TEVAOXALI
TEVA
oxaliplatina
Antineoplásico.
Apresentações.
Solução Injetável de 5 mg/ml.
Embalagem contendo 1 frasco-ampola de solução injetável com 10 mL - 50 mg.
Embalagem contendo 1 frasco-ampola de solução injetável com 20 mL - 100 mg.
Embalagem contendo 1 frasco-ampola de solução injetável com 28 mL - 140 mg.
Embalagem contendo 1 frasco-ampola de solução injetável com 40 mL - 200 mg.
USO INTRAVENOSO
USO ADULTO
Composição.
Cada mL da solução contém: oxaliplatina 5,0 mg. Excipientes q.s.p. 1 mL. (lactose monoidratada e água para injeção).
Indicações.
Tratamento do câncer colorretal metastático em associação às fluoropirimidinas. TEVAOXALI® (oxaliplatina) em combinação com 5-FU/FA e bevacizumabe é indicado para tratamento de primeira linha do câncer colorretal metastático.
TEVAOXALI® está indicado em combinação com fluoruracila e ácido folínico (leucovorin) (5-FU/FA) para o tratamento adjuvante de câncer colorretal em pacientes que sofreram ressecção completa do tumor primário, reduzindo o risco de recidiva tumoral.
Não fica indicado para os pacientes em estágio II sem características de alto risco.
Resultados de eficácia.
Em pacientes com câncer colorretal metastático, a eficácia de oxaliplatina (85 mg/m2 repetida a cada duas semanas) em associação com 5-fluoruracila/ácido folínico foi reportada em três estudos clínicos:
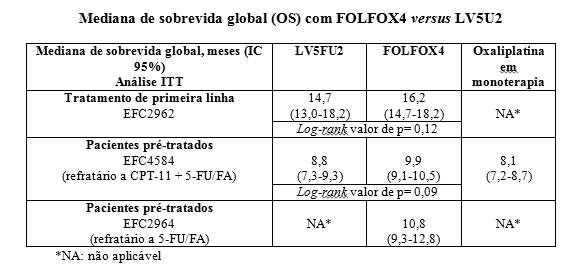
- no tratamento de primeira linha, os 2 braços comparativos do estudo de fase III EFC2962 randomizou 420 pacientes para receber 5-FU/FA isolado (LV5FU2, N=210) ou para receber a associação de oxaliplatina com 5-FU/FA (FOLFOX4, N=210);
- em pacientes pré-tratados, no estudo de fase III EFC4584, comparativo, de 3 braços, randomizou 821 pacientes refratários ao tratamento com irinotecano (CPT-11) + combinação de 5-FU/FA, para receber 5-FU/FA isolado (LV5FU2, N=275), oxaliplatina como agente único (n=275) ou associação de oxaliplatina com 5-FU/FA (FOLFOX4, N=271);
- por último, no estudo EFC2964, não-controlado, de fase II, incluiu pacientes refratários a 5-FU/FA isolado, que foram tratados com a associação de oxaliplatina e 5-FU/FA (FOLFOX4, N=57).
Os dois estudos clínicos randomizados, EFC2962, no tratamento de primeira linha e EFC4584, em pacientes pré-tratados, demonstraram uma taxa de resposta e um aumento na sobrevida livre de progressão (PFS)/tempo para progressão (TTP) significativamente mais elevadas quando comparado com o tratamento com 5-FU/FA isolado.

Em pacientes pré-tratados (EFC4584), que foram sintomáticos no estado basal, uma proporção mais elevada daqueles pacientes tratados com oxaliplatina e 5-FU/FA apresentaram uma melhora significativa destes sintomas relacionados à doença comparados com aqueles tratados com 5-FU/FA isolado (27,7% vs 14,6% p=0,0033).
Em pacientes não pré-tratados (EFC2962), não foi encontrada diferença estatisticamente significativa entre os grupos de tratamento para qualquer dimensão de qualidade de vida.
Entretanto, os números de qualidade de vida foram geralmente melhores no braço controle quanto à avaliação do status de saúde e dor global e piores no braço da oxaliplatina com relação à náusea/vômito.
No cenário adjuvante, o estudo MOSAIC comparativo, de fase III (EFC3313), randomizou 2246 pacientes (899 estágio II/Duke's B2 e 1347 estágio III/ Duke's C) após ressecção completa do tumor primário do câncer de cólon para receber tanto 5-FU/FA isolado (LV5FU2, N=1123) (B2/C = 448/675) quanto à associação de oxaliplatina com 5-FU/FA (FOLFOX4, N=1123 (B2/C) = 451/672).
O estudo demonstrou uma vantagem significativa global na sobrevida livre de doença em 3 anos para a associação de oxaliplatina com 5-FU/FA (FOLFOX4) sobre 5-FU/FA isolado (LV5FU2).
Sobrevida global (análise ITT):
No momento da análise de sobrevida livre de doença de até 3 anos, a qual foi o endpoint primário do estudo MOSAIC, 85,1% dos pacientes ainda estavam vivos no braço FOLFOX4 versus 83,8% no braço LV5FU. Isto explica uma redução global no risco de mortalidade de 10% a favor de FOLFOX4 não alcançar significância estatística (hazard ratio = 0,90).
Os valores foram de 92,2% versus 92,4% na subpopulação com estágio II (Duke's B2) (hazard ratio = 1,01) e de 80,4% versus 78,1% na subpopulação com estágio III (Duke's C) (hazard ratio = 0,87) para FOLFOX4 e LV5FU2, respectivamente.
Estudos europeus comparando tanto 5-FU/LV como oxaliplatina como agente único, mostraram que a combinação de oxaliplatina com 5-FU/LV apresentaram uma taxa de resposta tumoral significativamente melhorada, maior estabilização da doença, tempo mais longo para progressão do tumor e melhoria nos sintomas relacionados ao tumor.
NCI / Estudo N9741: Comparado ao regime IFL (irinotecano/5FU/FA), o regime de FOLFOX (oxaliplatina/5FU/FA) resultou em taxa de resposta tumoral significativamente aumentada, aumento do tempo para progressão do tumor, e acima de tudo, uma melhora na sobrevida global com um perfil de toxicidade mais favorável.
Câncer colorretal metastático (oxaliplatina/5-FU/FA/bevacizumabe):
A eficácia de oxaliplatina combinada com 5-FU/FA (FOLFOX) e bevacizumabe foi avaliada em 2 estudos clínicos, como quimioterapia de primeira linha (estudo TREE) e quimioterapia de segunda linha (estudo ECOG), em pacientes com câncer colorretal metastático.
O estudo TREE, um estudo randomizado, não comparativo de fase III, avaliou a combinação de FOLFOX/bevacizumabe (utilizando a dose padrão de bevacizumabe de 5 mg/kg de peso corpóreo, a cada duas semanas) (71 pacientes) e o regime FOLFOX isolado (49 pacientes). Na população de pacientes as-treated (pacientes que receberam o tratamento alocado na randomização), a taxa de resposta objetiva foi 52,1% e 40,8%, respectivamente, o tempo para progressão mediano (TTP, definido como sobrevida livre de progressão, PFS) foi 9,9 meses e 8,7 meses, respectivamente e a sobrevida global mediana (OS) foi 26,0 e 19,2 meses, respectivamente.
Os resultados do estudo NO16966 foram avaliados após seguimento mediano de 27,6 meses. A análise principal consistiu na comparação dos grupos tratados com bevacizumabe (n=699), em relação aos grupos tratados com placebo (n=701). Os resultados mostraram que o endpoint primário do estudo foi alcançado. Houve aumento estatisticamente significativo de 20,5% de sobrevida livre de progressão, quando a quimioterapia baseada em oxaliplatina foi combinada ao bevacizumabe (hazard ratio=0,83; intervalo de confiança de 97,5%=0,72 a 0,95; p=0,0023). A duração mediana da sobrevida livre de progressão aumentou de 8,0 meses quando a quimioterapia baseada em oxaliplatina foi combinada ao placebo, para 9,4 meses quando esse regime terapêutico foi combinado ao bevacizumabe.
O estudo ECOG 3200, um estudo randomizado, comparativo, de fase III, demonstrou na população de pacientes randomizados uma melhora significativa na taxa de resposta objetiva (22,2% vs 8,6%), sobrevida livre de progressão mediana (PFS, 7,5 vs 4,5 meses), sobrevida global mediana (OS, 13,0 vs 10,8 meses), com a combinação de FOLFOX/bevacizumabe (bevacizumabe na dose de 10 mg/kg de peso corpóreo, a cada duas semanas) (293 pacientes) comparado com o regime FOLFOX (292 pacientes).
1. Giacchetti S. et al.. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol, Jan 2000, 18 (1), 136-147.
2. De Gramont A. et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol., Aug 2000, 18 (16), 2938-2947. (EFC2962).
3. André T et al. Multicenter Phase II Study of Bimonthly High-Dose Leucovorin, Fluorouracil Infusion, and Oxaliplatin for Metastatic Colorectal Cancer Resistant to the Same Leucovorin and Fulorouracil Regimen. J. Clin. Oncol., 17:3560-3568, 1999. (EFC2964).
4. Goldberg et al. N9741: FOLFOX4 oxaliplatin (Oxal)/ 5-fluorouracil (5-FU)/ leucovorin (LV) or reduced dose R-IFL (CPT - 11 + 5-FU/LV) in advanced colorectal cancer (CRC): Final efficacy data from an intergroup study. Annual Meeting Proceedings American Society of Clinical Oncology, June 5-8, 2004, 275.
5. Andre, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004 Jun 3; 350(23): 2343-51.
6. André T, et al. Improved Overall Survival With Oxaliplatin, Fluorouracil, and Leucovorin As Adjuvant Treatment in Stage II or III Colon Cancer in the MOSAIC Trial. J Clin Oncol. 2009 May.
7. Extra JM, et al. Pharmacokinetics and safety profile of oxaliplatin. Semin Oncol. 1998, Apr; 25 (2 Suppl 5): 13 - 22.
8. Rothenberg ML et al. Superiority of oxaliplatin and fluorouracil-leucovorin compared with either therapy alone in patients with progressive colorectal cancer after irinotecan and fluorouracilleucovorin: Interim results of a phase III trial. J Clin Oncol, 21: 2059-2069, 2003. (EFC4584).
9. Bécouarn Y, et al. Phase II trial of oxaliplatin as first-line chemotherapy in metastatic colorectal cancer patients. Digestive Group of French Federation of Cancer Centers. J Clin Oncol. 1998 Aug; 16 (8): 2739 - 44.
10. Raymond E, et al. Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol. 1998 Oct; 9 (10): 1053 - 71.
11. Hochster H. S. et al. Safety and efficacy of oxaliplatin/fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer (mCRC): Final analysis of the TREE-Study. J. Clin. Oncology, 2006 ASCO Annual Meeting Proceedings Part I. Vol 24, No 18S (June 20 Supplement): 3510.
12. Saltz LB, Clarke S, Diaz-Rubio E, et al: Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26:2013-9, 2008.
Caract. farmacológicas.
Propriedades farmacodinâmicas
Agente citotóxico.
A oxaliplatina é um agente antineoplásico, pertencente a uma nova classe de compostos de platina, onde o átomo de platina é complexado com 1,2- diaminociclohexano ("DACH") e um grupo oxalato. A oxaliplatina é o único enantiômero, cis-[(1R,2R)-1,2-ciclohexanediamina-N,N'] oxalato (2-)-O,O'] platina.
A oxaliplatina exibe um amplo espectro da atividade citotóxica in vitro e atividade antitumor in vivo em vários sistemas de modelos tumorais, incluindo modelos de câncer colorretal nos seres humanos.
A oxaliplatina também demonstra atividades in vitro e in vivo em várias linhagens celulares resistentes à cisplatina.
Foi demonstrada atividade citotóxica sinérgica com 5-fluoruracila in vitro e in vivo.
Estudos do mecanismo de ação, embora não completamente elucidados, demonstram que os derivados hidratados resultantes da biotransformação da oxaliplatina interagem com DNA para formar ligações cruzadas na mesma fita e na fita complementar, resultando numa interrupção da síntese do DNA, levando às atividades citotóxicas e antitumorais.
Propriedades farmacocinéticas
A farmacocinética dos compostos ativos individuais não foi determinada. A farmacocinética da platina ultrafiltrável, representando uma mistura de todas as espécies de platina não-conjugada, ativa e inativa, seguida de duas horas de infusão de oxaliplatina a 130 mg/m2 a cada 3 semanas por 1 a 5 ciclos e oxaliplatina a 85 mg/m2 a cada 2 semanas por 1 a 3 ciclos, conforme descrito abaixo: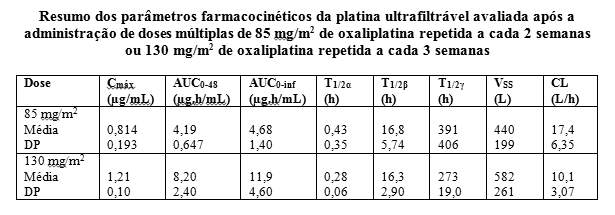
Valores médios de AUC0-48 e Cmáx calculados no ciclo 3 (85mg/m2) ou ciclo 5 (130 mg/m2)
Valores médios de AUC, Vss, CL e CLR0-48 calculados no ciclo 1
Valores de Cfinal, Cmáx, AUC, AUC0-48, Vss e CL calculados utilizando análise não-compartimental T1/2a , T1/2b eT1/2c: calculados utilizando análise compartimental (ciclos 1-3 associados)
Ao final de uma infusão de 2 horas, 15% da platina administrada é encontrada na circulação sistêmica e o restante, 85%, é rapidamente distribuído nos tecidos ou excretados na urina. A ligação irreversível às hemácias e plasma resulta na meia-vida destas matrizes que estão próximos ao movimento de regeneração natural das hemácias e da albumina sérica. Não foi observado acúmulo de platina no plasma ultrafiltrável após a infusão de 85 mg/m2 a cada 2 semanas ou 130 mg/m2 a cada 3 semanas e o estado de equilíbrio foi alcançado durante o ciclo 1 desta matriz. A variabilidade inter e intra-individuais é geralmente baixa.
In vitro, os metabólitos resultam a partir da degradação não-enzimática e não há evidência de metabolismo mediado pelo citocromo P450 do anel diaminociclohexano (DACH).
A oxaliplatina sofre biotransformação extensiva e o fármaco intacto não foi detectável no plasma ultrafiltrável no final de 2 horas de infusão. Vários metabólitos citotóxicos, incluindo as formas monocloro-, dicloro- e diaquo-DACH de espécies de platina foram identificadas na circulação sistêmica junto com um número de conjugados inativos nos pontos de tempo posteriores.
A platina é principalmente excretada na urina e seu clearance predomina durante 48 horas após a administração.
No 5° dia, aproximadamente 54% da dose total foi recuperada na urina e menos que 3% nas fezes. Foi observada uma redução significativa no clearance, de 17,6 ± 2,18 L/h para 9,95 ± 1,91 L/h nos pacientes com insuficiência renal junto com uma redução estatisticamente significativa no volume de distribuição de 330 ± 40,9 para 241 ± 36,1 L. Não foi avaliado o efeito da insuficiência renal severa no clearance da platina.
Pacientes com insuficiência renal
A disposição da oxaliplatina foi estudada em pacientes com variados níveis de função renal. A eliminação da oxaliplatina está significativamente correlacionada com o clearance de creatinina. O clearance total corpóreo da platina plasmática ultrafiltrável foi reduzido em pacientes com insuficiência renal, em 34% para insuficiência renal leve (CLcr = 50 a 80 mL/min), 57% para insuficiência renal moderada (CLcr = 30 a 49 mL/min) e 79% para insuficiência renal severa (CLcr < 30 mL/min), em comparação a pacientes com função renal normal (CLcr > 80 mL/min). Houve uma tendência de meias-vidas beta e gama aumentadas de platina plasmática ultrafiltrável com grau aumentado de insuficiência renal e principalmente no grupo com insuficiência renal severa.
Entretanto, os resultados não foram conclusivos devido à ampla variabilidade interpacientes e o pequeno número (4) de pacientes com insuficiência renal severa. A excreção urinária de platina e o clearance renal de platina plasmática ultrafiltrável também diminuíram com insuficiência renal (vide Posologia e Modo de usar e Advertência e Precauções).
Dados de segurança pré-clínica
Os órgãos alvos identificados nas espécies utilizadas durante os estudos pré-clínicos (camundongos, ratos, cães e/ou macacos) após doses únicas e repetidas, abrangeram medula óssea, sistema gastrintestinal, rins, testículos, sistema nervoso e coração. As toxicidades nos órgãos alvos observados em animais foram semelhantes àqueles observados com outros agentes de platina e com outras drogas citotóxicas interagindo com DNA e utilizados para tratar cânceres nos seres humanos, com a exceção dos efeitos provocados no coração. Os efeitos no coração, incluindo anormalidades eletrofisiológicas com fibrilação ventricular letal, foram somente observados nos cães.
A toxicidade cardíaca foi considerada como sendo específica para os cães, não apenas porque esta toxicidade foi somente observada nestes animais, mas também porque as doses semelhantes às doses letais nos cães (150 mg/m2) foram bem toleradas nos seres humanos. Os estudos pré-clínicos realizados nos sensores dos neurônios no rato sugeriram que os sintomas neurossensoriais agudos causados pela oxaliplatina poderiam estar ligados a uma interação com canais de sódio dependente de voltagem. A oxaliplatina é mutagênica e clastogênica nas células de mamíferos e foi demonstrada toxicidade embrio-fetal no rato. Embora os estudos referentes ao potencial carcinogênico não tenham sido realizados, a oxaliplatina é considerada como sendo provavelmente carcinogênica.
Contraindicações.
O uso de TEVAOXALI® (oxaliplatina) é contraindicado nos pacientes:
- que estejam amamentando;
- com histórico de hipersensibilidade à oxaliplatina e a outros derivados de platina;
- com mielossupressão (neutrófilos < 2 x 109/L e/ou contagem de plaquetas < 100 x 109/L) antes do primeiro ciclo de tratamento;
- neuropatia sensorial periférica com insuficiência funcional antes do primeiro ciclo de tratamento.
Este medicamento é contraindicado para uso por pacientes pediátricos.
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Advertências e precauções.
TEVAOXALI® (oxaliplatina) somente deve ser utilizado em unidades especializadas na administração de medicamentos oncológicos e deve ser administrado sob a supervisão de um médico capacitado, com experiência no uso de quimioterapia antineoplásica.
Devido à informação limitada de segurança em pacientes com insuficiência renal severa, a administração deve ser considerada após uma avaliação apropriada do risco/benefício para o paciente. Neste caso, a função renal deve ser rigorosamente monitorada e a dose inicial recomendada de oxaliplatina é 65 mg/m2 (vide Advertências e Precauções - Pacientes com insuficiência renal).
Os pacientes com histórico de reações alérgicas a produtos contendo platina devem ser monitorados quanto aos sintomas alérgicos. Reações alérgicas podem ocorrer durante qualquer ciclo. No caso de ocorrer reações do tipo anafilactoides em decorrência do TEVAOXALI®, deve-se interromper a infusão imediatamente e implementar tratamento sintomático apropriado. A reintrodução de TEVAOXALI® nestes pacientes é contraindicada.
No caso de extravasamento de TEVAOXALI®, a infusão deve ser interrompida imediatamente e deve ser implementado tratamento sintomático local padrão. Evite o uso de compressas frias em caso de extravasamento de TEVAOXALI®.
A neurotoxicidade sensorial periférica de TEVAOXALI® deve ser cuidadosamente monitorada, especialmente se administrada concomitantemente com outros medicamentos com toxicidade neurológica específica. Uma avaliação neurológica deve ser realizada antes de cada administração e depois periodicamente. No caso de ocorrer sintomas neurológicos (parestesia, disestesia), deve ser realizada a seguinte recomendação de ajuste na dose de TEVAOXALI®, baseado na duração e gravidade destes sintomas:
- se os sintomas persistirem por mais de 7 dias e forem desagradáveis, ou se a parestesia sem insuficiência funcional persistir até o próximo ciclo, a dose subsequente de TEVAOXALI® deve ser reduzida em 25%;
- se a parestesia com insuficiência funcional persistir até o próximo ciclo, o tratamento com TEVAOXALI® deve ser interrompido;
- se os sintomas melhorarem após a interrupção do tratamento com TEVAOXALI®, a re-introdução do tratamento pode ser considerada.
Para pacientes que desenvolvem disestesia faringolaríngea aguda (vide Reações Adversas), durante ou algumas horas após uma infusão de duas horas, a próxima infusão com TEVAOXALI® deve ser administrada durante um período de seis horas. Para prevenir disestesia, instrua o paciente a evitar exposição ao frio e a ingestão de alimentos e bebidas geladas ou frias durante ou algumas horas após a administração de TEVAOXALI®.
Sinais e sintomas de Síndrome de Leucoencefalopatia Posterior Reversível (RPLS, também conhecida como Síndrome de Encefalopatia Posterior Reversível - PRES) podem ser dor de cabeça, funcionamento mental alterado, convulsões, visão anormal desde turva até cegueira, associados ou não com hipertensão (vide Reações Adversas). O diagnóstico da Síndrome de Leucoenfalopatia Posterior Reversível é embasado mediante confirmação imaginológica do cérebro.
A toxicidade gastrintestinal, que se manifesta como náuseas e vômitos, permite uma terapia profilática e/ou terapêutica antiemética (vide Reações Adversas). A desidratação, íleo paralítico, obstrução intestinal, hipocalemia, acidose metabólica e até distúrbios renais podem estar associadas com diarreia/êmese severa, particularmente quando TEVAOXALI® é utilizado em associação com 5-fluoruracila (5-FU).
Se ocorrer toxicidade hematológica (evidenciados por valores de contagem sanguínea no estado basal, por exemplo: neutrófilos < 1,5 x 109/L ou plaquetas < 75 x 109/L) após um ciclo de tratamento, ou se mielossupressão estiver presente antes do início da terapia (1° ciclo), a administração do próximo ciclo ou do primeiro ciclo de tratamento deve ser adiada até que a contagem sanguínea retorne a níveis aceitáveis. Um hemograma completo com contagem diferencial de glóbulos brancos deve ser realizado antes de iniciar o tratamento e antes de cada ciclo subsequente.
Os pacientes devem ser adequadamente informados quanto ao risco de diarreia/êmese e neutropenia após administração concomitante de TEVAOXALI® e 5-fluoruracila (5-FU), de modo que contatem imediatamente seu médico para uma conduta apropriada.
Para administração concomitante de TEVAOXALI® e 5-fluoruracila (com ou sem ácido folínico), os ajustes de dose usuais para as toxicidades associadas ao 5-fluoruracila devem ser aplicados.
Se ocorrer diarreia severa/com risco de vida, neutropenia severa (neutrófilos < 1,0 x 109/L) e trombocitopenia severa (plaquetas < 50 x 109/L), o tratamento com TEVAOXALI® deve ser descontinuado até a melhora ou a recuperação, e a dose de TEVAOXALI® deve ser reduzida em 25% nos ciclos subsequentes, além de quaisquer reduções necessárias na dose do 5-fluoruracila.
Caso ocorram sintomas respiratórios inexplicados, tais como: tosse não produtiva, dispnéia, estertores crepitantes ou infiltrados pulmonares radiológicos, o tratamento com TEVAOXALI® deve ser interrompido até que as investigações pulmonares tenham eliminado a possibilidade de doença pulmonar intersticial (vide Reações Adversas).
No caso dos resultados de testes de função hepática anormais ou hipertensão portal que não resulte evidentemente de metástases hepática, casos muito raros de distúrbios vasculares hepáticos induzidos pelo fármaco devem ser considerados.
Para os detalhes de ajuste de dose de bevacizumabe, consulte as informações contidas na bula deste produto.
Incompatibilidades
- NÃO misture com qualquer outro produto na mesma bolsa de infusão ou não administre simultaneamente pela mesma linha de infusão.
- NÃO utilize em associação com soluções ou produtos alcalinos, em particular 5-fluoruracila (5-FU), soluções básicas, preparações de ácido folínico (FA) contendo trometamol como excipiente e sais de trometamol de outras substâncias ativas. Soluções ou produtos alcalinos afetarão desfavoravelmente a estabilidade da oxaliplatina.
- NÃO use agulhas ou equipamentos contendo partes de alumínio que podem entrar em contato com a solução. O alumínio pode degradar combinações de platina.
- NÃO use solução de cloreto de sódio ou outra solução contendo cloreto para diluir oxaliplatina.
Pacientes pediátricos
A oxaliplatina como agente único foi avaliada em 2 estudos de fase I (69 pacientes) e 2 estudos de fase II (166 pacientes). Um total de 235 pacientes pediátricos (7 meses - 22 anos de idade) com tumores sólidos foram tratados.
Num estudo de fase I/II, oxaliplatina foi administrada por infusão IV durante 2 horas nos dias 1, 8 e 15 a cada 4 semanas (ciclo 1), por um máximo de 6 ciclos, em 43 pacientes com tumores sólidos malignos refratários ou recaídos, principalmente neuroblastoma e osteossarcoma. Vinte e oito (28) pacientes pediátricos foram tratados no estudo de fase I com 6 níveis de dosagem iniciando a 40 mg/m2 e até 110 mg/m2. A toxicidade da dose-limitante (TDL) foi neuropatia sensorial periférica observada em 2
pacientes dos 3 pacientes tratados com 110 mg/m2. Deste modo, a dose recomendada (DR) foi estabelecida como 90 mg/m2 administrada por via IV nos dias 1, 8 e 15 a cada 4 semanas. Quinze (15) pacientes foram tratados com a dose recomendada de 90 mg/m2 por via IV obtidos a partir de um estudo de fase I e os principais eventos adversos observados foram: parestesia (60%, G3/4: 6,7%), febre (40%, G3/4: 6,7%) e trombocitopenia (40%, G3/4: 26,7%).
Num segundo estudo de fase I, oxaliplatina foi administrada em 26 pacientes pediátricos por infusão IV durante 2 horas no dia 1 a cada 3 semanas (ciclo 1) em 5 níveis de dosagem iniciando a 100 mg/m2 e até 160 mg/m2, por um máximo de 6 ciclos. No último nível de dosagem, oxaliplatina 85 mg/m2 foi administrada no dia 1 a cada 2 semanas, por um máximo de 9 doses. Os pacientes apresentavam tumores sólidos metastáticos ou irressecáveis principalmente neuroblastoma e ganglioneuroblastoma, para o qual o tratamento padrão não existe ou não é mais eficaz. A TDL foi neuropatia sensorial periférica observada em 2 pacientes tratados com 160 mg/m2 de oxaliplatina. A DR foi 130 mg/m2 a cada 3 semanas. Uma dose de 85 mg/m2 a cada 2 semanas também foi estabelecida como tolerável. Baseado nestes estudos, oxaliplatina 130 mg/m2 por infusão IV durante 2 horas no dia 1 a cada 3 semanas (ciclo 1) foi utilizada nos estudos de fase II subsequentes.
Em um estudo de fase II, 43 pacientes pediátricos com tumores do SNC embrionários recorrentes ou refratários foram tratados por um máximo de 12 meses na ausência de doença progressiva ou toxicidade inaceitável. Em pacientes < 10 Kg, a dose de oxaliplatina utilizada foi 4,3 mg/Kg. Os eventos adversos mais comuns relatados foram: leucopenia (67,4%, G3/4: 11,6%), anemia (65,1%, G3/4: 4,7%), trombocitopenia (65,1%, G3/4: 25,6%), vômito (65,1%, G3/4: 7,0%), neutropenia (58,1%, G3/4: 16,3%) e neuropatia sensorial periférica (39,5%, G3/4: 4,7%). Foi observada uma única resposta parcial (taxa de resposta objetiva: 2,3%).
Num segundo estudo de fase II, 123 pacientes pediátricos foram tratados por tumores sólidos recorrentes, sarcoma de Ewing ou PNET (tumor neuro-ectodérmico primitivo) periférico, osteossarcoma, rabdomiossarcoma e neuroblastoma, por um máximo de 12 meses ou 17 ciclos. Nos pacientes com idade inferior a 12 meses, a dose de oxaliplatina utilizada foi 4,3 mg/Kg. Os eventos adversos mais comuns relatados foram: neuropatia sensorial periférica (53,2%, G3/4: 14,9%), trombocitopenia (40,4%, G3/4: 25,5%), anemia (40,4%, G3/4: 14,9%), vômito (31,9%, G3/4: 0%), náusea (29,8%, G3/4: 2,1%) e TGO aumentada (25,5%, G3/4: 4,3%). Não foram observadas respostas.
Os parâmetros farmacocinéticos da platina ultrafiltrável foram avaliados em 105 pacientes pediátricos durante o primeiro ciclo. O clearance médio nos pacientes pediátricos, estimado pela análise da população farmacocinética, foi 4,70 L/h. A variabilidade inter-paciente do clearance da platina nos pacientes pediátricos com câncer foi 40,9%. Os parâmetros farmacocinéticos médios da platina ultrafiltrável foram Cmáx de 0,75 + 0,24 mcg/mL, AUC0-48 de 7,52 + 5,07 mcg.h/mL e AUCinf de 8,83 + 1,57 mcg.h/mL com 85 mg/m2 de oxaliplatina e Cmáx de 1,10 + 0,43 mcg/mL, AUC0-48 de 9,74 + 2,52 mcg.h/mL e AUCinf de 17,30 + 5,34 mcg.h/mL com 130 mg/m2 de oxaliplatina.
As análises farmacocinética/farmacodinâmica foram realizadas em 43 pacientes pediátricos que foram avaliáveis pelos estudos farmacocinéticos de fase II. Os resultados não sugeriram qualquer relação entre AUC e parâmetros de segurança testados como distúrbios gastrintestinais, distúrbios do sistema nervoso, distúrbios renal e urinário ou distúrbios hematológicos para esta população de pacientes pediátricos.
Não foi estabelecida a efetividade de oxaliplatina como agente único nas populações pediátricas descrita acima. A inclusão de pacientes em ambos os estudos de fase II foi interrompida devido à falta de resposta do tumor.
Gravidez e lactação
Até o momento não existem dados disponíveis com relação à segurança de oxaliplatina em mulheres grávidas. Baseado em dados pré-clínicos, o uso de oxaliplatina é provavelmente letal e/ou teratogênico ao feto humano na dose terapêutica recomendada e, portanto, não é recomendado durante a gravidez e deve ser somente considerado depois que a paciente for informada apropriadamente sobre os riscos ao feto e com consentimento da paciente.
Assim como com outros agentes citotóxicos, medidas contraceptivas efetivas devem ser tomadas em pacientes potencialmente férteis antes do início do tratamento quimioterápico com TEVAOXALI®.
Não foi estudada a passagem da oxaliplatina para o leite materno. A amamentação é contraindicada durante o tratamento com TEVAOXALI®.
Pacientes idosos
Não foi observado aumento de toxicidade severa quando oxaliplatina foi utilizada como agente único ou em associação com 5-fluoruracila (5-FU), em pacientes com idade superior a 65 anos. Consequentemente, não é necessário um ajuste na dose específico para pacientes idosos.
Pacientes com insuficiência renal
Em pacientes com câncer gastrointestinal com variados níveis de insuficiência renal, tratados com oxaliplatina (infusão intravenosa de duas horas, a cada duas semanas, por um máximo de 12 ciclos) em associação com 5-FU/FA (FOLFOX4), oxaliplatina demonstrou impacto clínico mínimo na função renal, conforme avaliado através do clearance médio de creatinina.
Os resultados de segurança foram similares entre os grupos de pacientes. Entretanto, a duração da exposição foi mais curta para pacientes com insuficiência renal. A exposição mediana foi de 4, 6 e 3 ciclos para pacientes com insuficiência renal leve, moderada e severa, respectivamente. Em pacientes com função renal normal, a exposição mediana foi de 9 ciclos. Mais pacientes descontinuaram o tratamento devido a eventos adversos em grupos com insuficiência renal. A dose inicial de oxaliplatina já foi reduzida para 65 mg/m2 para pacientes com insuficiência renal severa.
Em pacientes com função renal normal ou insuficiência renal leve a moderada, a dose recomendada de TEVAOXALI® é 85 mg/m2. Em pacientes com insuficiência renal severa, a dose inicial recomendada deve ser reduzida para 65 mg/m2.
Pacientes com insuficiência hepática
Um estudo de fase I com oxaliplatina em monoterapia por infusão IV durante 2 horas a cada 3 semanas, incluiu pacientes adultos com câncer com diferentes graus de insuficiência hepática (nenhuma severa).
A dose inicial de oxaliplatina foi baseada no grau da disfunção hepática, e foi então aumentada até 130 mg/m2 para qualquer grau de insuficiência hepática (nenhuma severa). De maneira geral, a gravidade e os tipos de toxicidade observados foram toxicidades esperadas com o uso de oxaliplatina (vide Reações Adversas). Não foi observada correlação entre o aumento da toxicidade total e a piora da função hepática. Não houve diferenças nas frequências dos eventos entre os diferentes grupos de tratamentos baseados no grau de insuficiência hepática.
Durante o desenvolvimento clínico, não foram realizados ajustes de dose específicos para pacientes com testes da função hepática anormais.
Alterações na capacidade de dirigir veículos e operar máquinas
Nenhum estudo sobre os efeitos na habilidade de dirigir veículos e operar máquinas foi realizado. Entretanto, o tratamento com oxaliplatina resultando em um aumento no risco de tontura, náusea e vômito e outros sintomas neurológicos que afetam a marcha e o equilíbrio podem levar a uma influência pequena ou moderada na habilidade de dirigir e operar máquinas.
As anormalidades na visão, em particular perda de visão transitória (reversível após a descontinuação do tratamento), podem afetar a habilidade do paciente de dirigir ou operar máquinas. Portanto, os pacientes devem ser prevenidos quanto ao potencial efeito destes eventos na habilidade de dirigir ou operar máquinas.
Atenção: Este medicamento contém açúcar (lactose).
Interações medicamentosas.
- medicamento-medicamento
Não foi observada alteração no nível de exposição ao 5-fluoruracila (5-FU) nos pacientes que receberam dose única de 85 mg/m2 de oxaliplatina imediatamente antes da administração de 5-fluoruracila.
In vitro, não foi observado deslocamento significativo da ligação da oxaliplatina às proteínas plasmáticas com os seguintes agentes: eritromicina, salicilatos, granisetrona, paclitaxel e valproato de sódio.
- medicamento-exame laboratorial
Não há dados disponíveis até o momento sobre a interferência da oxaliplatina em exames laboratoriais.
- medicamento-alimento
Não há dados disponíveis até o momento sobre a interação entre alimentos e a oxaliplatina.
Cuidados de armazenamento.
TEVAOXALI® (oxaliplatina) solução injetável deve ser mantido em sua embalagem original, em temperatura ambiente inferior a 25°C.
Após diluição em glicose 5%, foi demonstrada estabilidade química e física em uso por 24 horas entre + 2°C e + 8°C e por 6 horas a + 25°C.Do ponto de vista microbiológico, a preparação para infusão deve ser utilizada imediatamente.
Caso não seja utilizada imediatamente, as condições e tempo de armazenagem em uso antes da utilização são de responsabilidade do manipulador e normalmente não seria mais que 24 horas entre + 2°C e + 8°C a menos que a diluição tenha sido realizada em condições assépticas controladas e validada.
Prazo de validade: 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparado manter entre +2°C e +8°C por até 24 horas ou manter a +25°C por 6 horas.
Características físicas e organolépticas
TEVAOXALI® apresenta-se como uma solução límpida e incolor, contida em frasco-ampola.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
Somente deve ser administrado em adultos.
A dose recomendada de TEVAOXALI® (oxaliplatina) para câncer de cólon no cenário adjuvante é de 85 mg/m2 intravenosamente repetido a cada 2 semanas em associação com fluoropirimidinas por 12 ciclos (6 meses).
A dose recomendada de TEVAOXALI® para o tratamento do câncer colorretal metastático/avançado é de 85 mg/m2 intravenosamente repetido a cada 2 semanas até progressão da doença ou toxicidade inaceitável.
A dose administrada deve ser ajustada de acordo com a tolerabilidade de cada paciente (vide Advertências e Precauções).
TEVAOXALI® deve ser sempre administrado antes das fluoropirimidinas (5-FU).
TEVAOXALI® é administrado por infusão intravenosa (IV).
Instruções para administração:
Assim como com outros agentes citotóxicos, deve-se ter cautela ao manusear e preparar as soluções de oxaliplatina.
O manuseio de agentes citotóxicos por profissionais de saúde capacitados requer todas as precauções para garantir a proteção do manipulador e das pessoas que o cercam.
A preparação de soluções injetáveis de agentes citotóxicos deve ser realizada por pessoas especializadas e treinadas, com conhecimento do medicamento utilizado, em condições de garantir a proteção do ambiente e em particular a proteção do manipulador. É necessária uma área reservada para a preparação. É proibido fumar, comer ou beber nesta área. As pessoas devem estar com os equipamentos de segurança para manuseio apropriado, avental de manga longa, máscara de proteção, touca, óculos de proteção, luvas estéreis descartáveis, revestimento de proteção para a área de trabalho e sacos e conteineres para coleta dos resíduos.
Os excrementos e vômitos devem ser manuseados com cuidado.
Mulheres grávidas devem ser prevenidas a evitar o manuseio de agentes citotóxicos.
Qualquer frasco quebrado deve ser tratado com as mesmas precauções e deve ser considerado como resíduo contaminado. O resíduo contaminado deve ser incinerado em conteineres rígidos adequadamente rotulados.
Caso o concentrado de oxaliplatina, ou a solução para infusão entrar em contato com a pele, lave imediatamente e cuidadosamente com água.
Caso o concentrado de oxaliplatina ou a solução para infusão entrar em contato com membranas mucosas, lave imediatamente e cuidadosamente com água.
Modo de usar:
TEVAOXALI® deve ser sempre administrado antes das fluoropirimidinas (5-FU).
TEVAOXALI® deve ser utilizado por via intravenosa (IV).
A administração de TEVAOXALI® não requer hiperhidratação.
TEVAOXALI® diluído em 250 a 500 mL de solução de glicose a 5% (para que a concentração não seja inferior a 0,2 mg/mL) deve ser infundido por veia periférica ou linha central venosa ao mesmo tempo que a infusão intravenosa de ácido folínico em solução de glicose 5%, durante 2 a 6 horas, utilizando uma linha em Y imediatamente antes do local da infusão. Estes dois medicamentos não devem ser combinados na mesma bolsa de infusão. O ácido folínico (leucovorin) não deve conter trometamol como excipiente e deve apenas ser diluído utilizando solução isotônica de glicose 5%, nunca em soluções alcalinas ou soluções que contenham cloreto de sódio ou cloreto.
Faça uma inspeção visual antes da infusão. Apenas soluções límpidas sem partículas devem ser utilizadas.
A infusão de TEVAOXALI® deve sempre preceder a de 5-FU.
No caso de extravasamento, a administração deve ser interrompida imediatamente (vide Advertências e Precauções).
Quando utilizado em combinação com 5-FU/FA e bevacizumabe, TEVAOXALI® deve ser administrado após o bevacizumabe, mas antes da administração de 5-FU.
Não há estudos dos efeitos de TEVAOXALI® administrado por vias não recomendadas. Portanto, por segurança e para garantir a eficácia deste medicamento, a administração deve ser somente por via intravenosa.
Reações adversas.
1- Terapia combinada de oxaliplatina com 5-FU/FA (FOLFOX):
As frequências das reações adversas são definidas utilizando-se a seguinte convenção: muito comum (≥ 1/10), comum (≥ 1/100, < 1/10), incomum (≥ 1/100