TIVICAY PD
GLAXOSMITHKLINE
dolutegravir
Anti-retroviral.
Apresentações.
Comprimidos para suspensão de 5 mg em cartuchos com 60 comprimidos. A embalagem acompanha um copo dosador e uma seringa dosadora.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 4 SEMANAS (COM PESO SUPERIOR A 3 KG)
Composição.
Cada comprimido para suspensão de Tivicay PD 5 mg contém: dolutegravir 5 mg (equivalentes a 5,26 mg de dolutegravir sódico) excipientes* q.s.p. 1 comprimido para suspensão
*manitol, celulose microcristalina, povidona, amidoglicolato de sódio, água purificada, celulose microcristalina silicificada, crospovidona, sulfato de cálcio di-hidratado, sucralose, flavorizante creme de morango Permaseal PHS-132963, estearilfumarato de sódio e AquariusTM BP18237 Branco ou Opadry® OY-S-28876 Branco (hipromelose, macrogol e dióxido de titânio).
Informações técnicas.
1. INDICAÇÕES
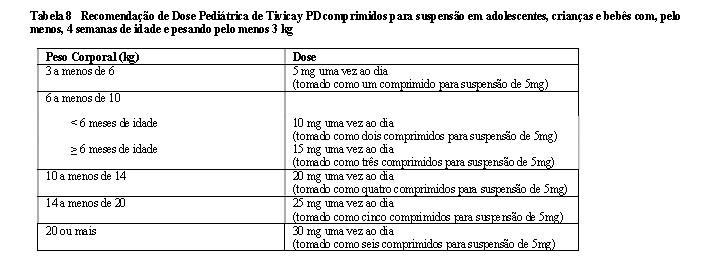
Tivicay PD é indicado para o tratamento da infecção pelo HIV (vírus da imunodeficiência humana) em combinação com outros agentes antirretrovirais em adultos e crianças com idade de, pelo menos, 4 semanas e com peso superior a 3 kg.
2. RESULTADOS DE EFICÁCIA
Indivíduos virgens de tratamento com antirretrovirais
A eficácia do dolutegravir em indivíduos vivendo com HIV e virgens de tratamento baseia-se nas análises dos dados de dois ensaios randomizados, internacionais, duplo-cegos e com controle ativo, 96 semanas do SPRING-2 (ING113086) e SINGLE (ING114467). Isto é suportado por dados de 96 semanas obtidos a partir do FLAMINGO (ING114915), um estudo aberto e controlado por substância ativa e dados adicionais da fase aberta do estudo SINGLE para 144 semanas. A eficácia de dolutegravir em combinação com lamivudina em adultos é suportada por dados do desfecho primário de 48 semanas de dois estudos idênticos de 148 semanas, randomizados, multicêntricos, duplo-cegos e de não-inferioridade GEMINI-1 (204861) e GEMINI-2 (205543). No estudo SPRING-2, 822 adultos vivendo com HIV-1, virgens de terapia antirretroviral (TARV), foram randomizados e receberam no mínimo uma dose de 50 mg de dolutegravir comprimidos revestidos uma vez ao dia ou uma dose de 400 mg de raltegravir duas vezes ao dia, ambos administrados com terapia dupla com ITRN em associação em dose fixa (abacavir/lamivudina [ABC/3TC] ou tenofovir/entricitabina [TDF/FTC]). No início do estudo, a idade mediana dos pacientes era de 36 anos, 14% eram do sexo feminino, 15% eram não brancos, 12% tinham coinfecção por hepatite B e/ou C e 2% eram classe C do CDC; essas características eram semelhantes entre os grupos de tratamento.
No estudo SINGLE, 833 indivíduos foram randomizados e receberam no mínimo uma dose de 50 mg de dolutegravir comprimidos revestidos uma vez ao dia com abacavir-lamivudina em associação em dose fixa (dolutegravir + ABC/3TC) ou de efavirenz-tenofovir-entricitabina em associação em dose fixa (EFV/TDF/FTC). No início do estudo, a idade mediana dos pacientes era de 35 anos, 16% eram do sexo feminino, 32% eram não brancos, 7% tinham coinfecção por hepatite C e 4% eram classe C do CDC; essas características eram semelhantes entre os grupos de tratamento.
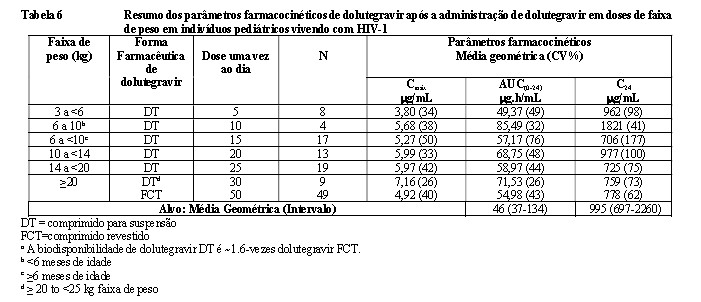
A Tabela 1 apresenta o desfecho primário e outros desfechos na semana 48 (inclusive os desfechos por covariáveis iniciais principais) dos estudos SPRING-2 e SINGLE.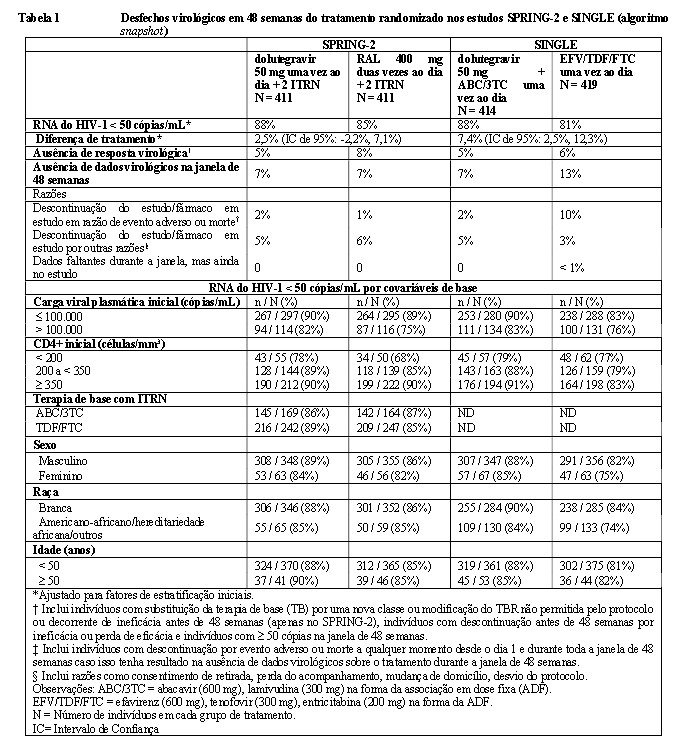
No estudo SPRING-2, durante 96 semanas, a supressão virológica (RNA do HIV-1 < 50 cópias/mL) no grupo do dolutegravir (81%) foi não inferior à ocorrida no grupo do raltegravir (76%). A variação mediana do número de células T CD4+ em relação ao início do estudo foi de 230 células/mm3 no grupo do Tivicay® e no grupo do raltegravir na semana 48. Na semana 96, a contagem foi de 276 células/mm3 no grupo do Tivicay® comparado a 264 células/mm3 no grupo recebendo raltegravir.
Na semana 48 do estudo SINGLE, a supressão virológica (RNA do HIV-1 < 50 cópias/mL) no braço de dolutegravir + ABC/3TC foi de 88%, superior à ocorrida no braço de EFV/TDF/FTC (81%), de acordo com a análise primária (p = 0,003). Na semana 96, a supressão virológica foi mantida, sendo superior no braço dolutegravir + ABC/3TC (80%) em comparação ao braço EFV/TDF/FTC (72%). A diferença do tratamento foi de 8.0 (2.3, 13.8), p = 0,006. A variação média ajustada do número de células T CD4+ em relação ao início do estudo foi de 267 células/mm3 no grupo do dolutegravir + ABC/3TC e de 208 células/mm3 no braço de EFV/TDF/FTC do estudo SINGLE em 48 semanas. A diferença ajustada e IC de 95% foi de 58,9 (33,4, 84,4), p < 0,001 (modelo de medidas repetidas ajustado para os fatores de estratificação iniciais: RNA do HIV-1 inicial e número inicial de células T CD4+, entre outros fatores). Essa análise foi especificada previamente e ajustada para multiplicidade. O tempo mediano até a supressão viral foi de 28 dias no grupo tratado com dolutegravir + ABC/3TC e de 84 dias no braço tratado com EFV/TDF/FTC do estudo SINGLE em 48 semanas (p < 0,0001). Essa análise foi especificada previamente e ajustada para multiplicidade. Na Semana 144 da fase aberta do estudo, a supressão virológica foi mantida e o braço dolutegravir + ABC/3TC (71%) foi superior ao braço EFV/TDF/FTC (63%). A diferença no tratamento foi 8,3% (2,0; 14,6). Tanto no estudo SPRING-2 quanto SINGLE as diferenças de tratamento relativas à supressão virológica (RNA do HIV-1 < 50 cópias/mL) foram comparáveis em todas as características de base (sexo, raça e idade). Durante as 96 semanas do estudo SINGLE e do estudo SPRING-2, não se isolaram mutações de resistência a INI nem resistência à terapia de base nos braços que continham dolutegravir. No estudo SPRING-2, houve falha terapêutica em quatro indivíduos do braço do raltegravir com mutações maiores associadas à ITRN e um indivíduo desenvolveu resistência ao raltegravir; no estudo SINGLE, houve falha terapêutica em seis indivíduos no braço de EFV/TDF/FTC com mutações associadas à resistência a ITRNN e um indivíduo desenvolveu uma mutação maior associada à ITRN.
No estudo FLAMINGO (ING114915), aberto e controlado por substância ativa, 484 adultos vivendo com HIV-1 e virgens de tratamento antirretroviral foram randomizados e tratados com 50mg de dolutegravir comprimidos revestidos uma vez ao dia ou com darunavir/ritonavir (DRV/r) 800mg/100mg uma vez ao dia, ambos administrados com dose fixa combinada de ITRN (ABC/3TC ou TDF/FTC). No início do estudo, a idade média dos pacientes era de 34 anos, 15% eram do sexo feminino, 28% eram não brancos, 10% tinham coinfecção por hepatite B e/ou C e 3% eram classe C do CDC. Estas características foram similares entre os grupos de tratamento. A supressão virológica (HIV-1 RNA < 50 cópias/mL) no grupo tratado com dolutegravir (90%) foi superior a do grupo tratado com DRV/r (83%) em 48 semanas. A diferença ajustada em proporção e IC de 95% foi de 7.1% (0.9, 13.2), p = 0,025. Na Semana 96, a supressão virológica do grupo tratado com dolutegravir (80%) foi superior ao grupo tratado com DRV/r (68%). Não foram observadas mutações emergentes de resistência primária ao tratamento com INI, IP ou ITRN nos indivíduos que receberam dolutegravir ou DRV+RTV.
Demonstrou-se resposta virológica mantida no estudo SPRING-1 (ING112276), no qual 88% dos pacientes tratados com 50 mg de dolutegravir comprimidos revestidos uma vez ao dia (n = 51) alcançaram RNA do HIV-1 < 50 cópias/mL em comparação com 72% dos pacientes no grupo tratado com efavirenz (n = 50) em 96 semanas. Não se isolaram mutações de resistência a INI nem resistência à terapia de base surgida durante o tratamento no grupo tratado com dolutegravir durante 96 semanas.
Nos estudos idênticos GEMINI-1 (204861) e GEMINI-2 (205543) de 148 semanas, randomizados, duplo-cegos, multicêntricos, de nãoinferioridade, 1433 indivíduos adultos vivendo com HIV-1 e virgens de tratamento com antirretrovirais foram randomizados e receberam um regime de tratamento duplo com dolutegravir 50 mg e lamivudina 300 mg uma vez ao dia ou dolutegravir 50 mg uma vez ao dia com dose fixa de TDF/FTC. Os indivíduos foram recrutados com RNA do HIV-1 plasmático de 1000 c/mL a ≤ 500.000 c/mL nos testes de triagem. Na análise dos dados reunidos de todos os pacientes na linha de base, a mediana da idade dos pacientes foi de 33 anos, sendo 15% do sexo feminino, 32% não brancos, 6% tinham coinfecção por hepatite C e 9% eram classe 3 do CDC; estas características foram semelhantes entre os grupos de tratamento.
A supressão virológica (RNA do HIV-1 < 50 cópias/mL) no grupo dolutegravir mais lamivudina [91% (dados reunidos)] não foi inferior ao grupo dolutegravir mais TDF/FTC [93% (dados reunidos)] na semana 48. A diferença ajustada na proporção e IC de 95% foi de -1,7% (-4,4; 1,1). Os resultados da análise de dados reunidos estavam em consonância com os dos estudos individuais, para os quais o desfecho primário foi atingido (diferença na proporção < 50 cópias/mL de RNA do HIV-1 plasmático na semana 48 com base no algoritmo Snapshot para dolutegravir mais lamivudina versus associação em dose fixa de dolutegravir e TDF/FTC). A diferença ajustada foi de -2,6% (IC de 95%: 6,7; 1,5) para GEMINI-1 e -0,7% (IC de 95%: -4,3; 2,9) para o GEMINI-2, com uma margem de não-inferioridade pré-especificada de 10%. Durante as 96 semanas, o grupo com dolutegravir mais 3TC (86% com RNA do HIV-1 < 50 cópias / mL [dados reunidos]) permaneceu não inferior ao grupo de dolutegravir mais TDF/FTC (90% com RNA do HIV-1 < 50 cópias / mL [dados reunidos]). A diferença ajustada em proporções e IC de 95% foi -3,4% (-6,7, 0,0). Os resultados da análise agrupada estavam em consonância com os dos estudos individuais, para os quais o desfecho secundário foi atingido (diferença na proporção < 50 cópias / mL de RNA do HIV-1 plasmático na semana 96 com base no algoritmo Snapshot para dolutegravir mais lamivudina versus associação em dose fixa de dolutegravir e TDF/FTC). As diferenças ajustadas de -4,9 (IC 95%: -9,8, 0,0) para GEMINI-1 e -1,8 (IC 95%: -6,4, 2,7) para GEMINI-2 estavam dentro da margem de não-inferioridade préespecificada de -10%. O aumento médio nas contagens de CD4 + células T foi de 269 células / mm3 no braço DTG + 3TC e 259 células / mm3 no braço DTG + FTC / TDF, na semana 96. Na semana 144 dos estudos GEMINI-1 e GEMINI-2, o grupo de dolutegravir mais 3TC (82% com RNA do HIV-1 < 50 cópias / mL [dados reunidos]) permaneceu não inferior ao grupo de dolutegravir mais TDF/FTC (84% com RNA do HIV-1 < 50 cópias / mL [dados reunidos]). Os resultados da análise agrupada estavam em consonância com os dos estudos individuais, para os quais o desfecho secundário foi atingido (diferença na proporção < 50 cópias / mL de RNA do HIV 1 plasmático na semana 144 com base no algoritmo Snapshot para dolutegravir mais lamivudina versus associação em dose fixa de dolutegravir e TDF/FTC). A diferença ajustada em proporções e IC de 95% foi de 1,8% (5,8, 2,1). As diferenças ajustadas de 3,6 (IC 95%: 9,4, 2,1) para GEMINI-1 e 0,0 (IC 95%: 5,3, 5,3) para GEMINI-2 estavam dentro da margem de não inferioridade pré-especificada de 10%. O aumento médio nas contagens de CD4 + células T foi de 302 células / mm3 no braço DTG + 3TC e 300 células / mm3 no braço DTG + FTC / TDF, na Semana 144. Até a semana 144 semanas dos estudos GEMINI-1 e GEMINI-2, nenhum indivíduo que atendeu aos critérios de retirada virológica confirmados, definidos pelo protocolo (CVW), apresentou integrase emergente ou substituições de resistência de classe de NRTI. Não existem dados disponíveis sobre o uso de dolutegravir mais lamivudina como regime de duas drogas em pacientes pediátricos.
Indivíduos previamente tratados com antirretrovirais (e virgens de tratamento com inibidor da integrase)
No estudo SAILING, duplo cego, multicêntrico, internacional (ING111762), 719 adultos vivendo com HIV-1, previamente tratados com TARV foram randomizados e tratados com 50 mg de dolutegravir comprimidos revestidos uma vez ao dia ou 400 mg de raltegravir duas vezes ao dia, com terapia de base (TB) escolhida pelo investigador constituída de até dois agentes (incluindo no mínimo um medicamento plenamente ativo). No início do estudo, a idade mediana dos pacientes era de 43 anos, 32% eram do sexo feminino, 50% eram não brancos, 16% tinham coinfecção por hepatite B e/ou C e 46% eram classe C do CDC. Todos os indivíduos tinham resistência a no mínimo duas classes de antirretrovirais (TARVs) e 49% tinham resistência a no mínimo três classes de TARVs no início do estudo. A Tabela 2 apresenta os desfechos na semana 48 (inclusive os desfechos por covariáveis iniciais principais) do estudo SAILING.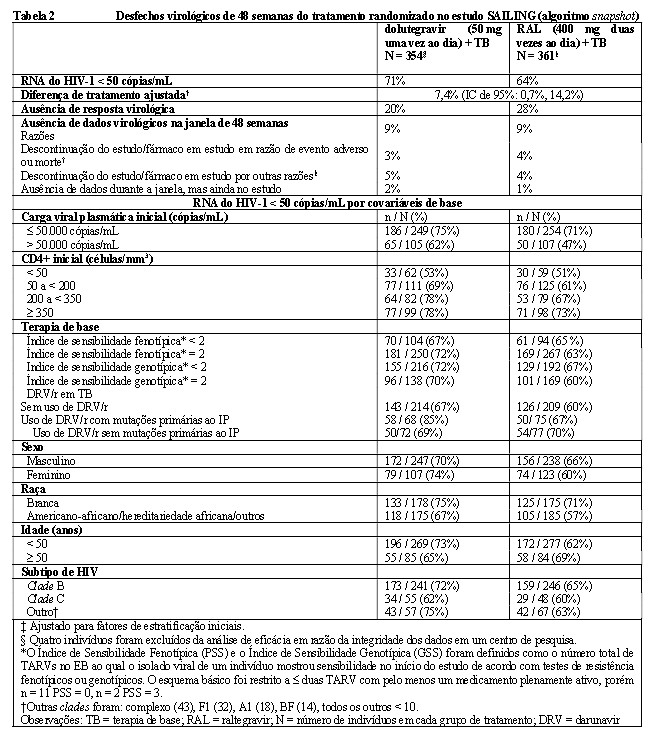
No estudo SAILING, a supressão virológica (RNA do HIV-1 < 50 cópias/mL) no braço de dolutegravir (71%) foi estatisticamente superior ao braço do raltegravir (64%), na semana 48 (p = 0,030). As diferenças de tratamento relativas à supressão virológica (RNA do HIV-1 < 50 cópias/mL) foram comparáveis em todas as características de base de sexo, raça e subtipo de HIV. As variações médias do número de células T CD4+ em relação ao início do estudo foram de 113 células/mm3 na semana 24 e 162 células/mm3 na semana 48, no grupo de dolutegravir e de 106 células/mm3 na semana 24 e 153 células/mm3 na semana 48, no grupo do raltegravir. Estatisticamente, menos pacientes apresentaram falha terapêutica com aparecimento de resistência no gene IN no braço de dolutegravir (4/354, 1%) que no de raltegravir (17/361, 5%) (p = 0,003).
Indivíduos resistentes a inibidores da integrase
No estudo piloto VIKING (ING112961) de coorte, sequencial, de braço único, aberto, multicêntrico, internacional, de fase IIb, foram inscritas duas coortes sequenciais de indivíduos com resistência a várias classes de fármacos, inclusive resistência a inibidores da integrase do HIV, para analisar a atividade antiviral de uma dose de 50 mg de dolutegravir comprimidos revestidos uma vez ao dia (n = 27) em comparação com a dose de 50 mg duas vezes ao dia (n = 24) depois de 10 dias de monoterapia funcional. As respostas foram maiores com a administração duas vezes ao dia (redução de 1,8 log10 em relação ao RNA do HIV inicial) que com a administração uma vez ao dia (redução de 1,5 log10 em relação ao valor inicial, diferença ajustada de 0,3 log10, p = 0,017). As taxas de resposta maiores com a administração duas vezes ao dia foram mantidas com o uso contínuo de dolutegravir e a otimização do esquema básico durante 48 semanas de tratamento (33% versus 71% < 50 cópias/mL, análise ITT-E TLOVR). Observou-se um perfil de segurança comparável entre as doses. Subsequentemente, o estudo VIKING-3 analisou o efeito de dolutegravir comprimidos revestidos, na dose de 50 mg duas vezes ao dia durante sete dias de monoterapia funcional, seguida por terapia de base otimizada e continuação do tratamento com dolutegravir duas vezes ao dia.
No estudo VIKING-3 (ING112574), de braço único, aberto, multicêntrico, adultos vivendo com HIV-1, previamente tratados com ARV, com falha virológica e evidência atual ou passada de resistência ao raltegravir e/ou elvitegravir, foram tratados com 50 mg de dolutegravir duas vezes ao dia com o esquema básico ineficaz em curso durante sete dias, mas com terapia de base otimizada a partir do dia 8. Foram incluídos 183 indivíduos, 133 com resistência a INI detectada por testes de triagem e 50 somente com histórico de evidência de resistência (e sem detecção de resistência nos testes de triagem). No início do estudo, a idade mediana dos pacientes era de 48 anos, 23% eram do sexo feminino, 29% eram não brancos e 20% tinham coinfecção por hepatite B e/ou C. A contagem mediana inicial de CD4+ era de 140 células/mm3, a duração mediana da TARV prévia era de 14 anos e 56% pertenciam à classe C do CDC. Os indivíduos apresentavam resistência a várias classes de TARV no início do estudo: 79% tinham mutações maiores relacionadas a ≥ 2 ITRN, 75% tinham mutações maiores relacionadas a ≥ 1 ITRNN e 71% tinham mutações maiores relacionadas a ≥ 2 IP; 62% tinham vírus não R5. A população com desfecho virológico (DV) excluiu os pacientes que interromperam o tratamento por motivos não relacionados à eficácia e aqueles com desvios maiores no protocolo (dose incorreta de dolutegravir, administração de medicamentos concomitantes proibidos). A população DV é um subconjunto da população ITT-E.
A variação média de RNA do HIV em relação ao nível inicial no dia 8 (desfecho primário) foi de 1,4 log10 (IC de 95% -1,3, -1,5 log10, p < 0,001). A resposta foi associada à via mutacional de INI no início do estudo, como mostra a Tabela 3.
Após a fase de monoterapia, quando possível, os indivíduos tiveram a oportunidade de otimizar a terapia de base. Dos 183 indivíduos que completaram 24 semanas de estudo ou que foram descontinuados antes do ponto de corte (cut-off) dos dados, 126 (69%) alcançaram < 50 cópias/mL de RNA na semana 24 (ITT-E, algoritmo de instantâneo [snapshot]). Indivíduos hospedando vírus com Q148 e outras mutações secundárias associadas a Q148 alcançaram menor resposta na semana 24. O índice de sensibilidade geral (OSS) à terapia base não foi associado à resposta na semana 24.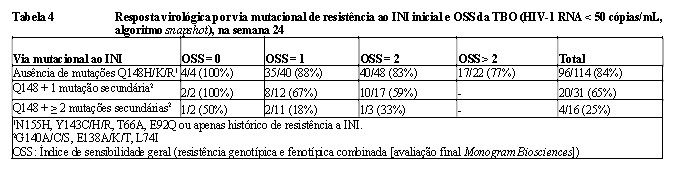
A taxa de resposta na semana 48 foi sustentada por 116/183 (63%) indivíduos com RNA do HIV-1 < 50 cópias/mL (ITT-E, algoritmo Snapshot). A resposta também foi sustentada durante a semana 48 em indivíduos portadores do vírus com Q148 e mutação secundária adicional associada ao Q148. A proporção dos indivíduos com RNA do HIV-1 < 50 cópias/mL na semana 48 foi de 88/113 (78%) para nenhuma mutação Q148, 13/31 (61%) para mutações Q148 + 1 e 4/16 (25%) para mutações secundárias Q148 + ≥2 (população VO, algoritmo Snapshot). A base da pontuação de suscetibilidade global (OOS) não foi associada com a resposta na semana 48.
A supressão virológica (HIV-1 RNA < 50 cópias/mL) foi comparável em todas as características basais (sexo, raça e idade). A variação mediana do número de células T CD4+ em relação ao início do estudo VIKING-3 baseado nos dados observados foi de 61 células/mm3 na semana 24 e 110 células/m³ na semana 48.
No estudo VIKING-4 (ING116529), multicêntrico, duplo-cego e controlado por placebo, 30 adultos vivendo com HIV-1, previamente tratados com TARV, com falha virológica, fazendo uso de um regime contendo inibidor da integrase e com resistência genotípica primária a INI detectada por testes de triagem foram randomizados e receberam uma dose de 50 mg de dolutegravir comprimidos revestidos duas vezes ao dia ou placebo mantendo o esquema básico ineficaz em curso por 7 dias. A partir do dia 8, todos os pacientes receberam abertamente dolutegravir com terapia de base otimizada. A mediana da idade dos pacientes no momento inicial foi de 49 anos, sendo 20% do sexo feminino, 58% não-brancos e 23% tendo coinfecção com hepatite B e/ou C. A mediana de CD4+ inicial foi de 160 células/mm3, a mediana de tempo de tratamento com TARV anterior foi de 13 anos e 63% eram classe C do CDC. No momento inicial, os indivíduos apresentaram resistência a TARVs de múltiplas classes: 80% tinham mutações a ITRN ≥2, 73% a ITRNN ≥1 e 67% a IP ≥2 IPs; 83% tinham vírus não-R5. Dezesseis dos trinta indivíduos (53%) portava o vírus Q148 no momento inicial. A comparação dos desfechos primários no dia 8 mostrou que dolutegravir foi superior ao placebo, com uma diferença média de tratamento ajustada para a variação de RNA do HIV em relação ao nível inicial de -1.2 log10 cópias/mL (IC de 95% -1,5, -0,8 log10, p < 0,001). As respostas obtidas no dia 8 desse estudo placebo-controlado foram consistentes com as observadas no estudo VIKING-3, incluindo as categorias de base de resistência à integrase. Na Semana 48, 12/30 (40%) dos indivíduos tinham RNA HIV-1 < 50 cópias/mL (ITT-E, algoritmo snapshot). Em uma análise combinada dos estudos VIKING-3 e VIKING-4 (n=186, população DV), a proporção de indivíduos com RNA HIV < 50 cópias/mL na Semana 48 foi 123/186 (66%). Para mutações não-Q148, a proporção de indivíduos com RNA HIV < 50 cópias/mL foi 96/126 (76%), para mutações Q148+1 mutação secundária foi de 22/41 (54%) e para Q148 + ≥2 mutações secundárias foi de 5/19 (26%).
Antiretroviral Pregnancy Registry (APR)
O APR recebeu relatórios de mais de 600 exposições a dolutegravir durante gestações que resultaram em nascimentos com vida, em Julho de 2019. Estes consistem em mais de 370 exposições durante o primeiro trimestre e mais de 230 exposições durante o segundo/terceiro trimestre, e incluíram 12 e 9 defeitos congênitos, respectivamente. A prevalência (IC 95%) de defeitos entre os bebês nascidos com vida e expostos ao dolutegravir no primeiro trimestre foi de 3,2% (1,7%,;5,5%) e no segundo/terceiro trimestre, de 3,8% (1,7%; 7,0%). Os dados disponíveis do APR não mostram nenhum aumento significativo no risco de defeitos congênitos maiores com o uso de dolutegravir em comparação com as taxas de base nas duas populações, com base em dois sistemas de vigilância [Programa de Defeitos Congênitos da área Metropolitana de Atlanta (Metropolitan Atlanta Congenital Defects Program) com 2,72 defeitos por 100 nascidos com vida e o Registro de Defeitos Congênitos do Texas (Texas Birth Defects Registry) com 4,17 por 100 nascidos com vida].
Crianças
Um estudo aberto, multicêntrico, de 48 semanas, em andamento, de fase I/II (P1093/ING112578) avaliou os parâmetros farmacocinéticos, a segurança, a tolerabilidade e a eficácia de Tivicay PD em terapias combinadas em bebês, crianças e adolescentes vivendo com HIV-1 e com idades ≥ 4 semanas a < 18 anos, sendo a maioria experimentada a tratamento.
Os resultados de eficácia (Tabela 05) incluem os participantes que receberam as doses recomendadas de comprimidos revestidos ou comprimidos para suspensão.
Referências Bibliográficas:
1. AKIL, B. et al. Dolutegravir versus placebo in subjects harbouring HIV-1 with integrase inhibitor resistance associated substitutions: 48week results from VIKING-4, a randomized study. Antivir Ther, 20(3): 343-8, 2015.
2. CAHN, P. et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet, 382(9893): 700-8, 2013.
3. CASTAGNA, A. et al. Dolutegravir in antiretroviral-experienced patients with raltegravir-and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis, 210(3): 354-62, 2014.
4. CLOTET, B. et al. Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 48 week results from the randomised open-label phase 3b study. Lancet, 383(9936): 2222-31, 2014.
5. ERON, JJ. et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24week results of the VIKING Study. J Infect Dis, 207(5): 740-8, 2013.
6. RAFFI, F. et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet, 381(9868): 735-43, 2013.
7. RAFFI, F. et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis, 13(11): 927-35, 2013.
8. VAVRO, C. et al. Durable efficacy and limited Integrase resistance in subjects receiving Dolutegravir after failing a prior INI regimen: Week 48 results from VIKING3. In: European Meeting on HIV and Hepatitis, 12, 2014, Barcelona.
9. VIANI, RM. et al. Safety, Pharmacokinetics and Efficacy of Dolutegravir in Treatment-experienced HIV-1 Infected Adolescents: Fortyeight-week Results from IMPAACT P1093. Pediatr Infect Dis J, 34(11): 1207-13, 2015.
10. WALMSLEY, SL. et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med, 369(19): 1807-18, 2013.
11. WALMSLEY, S. et al. Brief Report: Dolutegravir Plus Abacavir/Lamivudine for the Treatment of HIV-1 Infection in Antiretroviral Therapy-Naive Patients: Week 96 and Week 144 Results from the SINGLE Randomized Clinical Trial. J Acquir Immune Defic Syndr, 70(5): 515-9, 2015.
12. WIZNIA, A. et al. IMPAACT 1093: Dolutegravir in 6-to 12-year-old HIV-infected children: 48-week results. In: ANNUAL CONFERENCE ON RETROVIRUSES AND OPPORTUNISTIC INFECTIONS, 23, 2016, Boston, USA.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Código ATC
Grupo farmacoterapêutico: Antiviral para uso sistêmico. Antivirais para tratamento de infecções pelo HIV, combinações. Código ATC: J05AJ03
Mecanismo de ação
O dolutegravir inibe a integrase do HIV por ligação ao sítio ativo da integrase e bloqueio da etapa de transferência do filamento na integração do ácido desoxirribonucleico (DNA) do retrovírus, que é essencial para o ciclo de replicação do HIV. Os estudos bioquímicos de transferência de fita utilizando a HIV-1 integrase purificada e substrato de DNA pré-processado resultaram em valores IC50 de 2,7 nM e 12,6 nM. In vitro,
o dolutegravir dissocia-se lentamente do sítio ativo do complexo integrase-DNA de tipo selvagem (t ½ = 71 horas).
Efeitos farmacodinâmicos
Em um ensaio randomizado de determinação da dose, indivíduos vivendo com HIV-1 em monoterapia com dolutegravir apresentaram atividade antiviral rápida e dose-dependente, com declínio médio do RNA do HIV-1 entre o início do estudo e o dia 11 de 1,5, 2,0 e 2,5 log10 com as respectivas doses de 2 mg, 10 mg e 50 mg de dolutegravir uma vez ao dia. Essa resposta antiviral foi mantida durante três a quatro dias depois da última dose no grupo tratado com 50 mg.
Atividade antiviral em cultura celular
A EC50 do DTG foi de 0,51 nM nas células mononucleares do sangue periférico (CMSP) infectadas pela cepa BaL do HIV-1 e de 0,53 nM nas CMSP infectadas pela cepa NL432 do HIV-1. As EC50s das células MT-4 infectadas pela cepa IIIB do HIV-1 e incubadas com dolutegravir durante quatro ou cinco dias foram de 0,71 e 2,1 nM. Em um ensaio de sensibilidade da integrase viral que usou a região codificadora de integrase de 13 isolados da clade B clinicamente diferentes, a potência antiviral do dolutegravir foi semelhante à observada em cepas laboratoriais, com EC50 média de 0,52 nM. Quando avaliada em ensaios de CMSP contra um painel constituído de 24 isolados clínicos do HIV-1 (grupo M [clades A,B, C,D,E, Fe G] egrupo O)etrês isolados clínicos do HIV-2, a média geométrica de EC50 foi de 0,20 nM e os valores de EC50 variaram de 0,02 a 2,14 nM com o HIV-1, enquanto a média geométrica de EC50 foi 0,18 nM e os valores de IC50 variaram de 0,09 a 0,61 nM com isolados do HIV-2.
Atividade antiviral em combinação com outros agentes antivirais
Nenhum fármaco com atividade anti-HIV inerente teve ação antagonista do dolutegravir (as avaliações in vitro foram feitas pela técnica checkerboard em combinação com estavudina, abacavir, efavirenz, nevirapina, lopinavir, amprenavir, enfuvirtida, maraviroque, adefovir e raltegravir). Além disso, antivirais sem atividade anti-HIV inerente (ribavirina) não tiveram efeito aparente sobre a atividade do dolutegravir.
Efeito do soro humano e das proteínas séricas
Estudos in vitro sugeriram uma modificação de 75 vezes da EC50 do dolutegravir na presença de soro humano a 100% (pelo método de extrapolação), e a EC90 ajustada para proteína (PA-IC90) em CMSP foi estimada em 64 ng/mL. A concentração mínima de dolutegravir com uma dose única de 50 mg em indivíduos virgens de tratamento com inibidor da integrase foi de 1,20 mg/mL e, portanto, 19 vezes maior que a PA-EC90 estimada.
Resistência in vitro
Isolamento do HIV-1 de tipo selvagem: não se observaram vírus extremamente resistentes ao dolutegravir durante a passagem de 112 dias da cepa IIIB, com fold change (FC) máximo de 4,1 vezes nas populações de vírus resistentes após a passagem com substituições nas posições de IN conservadas S153Y e S153F. A passagem da cepa NL432 de HIV-1 selvagem na presença de dolutegravir selecionou as substituições E92Q (FC dos vírus da população de passagem = 3,1) e G193E (FC dos vírus da população de passagem = 3,2) no dia 56. A passagem adicional de vírus selvagens dos subtipos B, C e A/G na presença de DTG selecionou R263K, G118R e S153T.
Atividade anti-HIV contra cepas resistentes. Cepas resistentes a inibidores da transcriptase reversa e inibidores da protease: o dolutegravir mostrou potência equivalente contra dois clones mutantes de HIV-1 resistentes a inibidores não nucleosídeos da transcriptase reversa (ITRNN), três clones mutantes resistentes a inibidores nucleosídeos da transcriptase reversa (ITRN) e dois clones mutantes resistentes a inibidores da protease (IP) (um triplo e um sêxtuplo) em comparação com a cepa de tipo selvagem.
Cepas de HIV-1 resistentes a inibidores da integrase. Sessenta HIV-1 mutantes resistentes a inibidores da integrase (28 com uma substituição e 32 com duas ou mais substituições) foram produzidos a partir do vírus NL-432 selvagem por meio de mutagênese sítio-dirigida. O dolutegravir mostrou atividade anti-HIV (sensibilidade) com FC < 5 contra 27 de 28 vírus mutantes resistentes a inibidores da integrase com uma substituição, inclusive T66A/I/K, E92Q/V, Y143C/H/R Q148H/K/R e N155H, enquanto os números de vírus mutantes testados com FC < 5 foram de 17/28 com o raltegravir e 11/21 com o elvitegravir. Além disso, dos 32 vírus mutantes resistentes a inibidores da integrase com duas ou mais substituições, observou-se FC < 5 em 23 de 32 com dolutegravir, em 4 de 32 com raltegravir e em 2 de 25 testados com elvitegravir.
Cepas de HIV-2 resistentes a inibidores da integrase. HIV-2 mutantes sítio-dirigidos foram criados a partir de indivíduos vivendo com HIV2 e tratados com raltegravir que apresentaram falha virológica. Em geral, os FCs do HIV-2 observados foram semelhantes aos FCs do HIV-1 observados com vias mutacionais semelhantes. O FC com dolutegravir foi < 5 contra quatro HIV-2 (S163D, G140A/Q148R, A153G/N155H/S163G e E92Q/T97A/N155H/S163D); o FC com dolutegravir contra E92Q/N155H foi de 8,5 e contra G140S/Q148R foi de 17. O dolutegravir, o raltegravir e o elvitegravir tiveram igual atividade contra o HIV-2 mutante sítio-dirigido com S163D e contra o tipo selvagem; contra os demais HIV-2 mutantes o FC com raltegravir variou de 6,4 a 420 e o FC com elvitegravir variou de 22 a 640.
Isolados clínicos de indivíduos com falha virológica no tratamento com raltegravir. Trinta amostras de isolados clínicos com resistência genotípica e fenotípica ao raltegravir (FC mediano > 81) foram submetidas à avaliação da sensibilidade ao dolutegravir (FC mediano de 1,5) pelo teste PhenoSense da Monogram Biosciences. O FC mediano com dolutegravir de isolados que continham alterações em G140S + Q148H foi de 3,75; em G140S + Q148R foi de 13,3; em T97A + Y143R foi de 1,05 e em N155H foi de 1,37. A sensibilidade ao dolutegravir de 705 isolados resistentes ao raltegravir de pacientes previamente tratados com raltegravir foi analisada pelo teste PhenoSense de Monogram Biosciences. O dolutegravir apresenta FC ≤ 10 contra 93,9% dos 705 isolados clínicos, 16 (9%) de 184 isolados com a mutação de resistência INI Q148 + 1 e 25 (27%) de 92 isolados clínicos com a mutação de resistência INI Q148 + ≥ 2 tiveram aumento maior que 10 vezes.
Resistência in vivo: pacientes virgens de tratamento com inibidores da integrase
Nenhuma mutação de resistência a INI nem resistência à terapia de base com ITRN foi isolada durante o tratamento de pacientes virgens de tratamento com 50 mg de dolutegravir comprimidos revestidos uma vez ao dia, nos estudos SPRING-1, SPRING-2, SINGLE e FLAMINGO. No estudo SAILING com pacientes previamente tratados com antirretroviral (e virgens de tratamento com inibidores da integrase) (n = 354 no braço tratado com dolutegravir), observou-se, na semana 48, substituições a inibidores da integrase surgidas durante o tratamento em quatro de dezessete indivíduos com falha virológica recebendo dolutegravir. Desses quatro casos, dois pacientes apresentaram uma substituição singular de R263K na integrase, com FC máximo de 1,93, um paciente apresentou uma substituição polimórfica V151V/I na integrase, com FC máximo de 0,92 e um paciente apresentou mutações na integrase pré-existentes, caso onde foi considerado que este era experiente a integrase ou que foi infectado, no momento da transmissão, por um vírus resistente a integrase. (ver Estudos Clínicos, no item 2. Resultado de Eficácia).
Resistência in vivo: pacientes resistentes a inibidores da integrase
O estudo VIKING-3 avaliou a administração de dolutegravir (mais terapia de base otimizada) a indivíduos com resistência preexistente a INI. Trinta e seis indivíduos (36/183) apresentaram falha virológica definida por protocolo (PDVF) até a semana 24. Desses, 32 tiveram os dados iniciais e de resistência com PDVF pareados para análise e 17/32 (53%) apresentaram mutações surgidas durante o tratamento. As mutações surgidas durante o tratamento ou as combinações de mutações observadas foram L74L/M (n =1), E92Q (n = 2), T97A (n = 9 E138K/A/T (n = 8), G140S (n = 2), Y143H (n = 1), S147G (n = 1), Q148H/K/R (n = 4), N155H (n = 1) e E157E/Q (n=1). Quatorze dos 17 indivíduos que tinham vírus com mutações surgidas durante o tratamento abrigavam vírus com mutação na via Q148 presentes no início do estudo ou previamente. Cinco novos indivíduos experimentaram PDVF entre as semanas 24 e 48, e 2 desses cinco indivíduos tiveram mutações associadas ao tratamento. As mutações associadas ao tratamento ou a mistura de mutações observadas foram L74I (n=1), N155H (n=2). O Estudo VIKING-4 avaliou dolutegravir (mais terapia de base otimizada) em indivíduos com resistência genotípica primária a INIs nos testes de triagem em 30 indivíduos. As mutações emergentes ao tratamento observadas foram consistentes com aquelas observadas no estudo VIKING-3.
Efeitos no eletrocardiograma
Em um ensaio cruzado (cross-over), randomizado e controlado por placebo, administrou-se a 42 indivíduos saudáveis doses orais únicas de placebo, DTG em suspensão de 250 mg (exposições correspondentes a aproximadamente o triplo da dose de 50 mg uma vez ao dia em estado de equilíbrio) e moxifloxacino (400 mg, controle ativo) em sequência aleatória. O dolutegravir não prolongou o intervalo QTc por 24 horas após a dose. Após ajuste em função do valor inicial e do placebo, a alteração média máxima do QTc com base no método de correção de Fridericia (QTcF) foi de 1,99 ms (limite superior do IC de 95% unilateral: 4,53 ms).
Efeitos na função renal
O efeito de dolutegravir sobre o clearance de creatinina sérica (ClCr), a taxa de filtração glomerular (TFG) com uso de ioexol como marcador e o fluxo plasmático renal efetivo (FPRE) com uso de para-aminoipurato (PAH) como marcador foi avaliado em um estudo aberto, randomizado, de três braços, paralelo, controlado por placebo em 37 indivíduos saudáveis tratados com 50 mg de dolutegravir comprimidos revestidos uma vez ao dia (n = 12), 50 mg duas vezes ao dia (n = 13) ou placebo uma vez ao dia (n = 12) durante 14 dias. Observou-se pequena diminuição do ClCr com o dolutegravir na primeira semana de tratamento, compatível com o observado em estudos clínicos. O dolutegravir, nas duas doses, não teve efeito relevante sobre a TFG nem sobre o FPRE. Esses dados respaldam estudos in vitro sugestivos de que os pequenos aumentos da creatinina observados em estudos clínicos são causados por inibição não patológica do transportador de cátions orgânicos tipo 2 (OCT2) nos túbulos renais proximais, que medeia a secreção tubular de creatinina.
Propriedades Farmacocinéticas
A farmacocinética do dolutegravir é semelhante em indivíduos saudáveis e vivendo com HIV. A variabilidade farmacocinética (PK) do dolutegravir é de baixa a moderada. Em estudos de fase I em indivíduos saudáveis, o coeficiente de variação interindividual (CVb%) para AUC e Cmáx variou de ~20% a 40% e a Ct variou de 30% a 65% entre os estudos. A variabilidade PK interindividual do DTG foi maior em indivíduos vivendo com HIV que em indivíduos saudáveis. A variabilidade intraindividual (CVw%) é menor que a variabilidade interindividual. Os comprimidos revestidos e os comprimidos para suspensão não possuem a mesma biodisponibilidade. A biodisponibilidade relativa dos comprimidos para suspensão é aproximadamente 1,6 vezes maior em comparação aos comprimidos revestidos. Assim, uma dose de 30 mg de Tivicay PD administrada como seis comprimidos para suspensão de 5 mg terá exposição semelhante a uma dose de 50 mg de dolutegravir administrada como comprimido revestido. Do mesmo modo, uma dose de 25 mg de Tivicay PD administrada como cinco comprimidos para suspensão de 5 mg, proporcionará uma exposição comparável a uma dose de 40 mg de dolutegravir administrada como quatro comprimidos revestidos de 10 mg.
Absorção
O dolutegravir é absorvido rapidamente após a administração oral, com Tmáx mediano uma a três horas após a administração na forma de comprimido. A linearidade da farmacocinética do dolutegravir depende da dose e da formulação. Em geral, após a administração oral de formulações em comprimido o dolutegravir apresentou farmacocinética não linear com aumentos da exposição plasmática menores que os proporcionais à dose entre 2 e 100 mg; entretanto, o aumento da exposição ao dolutegravir parece proporcional à dose na faixa de 25 a 50 mg. Tivicay PD pode ser administrado acompanhado ou não de alimentos. Os alimentos aumentaram a extensão e reduziram a taxa de absorção do dolutegravir. A biodisponibilidade do dolutegravir depende do conteúdo da refeição: refeições com quantidade baixa, moderada e alta de gorduras aumentaram a AUC(0-¥) do dolutegravir de 33%, 41% e 66%, aumentaram Cmáx de 46%, 52% e 67% e prolongaram Tmáx de 2 horas em jejum para 3, 4 e 5 horas, respectivamente. Esses aumentos não têm importância clínica. A biodisponibilidade absoluta do dolutegravir não foi determinada.
Distribuição
De acordo com dados in vitro, o dolutegravir liga-se intensamente (cerca de 99,3%) às proteínas plasmáticas humanas. Estima-se que o volume aparente de distribuição (após administração oral da formulação em suspensão, Vd/F) seja de 12,5 L. A ligação do dolutegravir às proteínas plasmáticas foi independente da concentração. As razões de concentração de radioatividade relacionada ao fármaco no sangue total e no plasma variaram, em média, entre 0,441 e 0,535, indicando associação mínima da radioatividade com os componentes celulares do sangue. Estima-se que a fração livre de DTG no plasma seja d