TEVAGRASTIM
TEVA
filgrastim
Estimulante de granulócitos e macrófagos.
Apresentações.
Solução Injetável de 300 mcg/0,5 mL
Embalagem contendo 1 ou 5 seringas preenchidas de 0,5 mL com dispositivo de segurança de agulha
USO INTRAVENOSO E SUBCUTÂNEO
USO ADULTO
Composição.
Cada seringa preenchida contém: filgrastim 300,0 mcg. Excipientes q.s.p 0,5 mL. (ácido acético, hidróxido de sódio, sorbitol, polissorbato 80, água para injeção)
Indicações.
TEVAGRASTIM (filgrastim) é indicado para:
- Redução da duração da neutropenia e da incidência da neutropenia febril em pacientes tratados com quimioterapia citotóxica (com exceção da leucemia mielóide crônica e das síndromes mielodisplásicas).
- Redução da duração da neutropenia em pacientes sob terapia mieloablativa seguida de transplante de medula óssea que possam estar sob um risco aumentado de desenvolver neutropenia grave prolongada.
- Mobilização de células progenitoras do sangue periférico (CPSP).
- Aumento das contagens de neutrófilos e redução da incidência e da duração de eventos relacionados com infecções, em períodos longos de administração em pacientes com neutropenia congênita grave, cíclica, ou idiopática, com contagem absoluta de neutrófilos (CAN) de 0,5 x 109/L e histórico de infecções graves ou recorrentes.
Resultados de eficácia.
A eficácia e segurança clínica do TEVAGRASTIM (filgrastim) foram avaliadas em 3 estudos Fase III.
Câncer de mama
Estudo Fase III, multinacional, multicêntrico, randomizado e controlado de comparação entre TEVAGRASTIM (filgrastim) x comparador x placebo em 348 pacientes com câncer de mama tratados com quimioterapia citotóxica. Durante o estudo, os voluntários estavam sob tratamento quimioterápico de no máximo 4 ciclos de docetaxel 75 mg/m2 IV e doxorrubicina 60 mg/m2 IV no 1° dia.
Os resultados de eficácia basearam-se nas seguintes determinações:
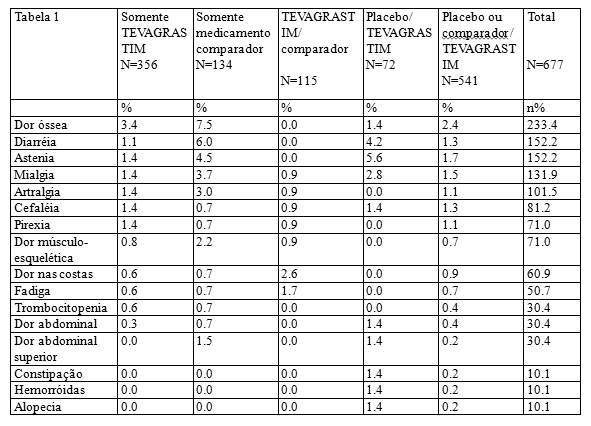
- Duração de neutropenia severa (DSN): a duração de neutropenia severa no ciclo 1 foi de 1.1 dias (faixa de 0 a 5) para pacientes tratados com o TEVAGRASTIM (filgrastim) e o medicamento comparador e 3.9 dias (faixa de 0 a 9) em pacientes que utilizaram o placebo. Os resultados foram similares no total de pacientes tratados, confirmando a comparabilidade de TEVAGRASTIM (filgrastim) e o medicamento comparador.
- Incidência de neutropenia febril (FN): No ciclo 1, a incidência observada ou definida em protocolo foi consideravelmente menor nos grupos tratados com TEVAGRASTIM (filgrastim) e o medicamento comparador do que nos grupos que receberam o placebo (12,1% x 12,5% x 36,1%). Não houve diferenças relevantes entre o TEVAGRASTIM (filgrastim) e o medicamento comparador na incidência de neutropenia febril no ciclo 1 ou em todos os demais ciclos.
- Contagem absoluta de neutrófilos (ANC): no ciclo 1, grupos que receberam o TEVAGRASTIM (filgrastim) e o medicamento comparador, apresentaram um aumento significativo após o 2° dia de tratamento atingindo contagem máxima no 3° dia de tratamento. Demonstraram, então, diminuição de ANC a 0,7 x 109/L no 7° dia e alcançaram novamente uma contagem máxima no 11° dia. No grupo que recebeu placebo, não houve aumento inicial de ANC, observou-se, ainda, diminuição constante do 2° dia, atingindo um nível consideravelmente baixo (0,2 x 109/L) no 11° dia.
No ciclo 1, o tempo médio de recuperação na contagem absoluta de neutrófilos foi similar nos grupos de que receberam o TEVAGRASTIM (filgrastim) e o medicamento comparador (8 dias) e consideravelmente maior no grupo que recebeu placebo (15 dias).
Nos ciclos 2 a 4, o ANC foi similar para todos os grupos (~1,0 x 109/L) e o tempo médio de recuperação na contagem absoluta de neutrófilos foi de 8 dias para todos os grupos.
Neste estudo de Fase III com pacientes de alto-risco ou com câncer de mama avançado, o TEVAGRASTIM (filgrastim) demonstrou ser superior ao placebo e de eficácia comparável ao medicamento comparador na redução da duração de neutropenia severa induzida pela quimioterapia, na contagem absoluta de neutrófilos e no tempo de recuperação na contagem absoluta de neutrófilos. O TEVAGRASTIM (filgrastim) e o medicamento comparador também demonstraram eficácia equivalente na redução da incidência de neutropenia febril quando comparados ao placebo.
Os resultados de eficácia resumidos estão demonstrados a seguir: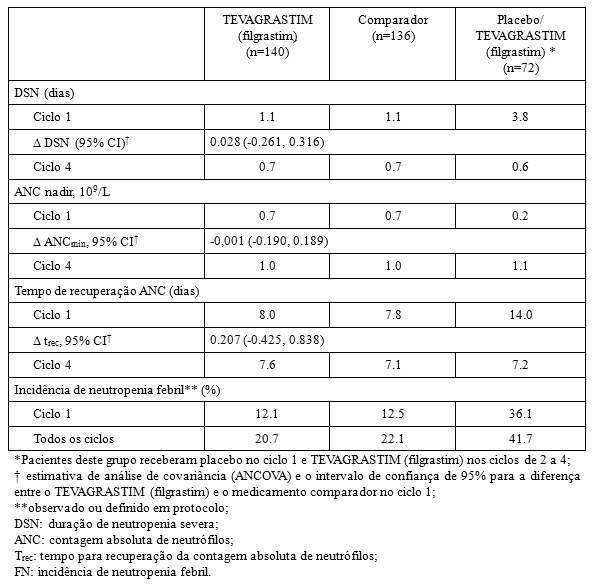
Câncer de pulmão
Estudo Fase III, multinacional, multicêntrico e randomizado, de comparação entre TEVAGRASTIM (filgrastim) x medicamento comparador em 240 pacientes com câncer de pulmão tratados com quimioterapia citotóxica. Durante o estudo, os voluntários estavam sob tratamento quimioterápico de no máximo 6 ciclos com derivados da platina. O regime de terapia mais comum utilizado foi cisplatina mais etoposídeo ou gencitabina em 49% e 15% dos pacientes, respectivamente. Outros regimes incluíram cisplatina mais vinorelbina e combinações de carboplatina com vinorelbina, etoposídeo, gencitabina ou paclitaxel.
Os resultados de eficácia basearam-se nas seguintes determinações:
- Duração de neutropenia severa (DSN): a duração de neutropenia severa no ciclo 1 foi de 0,5 dias para pacientes tratados com o TEVAGRASTIM (filgrastim) e 0,3 dias para os pacientes tratados com o medicamento comparador. A estimativa de análise de covariância (ANCOVA) entre o TEVAGRASTIM (filgrastim) e o medicamento comparador foi de 0,157 dias. O intervalo de confiança de 95% (- 0,114 a 0,428 dias) foi incluído na faixa pré-definida de equivalência (-1 a 1 dia), indicando que a duração de neutropenia severa (DSN) não foi diferente para o TEVAGRASTIM (filgrastim) e o medicamento comparador.
- Contagem absoluta de neutrófilos (ANC): o perfil de contagem absoluta de neutrófilos foi similar em todos os ciclos para o medicamento comparador e o TEVAGRASTIM (filgrastim). Houve um aumento inicial de ANC significativo atingindo contagem máxima no 5° dia e subseqüente diminuição nos 11° e 12° dias. ANC atingiu uma segunda contagem máxima no 14° dia e retornou próxima a contagem de base, gradualmente, até o 21° dia. A contagem absoluta de neutrófilos foi comparável entre os grupos que receberam o medicamento comparador e o TEVAGRASTIM (filgrastim) no ciclo 1 (2.1 vs. 2.9 x 109/L) e após migrarem do medicamento comparador para o TEVAGRASTIM (filgrastim) no ciclo 4 (2.3 vs. 3.2 x 109/L). No tempo médio de recuperação na contagem absoluta de neutrófilos houve diferenças mínimas entre os grupos no ciclo 1 (6.3 vs. 4.5 dias) que persistiu até o ciclo 4 quando o TEVAGRASTIM (filgrastim) foi administrado em ambos os grupos (6.4 vs. 4.5 dias).
- Incidência de neutropenia febril (FN): No ciclo 1, a incidência observada ou definida em protocolo foi de 15,0% no grupo que recebeu o TEVAGRASTIM (filgrastim) e 8,8% no grupo que recebeu o medicamento comparador. Estatisticamente, esta diferença é insignificante (p= 0.23). No ciclo 4, após os pacientes migrarem do medicamento comparador para o TEVAGRASTIM (filgrastim), a incidência de neutropenia febril foi de 4,3% e 3,3%, respectivamente, (p= 0.90). Nos outros ciclos, a incidência de neutropenia febril foi de 33,1% e 23,8% nos pacientes sob tratamento com o TEVAGRASTIM (filgrastim) e o medicamento comparador, respectivamente.
Neste estudo de Fase III com pacientes com câncer de pulmão sob tratamento quimioterápico citotóxico, a profilaxia primária de TEVAGRASTIM (filgrastim) e o medicamento comparador demonstraram eficácia e segurança equivalente. O perfil de contagem absoluta de neutrófilos, incluindo a duração de neutropenia severa foi similar entre ambos os medicamentos. Diferenças insignificantes entre o TEVAGRASTIM (filgrastim) e o medicamento comparador na incidência de neutropenia febril podem ser atribuídas às diferentes características dos pacientes.
Os resultados de eficácia resumidos estão demonstrados a seguir: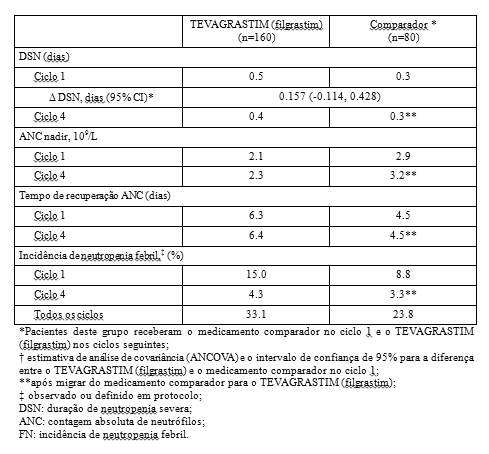
Linfomas Não-Hodgkin
Estudo Fase III, multinacional, multicêntrico e randomizado, de comparação entre TEVAGRASTIM (filgrastim) x medicamento comparador em pacientes com linfoma Não-Hodgkin. Durante o estudo, os voluntários estavam sob tratamento quimioterápico, de no máximo 6 ciclos, com ciclofosfamida, doxorrubicina, vincristina e prednisona (CHOP). Tratamento adicional com rituximab (anticorpo monoclonal anti-CD20) foi utilizado a critério de médico.
Os resultados de eficácia basearam-se nas seguintes determinações:
- Duração de neutropenia severa (DSN): a duração de neutropenia severa no ciclo 1 foi de 0,5 dias para pacientes tratados com o TEVAGRASTIM (filgrastim) e 0,9 dias para os pacientes tratados com o medicamento comparador. A estimativa de análise de covariância (ANCOVA) entre o TEVAGRASTIM (filgrastim) e o medicamento comparador foi de - 0,378 dias. O intervalo de confiança de 95% (- 0,837 a 0,081 dias) foi incluído na faixa pré-definida de equivalência (-1 a 1 dia), indicando que a duração de neutropenia severa (DSN) não foi diferente para o TEVAGRASTIM (filgrastim) e o medicamento comparador (p= 0.11). A duração de neutropenia severa (DSN), no ciclo 4, após os pacientes migrarem do TEVAGRASTIM (filgrastim) para o medicamento comparador foi de, respectivamente, 0,2 e 0,7 dias para o grupo que recebeu TEVAGRASTIM (filgrastim) e o grupo que recebeu o medicamento comparador.
- Contagem absoluta de neutrófilos (ANC): o perfil de contagem absoluta de neutrófilos foi similar em todos os ciclos para o medicamento comparador e o TEVAGRASTIM (filgrastim). No ciclo 1, houve um aumento inicial de ANC significativo atingindo contagem máxima no 4° dia e subseqüente diminuição no 9° dia. O ANC atingiu uma segunda contagem máxima no 11° dia e retornou próxima a contagem de base, gradualmente, até o 21° dia. A contagem absoluta de neutrófilos foi comparável entre os grupos que receberam o medicamento comparador e o TEVAGRASTIM (filgrastim) no ciclo 1 (1.7 vs. 1.1 x 109/L) e após migrarem do medicamento comparador para o v no ciclo 4 (2.1 vs. 1.8 x 109/L). O tempo médio de recuperação na contagem absoluta de neutrófilos nos pacientes que receberam o TEVAGRASTIM (filgrastim) e o medicamento comparador foi respectivamente de 6.0 e 6.7 dias no ciclo 1, e 4.9 e 6.1 dias no ciclo 4.
- Incidência de neutropenia febril (FN): No ciclo 1, a incidência observada ou definida em protocolo foi de 11,1% no grupo que recebeu o TEVAGRASTIM (filgrastim) e 20,7% no grupo que recebeu o medicamento comparador (p= 0.12). As taxas de incidência no ciclo 4 foram respectivamente 31.7 % e 41.4 % (p= 0.21).
Este estudo de Fase III foi conduzido em pacientes com linfoma Não-Hodgkin tratados com o regime quimioterápico CHOP, com ou sem rituximab. Os resultados do estudo confirmaram que a profilaxia primária com o TEVAGRASTIM (filgrastim) é tão eficaz quanto com o medicamento comparador na redução da duração de neutropenia severa e na incidência de neutropenia febril. O perfil de contagem absoluta de neutrófilos foi similar entre ambos os medicamentos no ciclo 1.
Os resultados de eficácia resumidos estão demonstrados a seguir: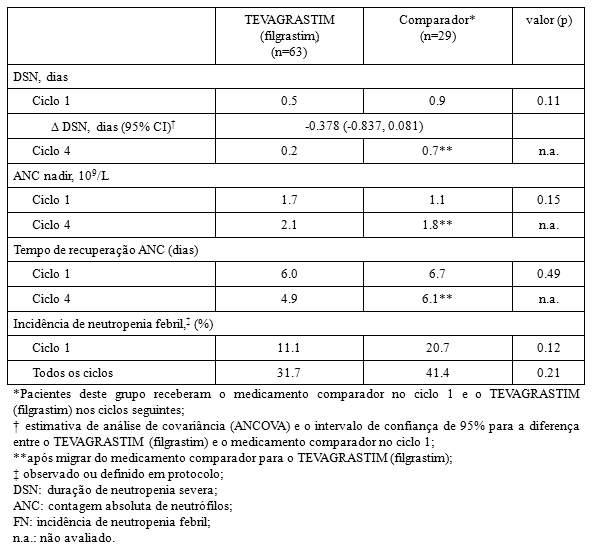
Caract. farmacológicas.
Propriedades farmacodinâmicas
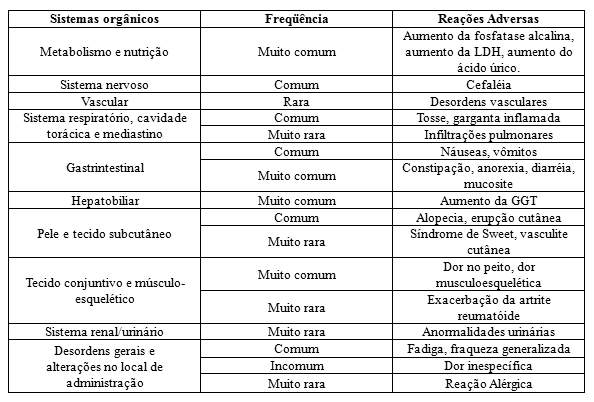
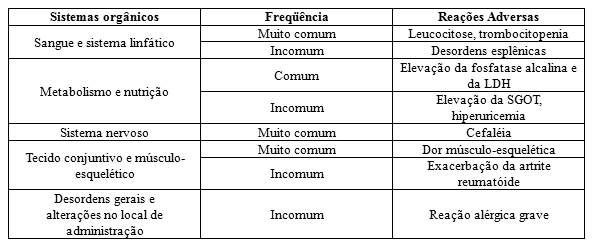
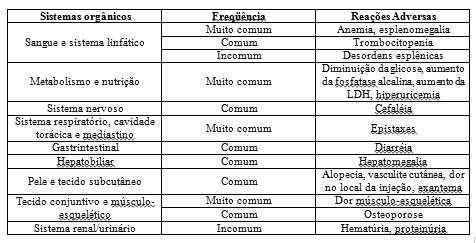
O filgrastim é uma glicoproteína que regula a produção e a liberação de neutrófilos funcionais da medula óssea. O filgrastim provoca, num período de 24 horas, um aumento significativo da contagem de neutrófilos no sangue periférico, com pequenos aumentos de monócitos. Em alguns pacientes com neutropenia crônica grave, o filgrastim pode também induzir um pequeno aumento do número de eosinófilos e basófilos circulantes relativamente aos valores de base; alguns destes pacientes podem apresentar eosinofilia ou basofilia antes do tratamento. Elevações nas contagens de neutrófilos são dose-dependentes nas doses recomendadas. Os neutrófilos produzidos em resposta ao filgrastim apresentam função normal ou aumentada, como demonstrado em testes de funções fagocítica e quimiostática. Após o término do tratamento com filgrastim, a contagem de neutrófilos circulantes diminui em 50% dentro de 1 a 2 dias e para níveis normais, dentro de 1 a 7 dias.
O uso de filgrastim em pacientes submetidos à quimioterapia citotóxica leva a reduções significativas na incidência, gravidade e duração da neutropenia e da neutropenia febril. O tratamento com filgrastim reduz significativamente a duração da neutropenia febril, a utilização de antibióticos e o tempo de hospitalização após quimioterapia de indução para leucemia mielóide aguda ou terapia mieloablativa seguida de transplante de medula óssea. A incidência de relatos de febre e infecções não foi reduzida em nenhum destes quadros clínicos. A duração da febre não diminuiu nos pacientes que receberam terapia mieloablativa seguida de transplante de medula óssea.
A utilização de filgrastim isoladamente ou após quimioterapia mobiliza as células progenitoras hematopoiéticas para o sangue periférico. Estas células progenitoras autólogas do sangue periférico (CPSPs) podem ser coletadas e infundidas após terapia citotóxica de dose elevada, em substituição ou em adição ao transplante de medula óssea. A infusão de CPSPs acelera a recuperação hematopoiética reduzindo a duração do risco de complicações hemorrágicas e a necessidade de transfusões de plaquetas.
Os receptores de CPSPs alogênicas mobilizadas com filgrastim tiveram uma recuperação hematológica significativamente mais rápida, levando a uma diminuição significativa do tempo de recuperação de plaquetas quando comparado com o transplante alogênico de medula óssea.
Previamente ao transplante de CPSPs alogênicas, a utilização de filgrastim para a mobilização de CPSP em doadores saudáveis permite um cultivo de 4 x 106 células CD34+ /kg de peso corporal do receptor na maioria dos doadores, após duas leucaféreses. Para estes doadores saudáveis é dada uma dose de 10 mcg/kg/dia, administrada por via subcutânea durante 4 a 5 dias consecutivos.
O uso de filgrastim em pacientes com neutropenia crônica grave (neutropenia congênita grave, neutropenia cíclica e neutropenia idiopática), induz um aumento sustentado das contagens absolutas de neutrófilos no sangue periférico e uma redução das infecções e eventos relacionados.
O filgrastim, assim como com outros fatores de crescimento hematopoiéticos, demonstrou in vitro propriedades estimuladoras sobre as células endoteliais humanas.
Propriedades Farmacocinéticas
O clearence do filgrastim, tanto após administração subcutânea como intravenosa, demonstrou seguir uma farmacocinética de primeira ordem. A meia-vida de eliminação sérica do filgrastim é de aproximadamente 3,5 horas com uma taxa de depuração de aproximadamente 0,6 mL/min/kg. A infusão contínua com filgrastim durante um período de até 28 dias, em pacientes em recuperação de transplante autólogo de medula óssea, não apresentou evidência de acumulação do fármaco e de meias-vidas comparáveis. Há uma correlação linear positiva entre a dose e a concentração sérica de filgrastim, se administrado por via intravenosa ou por via subcutânea. Após administração subcutânea das doses recomendadas, as concentrações séricas mantiveram-se acima dos 10 ng/mL, durante 8 a 16 horas. O volume de distribuição no sangue é aproximadamente de 150 mL/kg.
Contraindicações.
TEVAGRASTIM (filgrastim) não deve ser administrado em pacientes que apresentem sensibilidade ao filgrastim ou a qualquer componente da formulação.
TEVAGRASTIM (filgrastim) não deve ser administrado para o aumento de dose de quimioterapia citotóxica além dos regimes de dose estabelecidos, e em pacientes com neutropenia congênita grave (síndrome de Kostmann) com citogenética anormal.
Advertências e precauções.
A terapia com filgrastim só deve ser administrada em colaboração com um centro de oncologia, onde os profissionais tenham experiência no tratamento com filgrastim e em hematologia e onde tenha instalações diagnósticas necessárias. Os procedimentos de mobilização e de aférese devem ser realizados em colaboração com um centro de oncologia/hematologia com profissionais com experiência adequada neste campo e onde a monitorização das células progenitoras hematopoiéticas possa ser feita corretamente.
Advertências especiais
O filgrastim não deve ser utilizado para aumentar a dose de quimioterapia citotóxica além dos regimes posológicos estabelecidos.
O filgrastim não deve ser administrado a pacientes com neutropenia congênita grave (Síndrome de Kostman) com anormalidades citogenéticas.
Precauções especiais de utilização em doentes com leucemia mielóide aguda
Crescimento celular maligno
O filgrastim pode promover o crescimento de células mielóides in vitro, podendo ser igualmente observados efeitos similares em algumas células não mielóides in vitro.
Não foi estabelecida a segurança e eficácia da administração de filgrastim em pacientes com síndrome mielodisplásica ou leucemia mielóide crônica. Por este motivo, o filgrastim não é indicado para utilização nestas situações. Deve-se ter cuidado especial para distinguir o diagnóstico de transformação blástica de leucemia mielóide crônica da leucemia mielóide aguda.
Tendo em vista os dados limitados sobre a segurança e eficácia em pacientes com leucemia mielóide aguda secundária, filgrastim deve ser administrado com precaução.
Outras precauções especiais
A monitorização da densidade óssea pode ser indicada em pacientes com doença óssea osteoporótica submetidos a tratamento contínuo com filgrastim durante mais de 6 meses.
Foram relatadas reações adversas pulmonares raras, em especial pneumonia intersticial, após administração de filgrastim. Pacientes com um histórico recente de infiltrações pulmonares ou pneumonia poderão apresentar um risco aumentado. O aparecimento de sinais pulmonares, tais como tosse, febre e dispnéia, associados a sinais radiológicos de infiltração pulmonar e deterioração da função pulmonar podem ser sintomas preliminares indicativos de Síndrome de Dificuldade Respiratória do Adulto (SDRA). Nestes casos, a administração de filgrastim deve ser descontinuada e deve ser iniciado tratamento apropriado.
Precauções especiais em pacientes com câncer
Leucocitose
Foram observadas contagens de leucócitos iguais ou superiores a 100 x 109/L em menos de 5% dos pacientes que receberam filgrastim em doses superiores a 0,3 MUI (3 mcg)/kg/dia. Não foram relatados efeitos indesejáveis diretamente atribuídos a este grau de leucocitose. No entanto, devido aos riscos potenciais associados com a leucocitose grave, deve ser realizada uma contagem de leucócitos em intervalos regulares durante a terapia com filgrastim. Se a contagem de leucócitos exceder 50 x 109/L após o nadir esperado, a administração de filgrastim deve ser descontinuada imediatamente. No entanto, durante o período de administração de filgrastim para mobilização de CPSP, filgrastim deve ser descontinuado ou a sua dose reduzida caso a contagem de leucócitos seja superior a 70 x 109/L.
Risco associado com o aumento das doses de quimioterapia
Precauções especiais devem ser tomadas em pacientes tratados com doses elevadas de quimioterapia, dado que não está demonstrada uma melhora da resposta tumoral e porque a intensificação das doses de agentes quimioterápicos pode levar a um aumento das toxicidades, incluindo efeitos cardíacos, pulmonares, neurológicos e dermatológicos.
O tratamento isolado com filgrastim não previne a trombocitopenia nem a anemia devido à quimioterapia mielossupressora. Dada a possibilidade de receber doses mais elevadas de quimioterapia (por exemplo, doses máximas no esquema prescrito), o paciente pode ter um maior risco para trombocitopenia e anemia. É recomendada a monitorização regular do número de plaquetas e do hematócrito. Deve ser tomada uma precaução especial na administração de agentes quimioterápicos isolados ou em associação, que sejam conhecidos por provocarem trombocitopenia grave.
A utilização de células CPSP mobilizadas por filgrastim demonstrou reduzir a intensidade e a duração da trombocitopenia após quimioterapia mielossupressora ou mieloablativa.
Outras precauções especiais
Não foram estudados os efeitos de filgrastim em pacientes com redução substancial dos progenitores mielóides. O filgrastim atua primariamente nos precursores de neutrófilos para exercer o seu efeito no aumento da contagem de neutrófilos. Desse modo, em pacientes com redução do número de precursores de neutrófilos, a resposta pode ser diminuída (tais como os tratados com quimioterapia ou radioterapia extensiva, ou aqueles com medula óssea infiltrada por tumor).
O efeito do filgrastim na doença do enxerto versus hospedeiro não foi definido.
Precauções especiais em pacientes submetidos à mobilização das células progenitoras do sangue periférico
Mobilização
Não existem estudos comparativos aleatorizados e prospectivos entre os dois métodos recomendados de mobilização (filgrastim isolado ou em associação com quimioterapia mielossupressora) na mesma população de pacientes. O grau de variabilidade entre pacientes individuais e entre as análises laboratoriais de células CD34+ indica que a comparação direta entre os diferentes estudos é difícil. É ainda mais difícil recomendar um método ótimo. A escolha do método de mobilização deve ser considerada em relação aos objetivos gerais do tratamento para cada paciente individualmente.
Exposição anterior a fármacos citotóxicos
Pacientes que tenham sido submetidos previamente a terapia mielossupressora extensiva, podem não apresentar mobilização suficiente de CPSP para alcançar o nível mínimo recomendado (2,0 x 106 células CD34+ /kg) ou aceleração da recuperação das plaquetas.
Alguns agentes citotóxicos exibem toxicidades específicas aos progenitores das células hematopoiéticas e podem adversamente afetar a mobilização das células progenitoras.
Fármacos tais como o melfalan, a carmustina (BCNU) e a carboplatina, quando administrados durante longos períodos anteriores às tentativas de mobilização de células progenitoras podem reduzir o rendimento de células progenitoras. No entanto, a administração de melfalan, carboplatina ou BCNU concomitantemente com o filgrastim demonstrou ser eficaz na mobilização de células progenitoras.
Quando ocorre um transplante de células progenitoras do sangue periférico é aconselhável planejar o procedimento para mobilização das células-tronco no início do tratamento do paciente.
Deve-se ter especial atenção ao número de células progenitoras mobilizadas em tais pacientes antes da administração de doses elevadas de quimioterapia. Se o resultado de células for inadequado, medido pelos critérios acima mencionados, devem ser consideradas como alternativas outras formas de tratamento que não exijam suporte de células progenitoras.
Avaliação dos resultados de células progenitoras
Na avaliação do número de células progenitoras coletadas em pacientes tratados com filgrastim, deve ser prestada especial atenção ao método de quantificação. Os resultados da análise de fluxo citométrico do número de células CD34+ podem variar em função da metodologia específica utilizada e, conseqüentemente, as recomendações de números baseadas em estudos efetuados em outros laboratórios devem ser interpretadas com precaução.
A análise estatística da relação entre o número de células CD34+ infundidas e a taxa da recuperação plaquetária, após quimioterapia de dose elevada, indica uma relação complexa, mas contínua.
A recomendação de um valor mínimo de 2,0 x 106 de células CD34+/kg é baseada em trabalhos publicados sobre reconstituição hematológica adequada. Os valores superiores a este valor mínimo parecem estar relacionados com uma recuperação mais rápida, e os valores inferiores a uma recuperação mais lenta.
Precauções especiais em doadores saudáveis submetidos à mobilização de células progenitoras do sangue periférico
A mobilização de CPSP não proporciona um benefício clínico direto aos doadores saudáveis e deve ser apenas considerada com o objetivo de um transplante alogênico de células-tronco.
A mobilização de CPSP deve ser apenas considerada em doadores que cumpram critérios de elegibilidade clínicos e laboratoriais para a doação de células-tronco. Deve ser dada especial atenção aos valores hematológicos e às doenças infecciosas.
A segurança e eficácia do filgrastim não foram avaliadas em doadores saudáveis com menos de 16 anos ou mais de 60 anos de idade.
Foi observada uma trombocitopenia transitória (plaquetas < 100 x 109/L), após a administração de filgrastim e leucaférese, em 35% dos indivíduos estudados. Dentre estes, dois casos de plaquetas < 50 x 109/L foram relatados e atribuídos ao procedimento de leucaférese.
Se for necessária mais de uma leucaférese, deve ser prestada atenção especial aos doadores com plaquetas < 100 x 109/L antes da leucaférese; em geral as aféreses não devem ser efetuadas se os valores de plaquetas forem < 75 x 109/L.
As leucaféreses não devem ser efetuadas em doadores que tomam anticoagulantes ou que tenham problemas conhecidos de hemóstase.
A administração de filgrastim deve ser descontinuada ou a dose deve ser reduzida se a contagem de leucócitos for > 70 x 109/L.
Os doadores que receberam filgrastim para a mobilização de CPSP devem ser monitorizados até os valores hematológicos voltarem ao normal.
Doadores devem ser supervisionados por um longo período para a avaliação da segurança de filgrastim. Não pode ser excluído o risco da promoção de um clone mielóide maligno. É recomendado que o centro de aférese implemente um registro sistemático para acompanhamento dos doadores de células-tronco para assegurar a monitorização da segurança a longo prazo.
Após administração de filgrastim a doadores saudáveis e a pacientes, foram relatados casos freqüentes, mas geralmente assintomáticos de esplenomegalia e casos muito raros de ruptura esplênica. Alguns casos de ruptura esplênica foram fatais. Desta forma, as dimensões do baço devem ser cuidadosamente monitorizadas (por exemplo, exame clínico, ultrassom). Um diagnóstico de possível ruptura esplênica deve ser considerado em doadores e/ou pacientes que apresentem dor abdominal no quadrante superior esquerdo ou dor na extremidade do ombro.
Precauções especiais em receptores de CPSP alogênicas mobilizadas com filgrastim
Os dados atuais indicam que as interações imunológicas entre as CPSP alogênicas coletadas e o receptor podem estar associadas a um aumento do risco para doença aguda e crônica do enxerto versus hospedeiro quando comparado com o transplante de medula óssea.
Precauções especiais em pacientes com Neutropenia Crônica Grave
Contagem das células sangüíneas
As contagens de plaquetas devem ser monitorizadas com rigor, especialmente durante as primeiras semanas de terapia com filgrastim. Deve ser considerada a descontinuação intermitente ou a diminuição da dose de filgrastim em pacientes que desenvolvam trombocitopenia, ou seja, nível de plaquetas consistentemente < 100.000/mm3.
Ocorrem outras alterações de células sangüíneas, incluindo anemia e aumentos transitórios das células progenitoras mielóides que exigem monitorização rigorosa das contagens celulares.
Transformação em leucemia ou síndrome mielodisplásica
Deve-se ter precaução especial no diagnóstico das neutropenias crônicas graves para distingui-las de outras alterações hematopoiéticas, tais como anemia aplásica, mielodisplasia e leucemia mielóide.
Antes do tratamento, devem ser realizadas contagens totais das células sangüíneas com diferencial e contagem das plaquetas, e uma avaliação da morfologia da medula óssea e do cariótipo.
Em estudos clínicos em pacientes com neutropenia crônica grave tratados com filgrastim, foi observada uma baixa freqüência (aproximadamente 3%) de síndrome mielodisplásica (SMD) ou leucemia. Esta observação só foi feita em pacientes com neutropenia congênita. As leucemias e SMD são complicações naturais da doença e têm relação incerta com a terapia com filgrastim. Num subgrupo de aproximadamente 12% de pacientes com avaliações citogenéticas normais na linha de base, foram subseqüentemente encontradas anormalidades, incluindo monossomia 7, em avaliações de rotina repetidas. Se os pacientes com neutropenia crônica grave desenvolverem anormalidades citogenéticas, os riscos e benefícios do tratamento com filgrastim devem ser avaliados com cuidado; filgrastim deve ser descontinuado se ocorrer SMD ou leucemia.
Atualmente, ainda não é claro se o tratamento a longo prazo de pacientes com neutropenia crônica grave pode predispor os pacientes a anormalidades citogenéticas, SMD ou transformação leucêmica. É recomendada a realização de exames morfológicos e citogenéticos da medula óssea a intervalos regulares (aproximadamente a cada 12 meses).
Outras precauções especiais
Devem ser excluídas as causas de neutropenia transitória, tais como infecções virais.
A esplenomegalia é um efeito direto do tratamento com filgrastim. Trinta e um por cento (31%) dos pacientes em estudos revelaram esplenomegalia palpável. No início da terapia com filgrastim ocorreram aumentos do volume esplênico, medidos por radiografia, com tendência à estabilização. Foi observado que as reduções da dose diminuíam ou detinham a progressão da dilatação esplênica, e em 3% dos pacientes foi necessária uma esplenectomia. O tamanho do baço deve ser avaliado regularmente. A palpação abdominal deve ser suficiente para detectar os aumentos anormais do volume esplênico.
Ocorreu hematúria/proteinúria num pequeno número pacientes. Devem ser realizadas análises regulares de urina para monitorizar este efeito.
Não foram estabelecidas a segurança e eficácia em neonatos e em pacientes com neutropenia autoimune.
Populações especiais
Pacientes idosos
Os estudos clínicos com filgrastim incluíram um pequeno número de pacientes idosos, mas não foram realizados estudos específicos neste grupo de pacientes e, portanto, não podem ser feitas recomendações de dose específicas.
Pacientes com insuficiência hepática ou renal
Os estudos realizados com filgrastim em pacientes com insuficiência hepática ou renal revelaram que o perfil farmacocinético e farmacodinâmico é semelhante ao observado em indivíduos normais. Não é necessário qualquer ajuste de dose nestes casos.
Excipientes
TEVAGRASTIM (filgrastim) contém sorbitol. Pacientes com problemas hereditários raros de intolerância à frutose não devem utilizar este medicamento.
Gravidez e lactação
Não existem dados suficientes sobre a utilização de filgrastim em mulheres grávidas. Existem relatos na literatura em que se demonstra a passagem de filgrastim através da placenta em mulheres grávidas. Estudos em animais revelaram toxicidade reprodutiva. O risco potencial para os seres humanos é desconhecido. O filgrastim não deve ser utilizado durante a gravidez, a menos que seja claramente necessário.
É desconhecido se filgrastim é excretado no leite humano. A excreção de filgrastim no leite não foi estudada em animais. A decisão sobre continuar/interromper o aleitamento ou sobre continuar/interromper o tratamento com filgrastim deve ser tomada levando-se em consideração o benefício do aleitamento para o bebê e o benefício da terapia com filgrastim para a mulher.
Categoria C de risco na gravidez: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Interações medicamentosas.
Não foram ainda estabelecidas a segurança e a eficácia de filgrastim quando administrado no mesmo dia que a quimioterapia citotóxica mielossupressora. Tendo em vista a sensibilidade da rapidez da divisão das células mielóides à quimioterapia citotóxica mielossupressora, a utilização de filgrastim não é recomendada no período que decorre entre as 24 horas anteriores e às 24 horas posteriores à quimioterapia. Existem dados preliminares, obtidos a partir de um grupo pequeno de pacientes tratados concomitantemente com filgrastim e 5-fluoruracila, que indicam que a gravidade da neutropenia pode ser exacerbada.
Não foram ainda investigadas, em estudos clínicos, as possíveis interações com outros fatores de crescimento hematopoiéticos e com citocinas.
Dado que o lítio promove a liberação de neutrófilos, é provável que possa potencializar o efeito de filgrastim. Embora esta interação não tenha sido formalmente investigada e não exista qualquer evidência de que tal interação possa ser prejudicial.
Cuidados de armazenamento.
TEVAGRASTIM (filgrastim) apresenta prazo de validade de 24 meses a partir da data de fabricação, desde que conservado em temperatura entre 2°C e 8°C.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamentos com o prazo de validade vencido.
Para sua segurança mantenha o medicamento na embalagem original.
Quando diluído em solução de glicose a 5%, numa faixa entre 15 mg/mL e 60 mg/ mL, o produto se mantém estável por 24 horas à temperatura de 2 a 8°C e por mais 24 horas à temperatura de 25°C.
Quando diluído em solução de glicose a 5% e albumina sérica humana, numa faixa entre 2 mg/mL e 15 mg/ mL, o produto se mantém estável por 24 horas à temperatura de 2 a 8°C e por mais 24 horas à temperatura de 25°C.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
O medicamento TEVAGRASTIM (filgrastim) deve ser administrado uma vez ao dia, por injeção subcutânea ou por infusão intravenosa.
Para a injeção subcutânea:
1. Administrar o TEVAGRASTIM (filgrastim) aproximadamente no mesmo horário todos os dias.
2. Tirar a seringa preenchida do refrigerador e verificar a aparência do produto. A solução deve estar límpida, incolor e isenta de partículas.
3. Para menor desconforto da injeção, deixar a seringa preenchida por 30 minutos à temperatura ambiente ou segurar cuidadosamente a seringa por alguns minutos. Não aquecer o TEVAGRASTIM (filgrastim) de nenhuma outra maneira.
4. Segurar a seringa e retirar a tampa cuidadosamente sem girá-la. Não tocar na agulha ou empurrar o êmbolo, conforme figuras abaixo.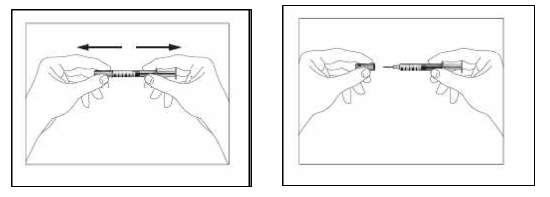
5. Segurar a seringa voltada para cima e empurrar o êmbolo para liberar o ar da seringa.
6. Limpar a área da injeção cuidadosamente com álcool e fazer uma prega na pele com o dedo indicador e o polegar, conforme indicado na figura abaixo.
7. Perfurar com a agulha no ângulo de 45°.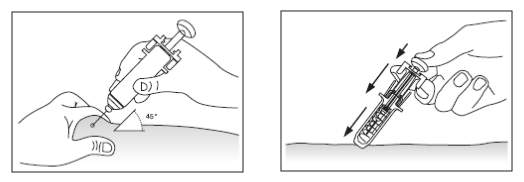
8. Perfurar com a agulha na região da coxa, abdômen ou parte posterior dos braços, conforme indicado nas figuras abaixo.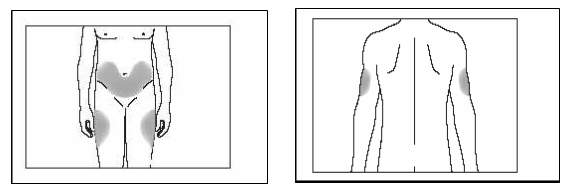
9. Puxar o êmbolo cuidadosamente para verificar se não houve perfuração de vaso sangüíneo. Se aparecer sangue na seringa, retire a agulha e tente em outro local.
10. Injetar a solução lenta e gradualmente, segurando a prega feita na pele.
11. Retirar a seringa ainda com o êmbolo pressionado.
12. Soltar o êmbolo. O dispositivo de segurança automaticamente cobrirá a agulha.
13. O conteúdo da seringa deve ser utilizado uma única vez.
Diluição
TEVAGRASTIM (filgrastim) não deve ser diluído em solução de cloreto de sódio.
Se houver necessidade, este medicamento pode ser diluído em solução de glicose a 5%. Não é recomendado diluir o produto a uma concentração final menor que 0,2 MUI (2 mg). A solução deve ser inspecionada visualmente antes do uso. Somente soluções límpidas e isentas de partículas podem ser utilizadas.
Para pacientes tratados com filgrastim diluído a uma concentração de 1,5 MUI (15 mg) / mL, deve ser adicionada albumina sérica humana para uma concentração final de 2 mg/mL.
Quando diluído em solução de glicose a 5%, numa faixa entre 15 mg/mL e 60 mg/ mL, o produto se mantém estável por 24 horas à temperatura de 2 a 8°C e por mais 24 horas à temperatura de 25°C.
Quando diluído em solução de glicose a 5% e albumina sérica humana, numa faixa entre 2 mg/mL e 15 mg/ mL, o produto se mantém estável por 24 horas à temperatura de 2 a 8°C e por mais 24 horas à temperatura de 25°C.
TEVAGRASTIM (filgrastim) não contém conservantes. Em virtude do possível risco de contaminação microbiana, as seringas devem ser de uso único.
TEVAGRASTIM (filgrastim) é compatível com vidro e plástico quando diluído numa solução de glicose.
Posologia
Quimioterapia citotóxica estabelecida
A dose recomendada de TEVAGRASTIM (filgrastim) é de 0,5 MUI (5 mcg)/kg/dia. A primeira dose de TEVAGRASTIM (filgrastim) não deve ser administrada nas 24 horas seguintes à quimioterapia citotóxica. TEVAGRASTIM (filgrastim) pode ser administrado por injeção subcutânea diária ou como infusão intravenosa diária quando diluído em solução de glicose a 5%, administrado durante 30 minutos. Na maioria dos casos é preferida a via subcutânea. Há alguns indícios, provenientes de um estudo de administração de dose única, de que a administração por via intravenosa pode encurtar a duração do efeito. A relevância clínica deste fato na administração de múltiplas doses é indeterminada. A escolha da via de administração depende da circunstância clínica específica de cada paciente. Em estudos clínicos aleatorizados foi utilizada uma dose subcutânea de 23 MUI (230 mcg)/m2/dia (4,0 a 8,4 mcg/kg/dia).
A administração diária de TEVAGRASTIM (filgrastim) deve continuar até que o nadir esperado de neutrófilos seja ultrapassado e a contagem de neutrófilos volte ao seu valor normal. Após a quimioterapia estabelecida para os tumores sólidos, linfomas e leucemias linfóides, a duração esperada do tratamento necessário para atingir estes valores é de aproximadamente 14 dias. Após o início e a consolidação do tratamento para leucemia mielóide aguda, a duração do tratamento poderá ser substancialmente superior (até 38 dias) depe