total respondeu por cerca de 93% da radioatividade plasmática total. Aproximadamente 78% e 11% da carga radioativa administrada foram recuperados nas fezes e na urina, respectivamente, ao longo de um período de coleta de 10 dias. Após 48 horas, os níveis plasmáticos de radioatividade eram indetectáveis.
Populações especiais
Pacientes pediátricos: a absorção e o metabolismo da ezetimiba são semelhantes em crianças e adolescentes (10 a 18 anos de idade) e adultos. Com base na ezetimiba total, não há diferenças farmacocinéticas entre adolescentes e adultos. Não estão disponíveis dados de farmacocinética na população pediátrica com menos de 10 anos de idade. A experiência clínica em pacientes pediátricos e adolescentes (idades entre 9 e 17 anos) é limitada aos pacientes com HFHo (Hipercolesterolemia familiar homozigótica) ou sitosterolemia.
Pacientes idosos: as concentrações plasmáticas da ezetimiba total são, aproximadamente, 2 vezes mais elevadas nos indivíduos idosos ( > 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens tratados com ezetimiba.
Portanto, não é necessário ajuste posológico para pacientes idosos.
Insuficiência hepática: após uma única dose de 10 mg da ezetimiba, a área sob a curva (AUC) média para a ezetimiba total aumentou aproximadamente 1,7 vezes em pacientes com insuficiência hepática leve (escore de Child-Pugh de 5 ou 6) em comparação com indivíduos sadios. Em um estudo de 14 dias no qual se administraram doses múltiplas (10 mg diariamente) a pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média da ezetimiba total aumentou aproximadamente 4 vezes no 1° dia e no 14° dia em comparação com o observado em indivíduos sadios. Não é necessário ajuste posológico para pacientes com insuficiência hepática leve. Uma vez que os efeitos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave (escore de Child-Pugh > 9) são desconhecidos, a ezetimiba não é recomendada para esses pacientes (vide "5. ADVERTÊNCIAS E PRECAUÇÕES).
Insuficiência renal: após a administração de uma única dose de 10 mg de ezetimiba a pacientes com doença renal grave (n = 8; clearance de creatinina médio < 30 mL/min/1,73 m2), a AUC média da ezetimiba total aumentou aproximadamente 1,5 vezes quando comparada àquela de indivíduos sadios (n = 9); esse resultado não é considerado clinicamente relevante. Não é necessário ajuste posológico para pacientes com disfunção renal.
Neste mesmo estudo, a exposição à ezetimiba total de um paciente submetido a transplante renal e que estava recebendo múltiplas medicações, inclusive ciclosporina, foi 12 vezes maior.
Sexo: As concentrações plasmáticas da ezetimiba total são discretamente mais elevadas ( < 20%) em mulheres em relação aos homens. A redução de LDL-C e o perfil de segurança são comparáveis entre homens e mulheres que receberam ezetimiba. Não é necessário, portanto, ajuste posológico com base no sexo.
Raça: Não foram demonstradas diferenças quanto à farmacocinética em negros e caucasianos com base em uma metanálise de estudos de farmacocinética.
Dados de segurança pré-clínica
Os dados pré-clínicos não revelam danos especiais em humanos, tendo como base estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico e toxicidade reprodutiva.
4. CONTRAINDICAÇÕES
TREZETE é contraindicado para:
• Pacientes com hipersensibilidade a qualquer dos componentes da medicação;
• Pacientes com doença hepática ativa; incluindo elevações persistentes e inexplicadas das transaminases séricas e qualquer elevação das transaminases séricas que exceda 3 vezes o limite superior do normal (LSN);
• Pacientes com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2);
• Pacientes com miopatia;
• Pacientes que fazem uso de ciclosporina;
• Durante a gravidez, na lactação, e para mulheres com potencial de engravidar que não estão usando métodos contraceptivos apropriados.
Categoria de risco na gravidez: X.
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Fígado: como para os inibidores da HMG-CoA redutase quando em uso isolado, TREZETE deve ser usado com cautela em pacientes que consomem quantidades excessivas de álcool e/ou que tenham uma história de doença hepática.
É recomendado que os testes de enzimas hepáticas sejam realizados antes e por 12 semanas após o início da terapia e no caso de qualquer elevação da dose, e depois periodicamente (por exemplo, semestralmente).
Uma vez que os efeitos da maior exposição à ezetimiba em pacientes com insuficiência hepática moderada ou grave são desconhecidos, TREZETE não é recomendado para uso nestes pacientes (vide "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Sistema musculoesquelético: foram relatados efeitos musculoesqueléticos, como mialgia, miopatia e, raramente, rabdomiólise em pacientes tratados com rosuvastatina isolada.
Em estudos clínicos, a incidência de CPK > 10 vezes o LSN (Limite Superior da Normalidade) foi de 0,2% para ezetimiba versus 0,1% para o placebo, e de 0,1% para ezetimiba coadministrado com uma estatina versus 0,4% para as estatinas isoladamente.
Assim como outros inibidores da HMG-CoA redutase, com a rosuvastatina, a frequência de rabdomiólise no uso pós-comercialização é maior com as doses mais altas administradas.
Houve relatos muito raros de uma miopatia necrotizante imunomediada caracterizada clinicamente por fraqueza muscular proximal persistente e elevação da creatinoquinase sérica durante o tratamento ou após a descontinuação de estatinas, incluindo a rosuvastatina. Testes neuromusculares e sorológicos adicionais podem ser necessários e tratamento com agentes imunossupressores podem ser requeridos.
Nos estudos com a rosuvastatina não houve evidência de aumento de efeitos musculoesqueléticos na administração concomitante com qualquer terapia. Entretanto, foi observado um aumento da incidência de miosite e miopatia em pacientes que estavam recebendo outros inibidores da HMG-CoA redutase junto com derivados do ácido fíbrico, incluindo genfibrozila, ácido nicotínico, antifúngicos do grupo azóis e antibióticos macrolídeos. Rabdomiólise foi relatada muito raramente na monoterapia com ezetimiba isolada e muito raramente com a adição de ezetimiba a agentes conhecidos por estarem associados ao risco aumentado de rabdomiólise. Todos os pacientes que iniciam a terapia com TREZETE devem ser alertados do risco de miopatia e devem relatar imediatamente qualquer dor, sensibilidade ou fraqueza muscular inexplicada. TREZETE deve ser imediatamente descontinuado se houver suspeita ou for comprovada a miopatia. A presença destes sintomas e um nível de creatina fosfoquinase (CPK) > 10 vezes o Limite Superior da Normalidade indica miopatia.
TREZETE deve ser prescrito com precaução em pacientes com fatores de predisposição para miopatia, tais como, insuficiência renal, idade avançada e hipotireoidismo, ou situações em que pode ocorrer um aumento nos níveis plasmáticos do medicamento (vide "3. CARACTERÍSTICAS FARMACOLÓGICAS" e "6. INTERAÇÕES MEDICAMENTOSAS").
O uso de TREZETE deve ser temporariamente interrompido em qualquer paciente com uma condição aguda grave sugestiva de miopatia ou que predispõe ao desenvolvimento de insuficiência renal secundária à rabdomiólise (por exemplo: sépsis; hipotensão; cirurgia de grande porte; trauma; alterações metabólicas, endócrinas e eletrolíticas graves; ou convulsões não controladas).
Diabetes mellitus: assim como com outros inibidores da HMG-CoA redutase, foi observado em pacientes tratados com rosuvastatina isolada um aumento dos níveis de HbA1c e de glicose sérica e, em alguns casos, estes aumentos podem exceder o limiar para o diagnóstico do diabetes, principalmente em pacientes com alto risco de para o desenvolvimento do diabetes mellitus (vide "9. REAÇÕES ADVERSAS").
Doença pulmonar intersticial: Foram relatados casos excepcionais de doença pulmonar intersticial com algumas estatinas, especialmente com terapia a longo prazo, podendo apresentar dispneia, tosse não produtiva e deterioração da saúde geral (fadiga, perda de peso e febre). Caso haja suspeita de que um paciente desenvolveu doença pulmonar intersticial, a terapia com Trezete deve ser descontinuada.
Raça: estudos de farmacocinética mostraram um aumento na exposição à rosuvastatina em pacientes descendentes asiáticos comparados
com pacientes caucasianos (vide "8. POSOLOGIA E MODO DE USAR" e "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Fibratos: a coadministração de TREZETE com fibratos, não foi estudada. Portanto, a coadministração de TREZETE e fibratos não é recomendada (vide "6. INTERAÇÕES MEDICAMENTOSAS").
Varfarina: Se TREZETE for acrescentado à terapia com varfarina ou outro anticoagulante cumarínico, a Razão Normalizada Internacional
(RNI) deve ser adequadamente monitorada (vide "6. INTERAÇÕES MEDICAMENTOSAS").
Idosos: A concentração plasmática da ezetimiba total é, aproximadamente, 2 vezes mais elevada nos indivíduos idosos ( > 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens que recebem ezetimiba. Como a idade avançada (≥ 65 anos) è um fator predisponente para miopatia por estatina, Trezete deve ser prescrito com cautela a idosos. Não é necessário, entretanto, ajuste posológico para pacientes idosos.
Crianças e adolescentes de 10 a 17 anos de idade: não há estudos disponíveis para comprovar a segurança de uso de TREZETE em população de crianças e adolescentes.
Efeito sobre a capacidade de dirigir veículos e operar máquinas: testes farmacológicos não revelaram evidências de efeito sedativo da rosuvastatina e da ezetimiba. A partir do perfil de segurança, não se espera que TREZETE afete a capacidade de dirigir veículos e operar máquinas.
Uso durante a gravidez e lactação: a segurança de TREZETE durante a gravidez e a lactação não foi estabelecida e seu uso é contraindicado durante a gravidez e lactação. Mulheres com potencial de engravidar devem usar métodos contraceptivos apropriados (vide "4. CONTRAINDICAÇÕES").
Este medicamento contém lactose (156 mg/comprimido para TREZETE 10 mg + 10 mg e 189,7 mg/comprimido para TREZETE 20 mg + 10 mg), portanto deve ser usado com cautela por pacientes com intolerância à lactose.
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de medicamentos coadministrados sobre a rosuvastatina
Dados in vitro e in vivo indicam que a rosuvastatina não tem interação clinicamente significativa com o citocromo P450 (como um substrato, inibidor ou indutor). A rosuvastatina é um substrato para determinadas proteínas transportadoras, incluindo o transportador hepático de captação OATP1B1 e o transportador de efluxo BCRP. A administração concomitante de rosuvastatina com medicamentos que são inibidores destas proteínas transportadoras pode resultar em maior concentração plasmática de rosuvastatina e maior risco de miopatia.
Interações que requerem ajuste da dose de rosuvastatina: Quando é necessária a coadministração de TREZETE com outros medicamentos que conhecidamente aumentam a exposição à rosuvastatina, a dose de TREZETE deve ser ajustada. Se o aumento esperado na exposição (AUC) for de aproximadamente 2 vezes ou mais, deve-se avaliar a possibilidade de iniciar o tratamento com ezetimiba e rosuvastatina separadamente, iniciando-se com uma dose de 5 mg uma vez dia de rosuvastatina e passando-se para a combinação fixa posteriormente, ajustando a dose máxima diária de rosuvastatina.
Interação com outros medicamentos
Antiácidos: a administração simultânea de rosuvastatina com uma suspensão de antiácido contendo hidróxido de alumínio e hidróxido de magnésio resultou em diminuição da concentração plasmática da rosuvastatina de aproximadamente 50%.
Este efeito foi reduzido quando o antiácido foi administrado 2 horas após rosuvastatina. A relevância clínica desta interação não foi estudada.
Ácido fusídico: estudos de interação com rosuvastatina e ácido fusídico não foram conduzidos. Assim como com outras estatinas, eventos musculares relacionados incluindo rabdomiólise foram relatados na experiência pós-comercialização com a administração concomitante de rosuvastatina e ácido fusídico. Os pacientes devem ser rigorosamente monitorados e a suspensão temporária do tratamento com rosuvastatina pode ser apropriada.
Efeito da rosuvastatina sobre medicamentos coadministrados
Varfarina: a farmacocinética da varfarina não é significativamente afetada após a coadministração com rosuvastatina. Entretanto, como com outros inibidores da HMGCoA redutase, a coadministração de rosuvastatina e varfarina pode resultar em um aumento da Razão Normalizada Internacional (RNI)) em comparação com a varfarina isoladamente. Em pacientes em tratamento com antagonistas da vitamina K, recomenda-se a monitorização da RNI, tanto no início quanto no término do tratamento com rosuvastatina ou após ajuste de dose.
Fenofibratos/derivados do ácido fíbrico: embora nenhuma interação farmacocinética entre rosuvastatina e fenofibrato tinha sido observada, uma interação farmacodinâmica pode ocorrer. A genfibrozila, o fenofibrato e outros ácidos fíbricos, incluindo o ácido nicotínico, podem aum