TECVAYLI
JANSSEN-CILAG
teclistamabe
Anticorpo biespecífico IgG4-PAA.
Apresentações.
Solução injetável de 10 mg/mL de teclistamabe em embalagem com 1 frasco-ampola de 3 mL. Solução injetável de 90 mg/mL de teclistamabe em embalagem com 1 frasco-ampola de 1,7 mL.
USO SUBCUTÂNEO
USO ADULTO
Composição.
Cada frasco-ampola de 3 mL contém 30 mg de teclistamabe (10 mg/mL). Cada frasco-ampola de 1,7 mL contém 153 mg de teclistamabe (90 mg/mL).
Excipientes: acetato de sódio tri-hidratado, ácido acético glacial, sacarose, polissorbato 20, edetato dissódico di-hidratado e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
TecvayliTM é indicado para o tratamento de pacientes adultos com mieloma múltiplo recidivado ou refratário que receberam pelo menos três terapias anteriores, incluindo um inibidor de proteassoma, um agente imunomodulador e um anticorpo monoclonal antiCD38.
2. RESULTADOS DE EFICÁCIA
A eficácia de TecvayliTM em monoterapia foi avaliada em pacientes com mieloma múltiplo recidivado ou refratário em um estudo de fase 1/2 de braço único, aberto, multicêntrico (MajesTEC-1). O estudo incluiu pacientes que receberam pelo menos três terapias anteriores, incluindo um inibidor de proteassoma, um agente imunomodulador e um anticorpo monoclonal anti-CD38. O estudo excluiu pacientes que sofreram AVC ou convulsão nos últimos 6 meses e pacientes com pontuação de desempenho do Eastern Cooperative Oncology Group (ECOG PS) ≥2, leucemia de células plasmáticas, envolvimento ativo do SNC conhecido ou sinais clínicos de envolvimento meníngeo de mieloma múltiplo, ou história ativa ou documentada de doença autoimune com exceção de vitiligo, diabetes tipo 1 e tireoidite autoimune prévia.
Os pacientes receberam doses iniciais de 0,06 mg/kg e 0,3 mg/kg de TecvayliTM administradas por via subcutânea, seguidas pela dose de tratamento de TecvayliTM de 1,5 mg/kg, administradas por via subcutânea uma vez por semana a partir daí, e até a progressão da doença ou toxicidade inaceitável (ver item 8. Posologia e modo de usar). A duração mediana entre a dose para escalonamento 1 e a dose para escalonamento 2 foi de 2,9 (intervalo: 2-7) dias. A duração mediana entre a dose para escalonamento 2 e a dose de tratamento inicial foi de 3,1 (intervalo: 2-9) dias. Os pacientes foram hospitalizados para monitoramento por pelo menos 48 horas após a administração de cada dose do cronograma de doses para escalonamento de TecvayliTM.
A população de eficácia incluiu 165 pacientes. A idade mediana foi de 64 anos (intervalo: 33-84) com 15% dos indivíduos ≥75 anos de idade; 58% eram do sexo masculino; 81% eram brancos, 13% eram negros, 2% eram asiáticos. O International Staging System (ISS) no início do estudo era de 52% no Estágio I, 35% no Estágio II e 12% no Estágio III. Citogenética de alto risco (presença de del(17p), t(4;14) ou t(14;16)) estava presente em 26% dos pacientes. Dezessete por cento dos pacientes apresentavam plasmocitomas extramedulares.
A mediana de tempo desde o diagnóstico inicial de mieloma múltiplo até a inscrição foi de 6 (intervalo: 0,8-22,7) anos. O número mediano de terapias anteriores foi 5 (intervalo: 2-14), com 23% dos pacientes que receberam 3 terapias anteriores. Oitenta e dois por cento dos pacientes receberam previamente transplante autólogo de células-tronco e 4,8% dos pacientes receberam transplante alogênico prévio. Setenta e oito por cento dos pacientes eram triplo-refratários (refratário ao inibidor de proteassoma, a um agente imunomodulador e a um anticorpo monoclonal anti-CD38).
Os resultados de eficácia foram baseados na taxa de resposta global, conforme determinado pela avaliação do Comitê de Revisão Independente (IRC), usando os critérios do International Myeloma Working Group (IMWG) 2016 (ver Tabela 1).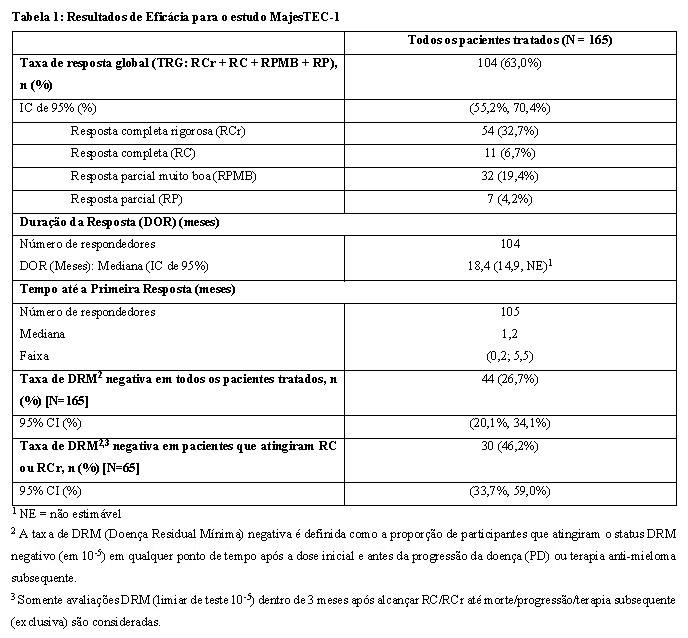
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
O teclistamabe é um anticorpo biespecífico IgG4-PAA completo que tem como alvo o receptor CD3, expresso na superfície de células T e o antígeno de maturação de células B (BCMA), que é expresso na superfície das células da linhagem B de mieloma múltiplo malignas, bem como células B em estágio avançado e células plasmáticas. Com os seus duplos sítios de ligação, o teclistamabe é capaz de fazer com que células T CD3+ fiquem em estreita proximidade com células BCMA+, resultando em ativação de células T e subsequentes lise e morte de células BCMA+, que é mediado pela secreção de perforina e várias granzimas armazenadas nas vesículas secretórias de células T citotóxicas. Esse efeito ocorre sem consideração da especificidade de receptores de células T ou dependência de moléculas do complexo principal de histocompatibilidade (MHC) de Classe I na superfície das células apresentadoras de antígenos.
Efeitos farmacodinâmicos
Dentro do primeiro mês de tratamento com teclistamabe, foram observadas ativação e redistribuição de células T, redução de células B e indução de citocinas séricas. Dentro de um mês, a maioria dos respondedores apresentou redução no BCMA solúvel e uma maior redução no BCMA solúvel foi observada em pacientes com respostas mais profundas ao teclistamabe.
Imunogenicidade
A incidência observada de anticorpos antidrogas é altamente dependente da sensibilidade e especificidade do ensaio. As diferenças nos métodos de ensaio impedem comparações significativas da incidência de anticorpos antidrogas nos estudos descritos abaixo com a incidência de anticorpos antidrogas em outros estudos, incluindo os de teclistamabe ou de outros produtos com teclistamabe. Durante o acompanhamento do estudo MajesTEC-1 (27 meses até o momento), 1/186 (0,5%) dos pacientes tratados com TecvayliTM subcutâneo em várias dosagens desenvolveu anticorpos anti-teclistamabe. Devido à baixa ocorrência de anticorpos antidrogas, o efeito desses anticorpos na farmacocinética, farmacodinâmica, segurança e/ou eficácia dos produtos de teclistamabe é desconhecido.
Efeito sobre o intervalo QT/QTc e a eletrofisiologia cardíaca
Com a dose de tratamento recomendada (1,5 mg/kg) de TecvayliTM, não foi observado nenhum prolongamento do QTc clinicamente relevante.
Propriedades Farmacocinéticas
A Cmax e ASCtau de teclistamabe após a primeira dose de tratamento subcutâneo aumentam proporcionalmente ao longo de um intervalo de dosagem de 0,08 mg/kg a 3 mg/kg (0,05 a 2 vezes a dose de tratamento recomendada aprovada). Noventa por cento da exposição no estado estacionário foi alcançada após 12 doses semanais de tratamento. A taxa média de acumulação entre a primeira e a 13ª dose semanal de tratamento de teclistamabe 1,5 mg/kg foi de 4,2 vezes para Cmax, 4,1 vezes para Ctrough e 5,3 vezes para ASCtau.
Cmax, Ctrough e ASCtau de teclistamabe são apresentados na Tabela 2.
Absorção
A biodisponibilidade média de teclistamabe foi de 72% quando administrado por via subcutânea. A mediana (faixa) de Tmax de teclistamabe após a primeira e 13ª doses de tratamento foi de 139 (19 a 168) horas e 72 (24 a 168) horas, respectivamente.
Distribuição
O volume médio (coeficiente de variação [CV]%) de distribuição de teclistamabe foi de 5,63 L (29%).
Eliminação
A depuração de teclistamabe diminui ao longo do tempo, com uma redução média (CV%) máxima desde o início até à 13ª dose de tratamento de 40,8% (56%). A depuração média geométrica (CV%) é de 0,472 L/dia (64%) na 13ª dose de tratamento. Espera-se que os pacientes que descontinuam o teclistamabe após a 13ª dose de tratamento tenham uma redução de 50% da Cmax na concentração de teclistamabe em um tempo mediano (5° a 95° percentil) de 15 (7 a 33) dias após Tmax e uma redução de 97% da Cmax em concentração de teclistamabe em um tempo mediano de 69 (32 a 163) dias após o Tmax.
Populações específicas
O volume de distribuição e a depuração do teclistamabe aumentam com o aumento do peso corporal (41 kg a 139 kg).
Não houve diferenças clinicamente significativas na farmacocinética do teclistamabe com base na idade (24 a 84 anos), sexo, raça (branca, negra ou afro-americana), etnia (hispânica/latina, não hispânica/latina), insuficiência renal leve ou moderada (taxa de filtração glomerular estimada [eGFR] pelo método de Modificação da Dieta na Doença Renal [MDRD]: 30 a 89 mL/min), ou insuficiência hepática leve (bilirrubina total menor ou igual ao limite superior do normal [LSN] com AST superior ao LSN ou bilirrubina total superior a 1 a 1,5 vezes o LSN com qualquer AST). Os efeitos da insuficiência renal grave (eGFR inferior a 30 mL/min) ou insuficiência hepática moderada a grave (bilirrubina total superior a 1,5 vezes o LSN com qualquer AST) na farmacocinética de teclistamabe são desconhecidos.
Estudos de interação medicamentosa
Não foram conduzidos estudos clínicos avaliando o potencial de interação medicamentosa do teclistamabe.
Informações Não Clínicas
Com base na expressão de BCMA, o teclistamabe se direciona especificamente a células BCMA+, dessa maneira reduz potenciais efeitos em outras linhagens celulares.
Carcinogenicidade e Mutagenicidade
Não foi realizado nenhum estudo de genotoxicidade ou carcinogenicidade para avaliar o potencial carcinogênico ou genotóxico do teclistamabe.
Toxicologia Reprodutiva
Não foi conduzido nenhum estudo de toxicidade reprodutiva e de desenvolvimento em animais para avaliar os potenciais efeitos do teclistamabe.
Fertilidade
Não foram conduzidos estudos para avaliar os efeitos do teclistamabe sobre a fertilidade em machos ou fêmeas. No estudo de toxicidade de doses repetidas de 5 semanas em macacos cynomolgus, não houve nenhum efeito notável nos órgãos reprodutores de machos e fêmeas com doses de até 30 mg/kg/semana (aproximadamente 22 vezes a dose máxima recomendada em humanos com base na exposição AUC) por via intravenosa por cinco semanas.
4. CONTRAINDICAÇÕES
O Tecvayli™ é contraindicado em pacientes com hipersensibilidade conhecida ao teclistamabe ou a qualquer um dos componentes na formulação.
5. ADVERTÊNCIAS E PRECAUÇÕES
Os dados descritos nas Advertências e Precauções refletem o perfil de segurança de 165 pacientes com mieloma múltiplo recidivado ou refratário que receberam o regime de dose recomendado de monoterapia com TecvayliTM por via subcutânea no estudo MajesTEC-1.
Síndrome de liberação de citocinas (SLC)
TecvayliTM pode causar síndrome de liberação de citocinas (SLC), incluindo reações com risco de vida ou fatais (ver item 9. Reações Adversas). No estudo clínico, a SLC ocorreu em 72% dos pacientes que receberam TecvayliTM na dose recomendada, com SLC de Grau 1 ocorrendo em 50% dos pacientes, Grau 2 em 21% e Grau 3 em 0,6%. A SLC recorrente ocorreu em 33% dos pacientes. A maioria dos pacientes apresentou SLC após a dose para escalonamento 1 (42%), dose para escalonamento 2 (35%) ou a dose inicial de tratamento (24%). Menos de 3% dos pacientes desenvolveram a primeira ocorrência de SLC após doses subsequentes de TecvayliTM. O tempo mediano até ao início da SLC foi de 2 (intervalo: 1 a 6) dias após a dose mais recente com uma duração mediana de 2 (intervalo: 1 a 9) dias. Os sinais e sintomas clínicos da SLC incluíram, mas não se limitaram a, febre, hipóxia, calafrios, hipotensão, taquicardia sinusal, dor de cabeça e elevação das enzimas hepáticas (elevação de aspartato aminotransferase e alanina aminotransferase).
Inicie a terapia de acordo com o cronograma de doses para escalonamento de TecvayliTM para reduzir o risco de SLC (ver item 8. Posologia e modo de usar). Administre medicamentos pré-tratamento para reduzir o risco da SLC e monitore os pacientes após a administração de TecvayliTM (ver item 8. Posologia e modo de usar). Ao primeiro sinal da SLC, avalie imediatamente o paciente para internação. Administre cuidados de suporte com base na gravidade e considere o manejo adicional de acordo com as diretrizes práticas atuais. Suspender ou descontinuar permanentemente TecvayliTM com base na gravidade (ver item 8. Posologia e modo de usar).
Toxicidades neurológicas
TecvayliTM pode causar toxicidade neurológica grave ou com risco de vida, incluindo Síndrome de Neurotoxicidade Associada a Células Efetoras Imunes (ICANS) (ver item 9. Reações adversas). No estudo clínico, ocorreu toxicidade neurológica em 57% dos pacientes que receberam TecvayliTM na dose recomendada, com toxicidade neurológica de Grau 3 ou 4 ocorrendo em 2,4% dos pacientes. As toxicidades neurológicas mais frequentes foram cefaleia (25%), disfunção motora (16%), neuropatia sensorial (15%) e encefalopatia (13%). Com acompanhamento mais longo, convulsões de Grau 4 e síndrome de Guillain-Barré fatal (um paciente cada) ocorreram em pacientes que receberam TecvayliTM. No estudo clínico, ICANS foi relatado em 6% dos pacientes que receberam TecvayliTM na dose recomendada (ver item 9. Reações adversas). ICANS recorrente ocorreu em 1,8% dos pacientes. A maioria dos pacientes experimentou ICANS após a dose para escalonamento 1 (1,2%), dose para escalonamento 2 (0,6%) ou a dose inicial do tratamento (1,8%). Menos de 3% dos pacientes desenvolveram a primeira ocorrência de ICANS após doses subsequentes de TecvayliTM. A mediana de tempo para o início do ICANS foi de 4 (intervalo: 2 a 8) dias após a dose mais recente, com uma duração mediana de 3 (intervalo: 1 a 20) dias. As manifestações clínicas mais frequentes da ICANS relatadas foram estado de confusão e disgrafia. O início da ICANS pode ser concomitante com a SLC, após a resolução da SLC ou na ausência da SLC. Monitore os pacientes quanto a sinais e sintomas de toxicidade neurológica durante o tratamento. Ao primeiro sinal de toxicidade neurológica, incluindo ICANS, avalie imediatamente o paciente e forneça terapia de suporte com base na gravidade. Suspender ou descontinuar permanentemente TecvayliTM com base na gravidade de acordo com as recomendações e considerar o manejo adicional de acordo com as diretrizes práticas atuais (ver item 8. Posologia e modo de usar). Devido ao potencial de toxicidade neurológica, os pacientes que recebem TecvayliTM correm o risco de depressão do nível de consciência (ver item 9. Reações Adversas). Aconselhe os pacientes a se absterem de dirigir ou operar máquinas pesadas ou potencialmente perigosas durante e por 48 horas após a conclusão do cronograma de doses para escalonamento de TecvayliTM e no caso de aparecimento de qualquer sintoma de toxicidade neurológica até a resolução da toxicidade neurológica (ver item 8. Posologia e modo de usar).
Hepatotoxicidade
TecvayliTM pode causar hepatotoxicidade, incluindo fatalidades. Em pacientes que receberam TecvayliTM na dose recomendada no estudo clínico, houve um caso fatal de insuficiência hepática. Aspartato aminotransferase (AST) elevada ocorreu em 34% dos pacientes, com elevações de Grau 3 ou 4 em 1,2%. Alanina aminotransferase (ALT) elevada ocorreu em 28% dos pacientes, com elevações de Grau 3 ou 4 em 1,8%. Ocorreu aumento da bilirrubina total em 6% dos pacientes com elevações de Grau 3 ou 4 em 0,6%. A elevação das enzimas hepáticas pode concomitante ou não à SLC. Monitore as enzimas hepáticas e a bilirrubina no início e durante o tratamento conforme clinicamente indicado. Suspenda TecvayliTM ou considere a descontinuação permanente de TecvayliTM com base na gravidade (ver item 8. Posologia e Modo de Usar).
Infecções
TecvayliTM pode causar infecções graves, com risco de vida ou fatais. Em pacientes que receberam TecvayliTM na dose recomendada no ensaio clínico, infecções graves, incluindo infecções oportunistas, ocorreram em 30% dos pacientes, com infecções de Grau 3 ou 4 em 35%, e infecções fatais em 4,2% (ver item 9. Reações Adversas). Monitore os pacientes quanto a sinais e sintomas de infecção antes e durante o tratamento com TecvayliTM e trate adequadamente. Administre antimicrobianos profiláticos de acordo com as diretrizes locais (ver item 8. Posologia e Modo de Usar). Suspenda TecvayliTM ou considere a descontinuação permanente de TecvayliTM com base na gravidade (ver item 8. Posologia e Modo de Usar). Monitore os níveis de imunoglobulina durante o tratamento com TecvayliTM e trate de acordo com diretrizes, incluindo precauções contra infecções e profilaxia antibiótica ou antiviral (ver item 8. Posologia e Modo de Usar).
Reativação do Vírus da Hepatite B
Reativação do vírus da hepatite B pode ocorrer em pacientes tratados com medicamentos dirigidos contra células B e, em alguns casos, pode resultar em hepatite fulminante, insuficiência hepática e óbito.
Pacientes com evidências de sorologia positiva para HBV devem ser monitorados quanto a sinais clínicos e laboratoriais de reativação do HBV enquanto estiverem recebendo TecvayliTM e por pelo menos seis meses após o final do tratamento.
Em pacientes que desenvolverem reativação do HBV enquanto estiverem recebendo TecvayliTM, suspenda o tratamento com TecvayliTM conforme indicado na tabela 4 e maneje de acordo com as diretrizes institucionais locais (ver item 8. Posologia e modo de usar - Modificações posológicas).
Hipogamaglobulinemia
Hipogamaglobulinemia foi relatada em pacientes recebendo TecvayliTM (ver item 9. Reações Adversas). Monitore os níveis de imunoglobulinas durante o tratamento com TecvayliTM e trate de acordo com as diretrizes institucionais locais, incluindo precauções para infeção, profilaxia com antibiótico ou antiviral.
Vacinas
A resposta imunológica a vacinas pode ser reduzida enquanto o TecvayliTM estiver sendo administrado. A segurança da imunização com vacinas virais vivas durante ou após o tratamento com TecvayliTM não foi estudada. A administração de vacinas de vírus vivos não é recomendada por pelo menos 4 semanas antes do início do tratamento, durante o tratamento e pelo menos 4 semanas após o tratamento.
Neutropenia
TecvayliTM pode causar neutropenia e neutropenia febril. Em pacientes que receberam TecvayliTM na dose recomendada no ensaio clínico, ocorreu redução na contagem de neutrófilos em 84% dos pacientes, com ocorrências de Grau 3 ou 4 em 56%. Neutropenia febril ocorreu em 3% dos pacientes (ver item 9. Reações Adversas). Monitore contagens de todas as células sanguíneas no início e periodicamente durante o tratamento e forneça cuidados de suporte de acordo com as diretrizes institucionais locais. Monitore pacientes com neutropenia quanto a sinais de infecção. Suspenda TecvayliTM com base na gravidade (ver item 8. Posologia e Modo de Usar).
Hipersensibilidade e outras reações da administração
TecvayliTM pode causar reações sistêmicas relacionadas à administração e reações locais no local da injeção.
Reações Sistêmicas
Em pacientes que receberam TecvayliTM na dose recomendada no estudo clínico, 1,2% apresentaram reações sistêmicas relacionadas à administração, que incluíram pirexia recorrente de Grau 1 e língua inchada de Grau 1.
Reações Locais
Em pacientes que receberam TecvayliTM na dose recomendada no estudo clínico, reações no local da injeção ocorreram em 35% dos pacientes, com reações de Grau 1 no local da injeção em 30% e Grau 2 em 4,8%. Suspenda TecvayliTM ou considere a descontinuação permanente de TecvayliTM com base na gravidade (ver item 8. Posologia e Modo de Usar).
Efeitos sobre a capacidade de dirigir e operar máquinas
Em razão do potencial para toxicidade neurológica, pacientes que recebem TecvayliTM estão em risco de nível diminuído de consciência. Os pacientes devem evitar dirigir ou operar máquinas pesadas ou potencialmente perigosas durante e por 48 horas depois de concluir o cronograma de doses para escalonamento de TecvayliTM e no caso de novo aparecimento de quaisquer sintomas neurológicos até a resolução da toxicidade neurológica (tabela 3) (ver item 8. Posologia e modo de usar).
Gravidez, amamentação e fertilidade
Gravidez (Categoria C)
Não há dados disponíveis sobre o uso de TecvayliTM em mulheres grávidas ou dados em animais para avaliar o risco do TecvayliTM na gravidez. Sabe-se que a IgG humana atravessa a placenta após o primeiro trimestre de gravidez. Portanto, o teclistamabe tem o potencial para ser transmitido da mãe para o feto em desenvolvimento. O TecvayliTM não é recomendado para mulheres que estiverem grávidas. O TecvayliTM está associado a hipogamaglobulinemia; portanto, deve ser considerada a avaliação dos níveis de imunoglobulinas em recém-nascidos de mães tratadas com TecvayliTM.
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Amamentação
Não se sabe se o teclistamabe é excretado no leite humano ou animal, se afeta bebês amamentados ou afeta a produção de leite. Em razão do potencial para reações adversas graves do TecvayliTM em bebês amamentados, aconselhe as pacientes a não amamentar durante o tratamento com TecvayliTM e por pelo menos cinco meses após a última dose.
Mulheres e homens com potencial reprodutivo
Exames de gravidez
No caso de mulheres com potencial para engravidar deve-se verificar se estão grávidas antes do início do tratamento com TecvayliTM.
Contracepção
Aconselhe mulheres com potencial reprodutivo a usar um método de contracepção eficaz durante o tratamento e por cinco meses após a dose final de TecvayliTM. Aconselhe pacientes do sexo masculino com uma parceira do sexo feminino com potencial reprodutivo a usar um método de contracepção eficaz durante o tratamento e por três meses após a última dose de TecvayliTM.
Fertilidade Não há dados sobre o efeito do TecvayliTM sobre a fertilidade. Os efeitos do TecvayliTM sobre a fertilidade de machos e fêmeas não foram avaliados em estudos em animais.
Atenção: Este medicamento contém açúcar, portanto, deve ser usado com cautela em pessoas com diabetes.
6. INTERAÇÕES MEDICAMENTOSAS
Não foi realizado nenhum estudo de interação medicamentosa com o TecvayliTM. A liberação inicial de citocinas associada ao início do tratamento com TecvayliTM pode suprimir enzimas do CYP450. Com base na modelagem farmacocinética de base fisiológica (PBPK), prevê-se que o maior risco de interação medicamentosa seja a partir do início do cronograma de dose de escalonamento de TecvayliTM até 7 dias após a primeira dose de tratamento ou durante um evento de SLC. Durante este período de tempo, monitore quanto a toxicidade ou monitore as concentrações do medicamento (p. ex., ciclosporina) em pacientes que estão recebendo concomitantemente substratos da CYP450 com índice terapêutico estreito. A dose do medicamento concomitante deve ser ajustada conforme necessário.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
O TecvayliTM deve ser armazenado sob refrigeração de 2°C a 8°C. Armazene na embalagem original a fim de proteger da luz. Não congele. Mantenha fora da vista e do alcance de crianças. O prazo de validade do TecvayliTM é de 18 meses desde a data da sua fabricação.
Aspecto físico
O TecvayliTM é uma solução para injeção livre de conservantes incolor a amarelo-clara.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use o medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
Posologia - Adultos (maiores de 18 anos de idade)
O TecvayliTM deve ser administrado somente por injeção subcutânea.
Administre medicamentos pré-tratamento antes de cada dose do cronograma de doses para escalonamento de TecvayliTM (ver item
8. Posologia e modo de usar - Medicamentos pré-tratamento).
Cronograma de doses para escalonamento
Administre o TecvayliTM de acordo com o cronograma de doses para escalonamento na tabela 3 para reduzir a incidência e a severidade de síndrome de liberação de citocinas (SLC). Devido ao risco da SLC e toxicidade neurológica, incluindo ICANS, recomenda-se que os pacientes sejam hospitalizados por 48 horas após administração de todas as doses dentro do cronograma de doses de escalonamento de TecvayliTM (ver item 8. Posologia e Modo de Usar e 5. Advertências e Precauções). O não cumprimento das doses ou esquema posológico recomendados para o início da terapia ou o reinício da terapia após atrasos de dose pode resultar em aumento da frequência e severidade de eventos adversos relacionados ao mecanismo de ação, particularmente síndrome de liberação de citocinas (ver item 8. Posologia e modo de usar -Modificações posológicas e ver item
5. Advertências e Precauções - Síndrome de Liberação de Citocinas).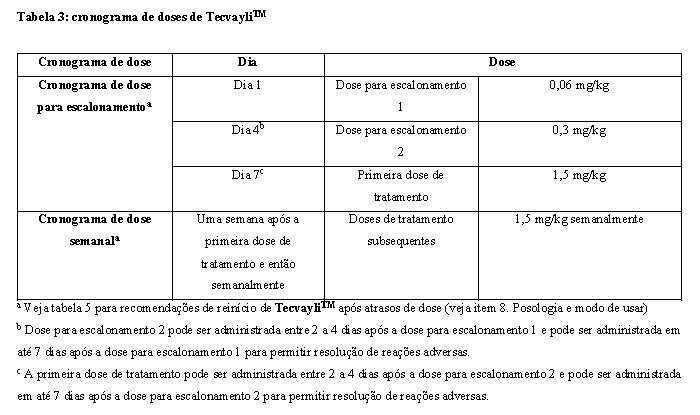
Posologia recomendada
A posologia recomendada de TecvayliTM consiste em uma dose de 1,5 mg/kg de peso corporal real administrada uma vez por semana após a conclusão do cronograma de doses para escalonamento (veja a tabela 3). O TecvayliTM deve ser continuado até a de progressão da doença ou toxicidade inaceitável. Para orientações em relação ao reinício da terapia com TecvayliTM após atrasos de dose (ver item 8. Posologia e modo de usar - Reinício do TecvayliTM após atrasos de dose).
Medicamentos pré-tratamento
Administre os medicamentos pré-tratamento a seguir de 1 a 3 horas antes de cada dose do cronograma de doses para escalonamento de TecvayliTM para reduzir o risco de síndrome de liberação de citocinas (ver item 5. Advertências e Precauções -Síndrome de Liberação de Citocinas e ver item 9. Reações Adversas).
• Corticosteroide (dexametasona oral ou intravenosa, 16 mg)
• Anti-histamínico (difenidramina oral ou intravenosa, 50 mg, ou equivalente)
• Antipiréticos (acetaminofeno oral ou intravenoso, 650 mg a 1000 mg, ou equivalente) A administração dos medicamentos pré-tratamento pode ser necessária antes da administração das doses subsequentes de TecvayliTM nos seguintes pacientes:
• Pacientes que repetem as doses do cronograma de escalonamento de doses de TecvayliTM após um atraso de dose (ver item 8. Posologia e modo de usar).
• Pacientes que apresentaram SLC após a dose prévia de TecvayliTM (ver item 8. Posologia e modo de usar).
Profilaxia para reativação do vírus do herpes-zóster
Antes do início do tratamento com TecvayliTM, deve ser considerada a profilaxia com antiviral para a prevenção de reativação do vírus do herpes-zóster, de acordo com as diretrizes institucionais locais.
Modificações posológicas
Não pule doses para escalonamento de TecvayliTM.
Reduções de dose de TecvayliTM não são recomendadas.
Atrasos de dose podem ser necessários para manejar toxicidades relacionadas ao TecvayliTM (ver item 5. Advertências e Precauções).
Veja a tabela 4 para as medidas recomendadas para reações adversas após a administração de TecvayliTM.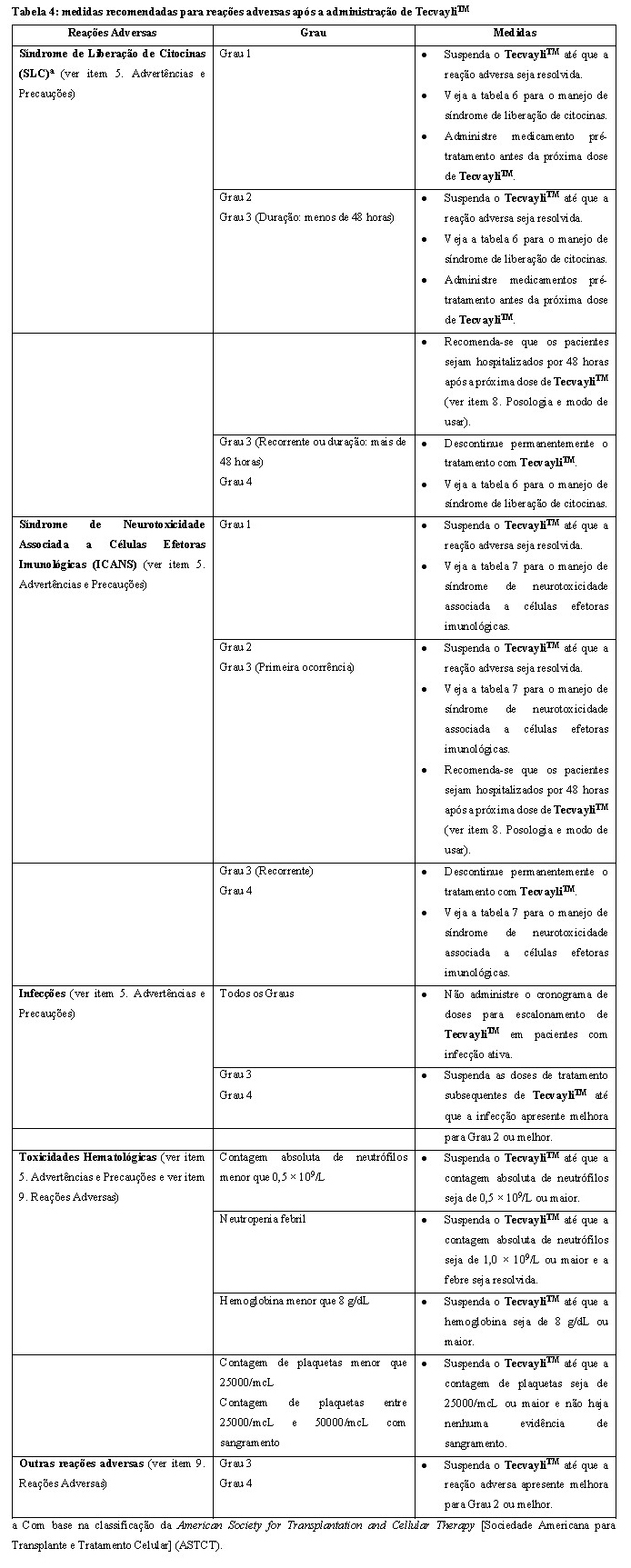
Reinício do TecvayliTM após atrasos de dose
Se uma dose de TecvayliTM for atrasada, reinicie a terapia com base nas recomendações listadas na tabela 5 e retome o cronograma de tratamento apropriadamente (ver item 8. Posologia e modo de usar -Posologia - Adultos (maiores de 18 anos de idade)). Administre medicamentos pré-tratamento conforme indicado. Devido ao risco da SLC e toxicidade neurológica, incluindo ICANS, recomenda-se que os pacientes sejam hospitalizados por 48 horas após a administração de todas as doses dentro do cronograma de doses para escalonamento de TecvayliTM (ver item 8. Posologia e modo de usar e 5. Advertências e Precauções).
Manejo de reações adversas severas Síndrome de liberação de citocinas (SLC)
As recomendações do protocolo para o manejo e prevenção da SLC no estudo MajesTEC-1 incluíram o uso de tocilizumabe e corticosteroides com base na gravidade dos sintomas. No estudo MajesTEC-1, a maioria dos eventos de SLC foi de Grau 1 (50%, 83/165) e Grau 2 (21%, 35/165). Menos de um por cento (0,6%, 1/165) dos eventos de SLC foram de Grau 3 e não ocorreram eventos de Grau 4 ou fatais. Um terço (33%, 55/165) dos pacientes apresentou mais de um evento de SLC. Nenhum paciente necessitou de descontinuação do tratamento para SLC, todos os eventos foram resolvidos. Tocilizumabe, corticosteroides e tocilizumabe em combinação com corticosteroides foram usados para tratar 32%, 11% e 3% dos eventos de SLC, respectivamente. Quarenta e cinco pacientes receberam tocilizumabe para seu primeiro evento de SLC, dos quais 9 pacientes (20%) tiveram um evento de SLC subsequente. Para os pacientes que não receberam tocilizumabe em seu primeiro evento, a recorrência da SLC ocorreu em 46 de 74 pacientes (62,2%). Com base nesses dados, o tocilizumabe pode ser usado para controlar a SLC conforme descrito na Tabela 6.
Identifique a SLC com base na apresentação clínica (ver item 5. Advertências e Precauções -Síndrome de Liberação de Citocinas). Avalie e trate outras causas de febre, hipóxia e hipotensão. Se houver suspeita de SLC, suspenda o TecvayliTM até que a reação adversa seja resolvida (veja a tabela 4) e maneje de acordo com as recomendações na tabela 6. Administre cuidados de suporte para SLC (incluindo, entre outros, agentes antipiréticos, suporte com líquidos intravenosos, vasopressores, oxigênio suplementar, etc.), conforme apropriado. Considere exames laboratoriais para monitoramento quanto a coagulação intravascular disseminada (DIC), parâmetros de hematologia, bem como funções pulmonar, cardíaca, renal e hepática.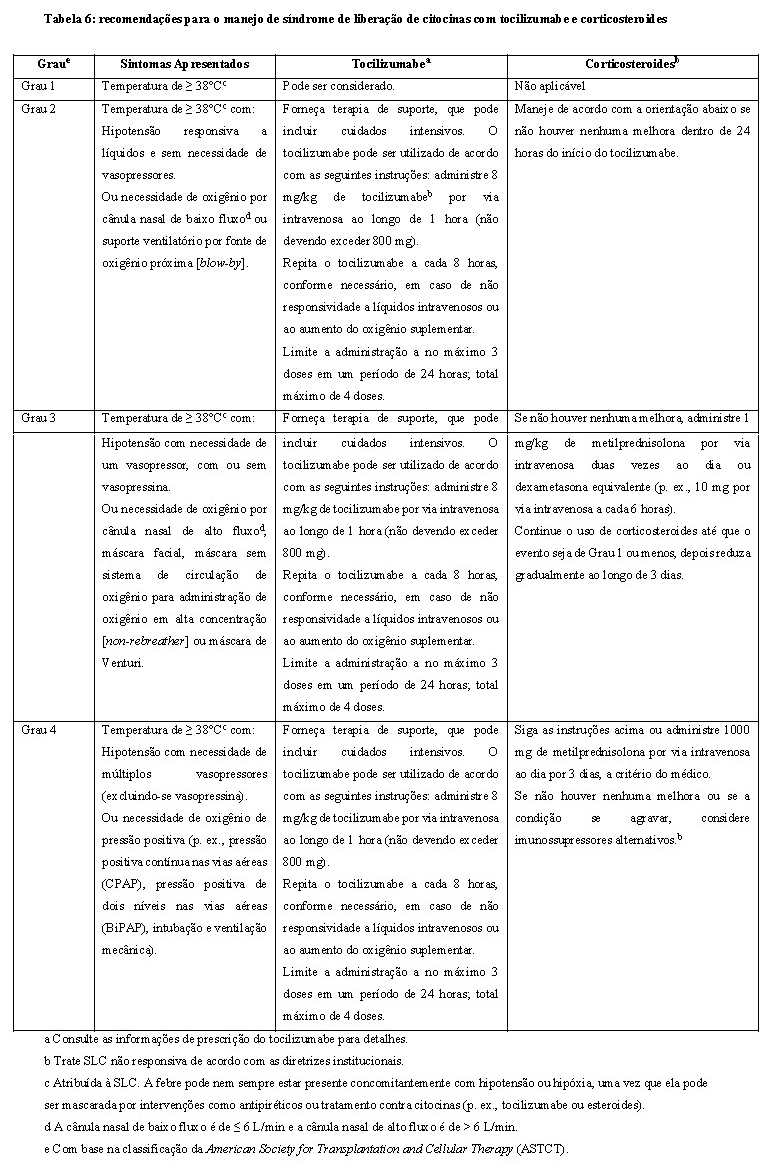
Toxicidades neurológicas
O manejo geral para toxicidade neurológica (p. ex., Síndrome de Neurotoxicidade Associada a Células Efetoras Imunológicas (ICANS), concomitante ou não à SLC) é resumido na tabela 7. Ao primeiro sinal de toxicidade neurológica, incluindo ICANS, considere uma avaliação neurológica. Descarte outras causas de sintomas neurológicos. Forneça cuidados intensivos e tratamento de suporte para toxicidades neurológicas severas ou de risco à vida (ver item 5. Advertências e Precauções - Toxicidades neurológicas). Suspenda o TecvayliTM conforme indicado na tabela 4.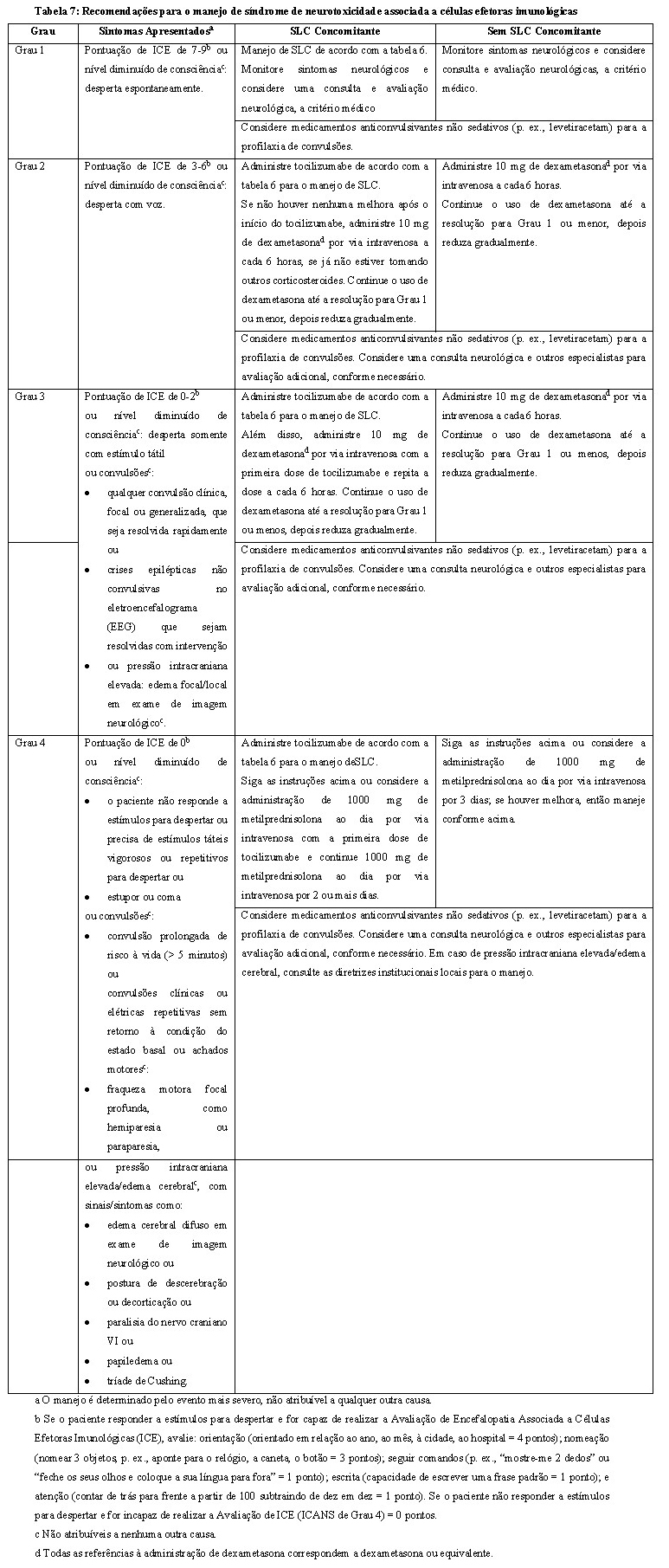
Populações especiais
Pacientes pediátricos (pacientes com até 17 anos de idade) A segurança e a eficácia do TecvayliTM não foram estabelecidas em pacientes pediátricos com até 17 anos de idade. Não está disponível nenhum dado.
Idosos (acima de 65 anos de idade)
Dos 165 pacientes tratados com TecvayliTM no estudo MajesTEC-1 com a dose recomendada, 48% tinham 65 anos de idade ou mais e 15% tinham 75 anos de idade ou mais. Não foi observada nenhuma diferença global na segurança ou na efetividade entre esses pacientes e os mais jovens. Não é necessário nenhum ajuste de dose (ver item 3. Características Farmacológicas -Propriedades Farmacocinéticas).
Insuficiência renal
Não foram conduzidos estudos formais com TecvayliTM em pacientes com insuficiência renal. Com base em análises farmacocinéticas populacionais, não é recomendado nenhum ajuste de dose para pacientes com insuficiência renal leve ou moderada (ver item 3. Características Farmacológicas -Propriedades Farmacocinéticas).
Insuficiência hepática
Não foram conduzidos estudos formais com TecvayliTM em pacientes com insuficiência hepática. Com base em análises farmacocinéticas populacionais, não é recomendado nenhum ajuste de dose para pacientes com insuficiência hepática leve (ver item 3. Características Farmacológicas -Propriedades Farmacocinéticas).
Administração
É muito importante que as instruções para a preparação e a administração fornecidas nesta seção sejam seguidas estritamente para minimizar potenciais erros de administração com o frasco-ampola de 10 mg/mL de TecvayliTM e o frasco-ampola de 90 mg/mL de TecvayliTM.
TecvayliTM deve ser administrado somente via injeção subcutânea. Não administre TecvayliTM por via intravenosa. TecvayliTM deve ser administrado por um profissional de saúde com equipe médica adequadamente treinada e equipamentos médicos apropriados para manejar reações severas, incluindo síndrome de liberação de citocinas (ver item 5. Advertências e Precauções -Síndrome de Liberação de Citocinas). Esta administração deve ocorrer em ambiente hospitalar, ou seja, em hospitais, ambulatórios ou clínicas médicas. O frasco-ampola de 10 mg/mL de TecvayliTM e o frasco-ampola de 90 mg/mL de TecvayliTM são apresentados na forma de solução para injeção pronta para uso sem necessidade de diluição antes da administração. Frascos-ampola de TecvayliTM de diferentes concentrações não devem ser combinados para obter a dose de tratamento. Use técnica asséptica para preparar e administrar TecvayliTM.
Preparação do TecvayliTM
• Verifique a dose prescrita para cada injeção de TecvayliTM. Para minimizar erros, use as tabelas a seguir para preparar a injeção de TecvayliTM.
- Use a tabela 8 para determinar a dose total, o volume de injeção e o número de frascos-ampola necessários com base no peso corporal real do paciente para a dose para escalonamento 1 usando TecvayliTM 10 mg/mL.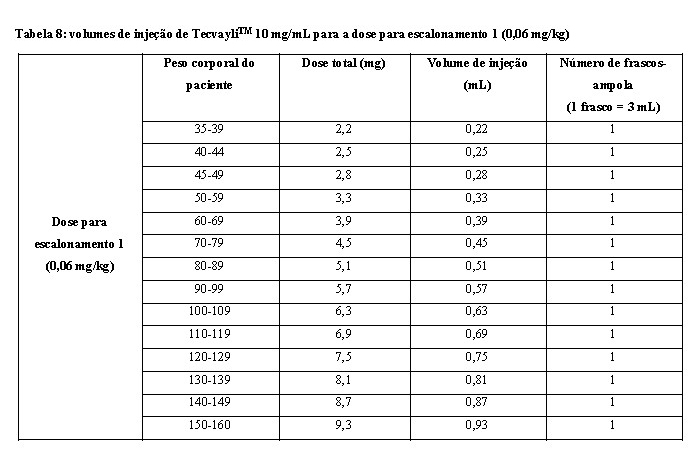
- Use a tabela 9 para determinar a dose total, o volume de injeção e o número de frascos-ampola necessários com base no peso corporal real do paciente para a dose para escalonamento 2 usando TecvayliTM 10 mg/mL.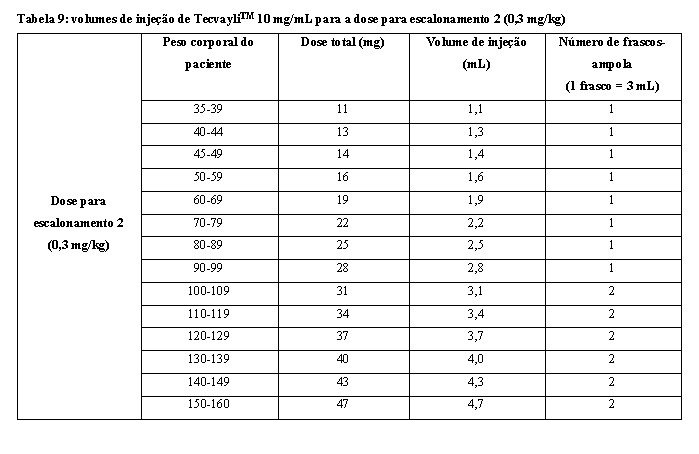
- Use a tabela 10 para determinar a dose total, o volume de injeção e o número de frascos-ampola necessários com base no peso corporal real do paciente para a dose de tratamento usando TecvayliTM 90 mg/mL.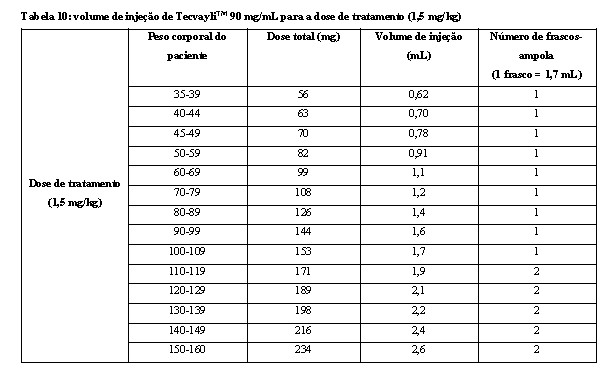
• Remova o frasco-ampola de TecvayliTM de concentração apropriada do armazenamento refrigerado (2°C-8°C) e equilibre à temperatura ambiente (15°C-30°C), conforme necessário, por pelo menos 15 minutos. Não aqueça TecvayliTM de nenhuma outra maneira.
• Uma vez equilibrado, gire delicadamente o frasco-ampola por aproximadamente 10 segundos para misturar. Não agite.
• Retire o volume de injeção necessário de TecvayliTM do(s) frasco(s)-ampola para uma seringa de tamanho apropriado usando uma agulha de transferência.
-Cada volume de injeção não deve exceder 2,0 mL. Divida doses que precisarem de mais de 2,0 mL igualmente em múltiplas seringas.
• TecvayliTM é compatível com agulhas de injeção de aço inoxidável e com material de seringas de polipropileno ou policarbonato.
• Substitua a agulha de transferência por uma agulha de tamanho apropriado para a injeção.
• Inspecione visualmente o TecvayliTM quanto a material particulado e descoloração antes da administração. Não use se a solução estiver descolorida, ou turva, ou se partículas estranhas estiverem presentes.
• TecvayliTM solução para injeção é incolor a amarelo-claro.
Administração de TecvayliTM
• Injete o volume necessário de TecvayliTM no tecido subcutâneo do abdome (local da injeção preferencial). Alternativamente, TecvayliTM pode ser injetado no tecido subcutâneo em outros locais (p. ex., coxa). Se múltiplas injeções forem necessárias, as injeções de TecvayliTM devem ser realizadas a uma distância de pelo menos 2 cm uma da outra.
• Não injete em tatuagens ou cicatrizes ou em áreas em que a pele estiver vermelha, machucada, sensível, dura ou não intacta.
• Qualquer produto medicinal não usado ou material residual deve ser descartado de acordo com as exigências locais.
Armazenamento
• Se TecvayliTM não for usado imediatamente, armazene a 2-8°C ou em temperatura ambiente por no máximo 20 horas. Descarte após 20 horas, se não for usado.
Monitoramento
• Devido ao risco de SLC e toxicidade neurológica, incluindo ICANS, recomenda-se que os pacientes sejam hospitalizados por 48 horas após a administração de todas as doses do cronograma de escalonamento de doses de TecvayliTM (ver item
8. Posologia e modo de usar e 5. Advertências e Precauções).
9. REAÇÕES ADVERSAS
Como os ensaios clínicos são conduzidos em condições muito variadas, as taxas de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser comparadas diretamente com as taxas nos ensaios clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
Mieloma Múltiplo Recidivado/Refratário
MajesTEC-1
A segurança de TecvayliTM foi avaliada no estudo MajesTEC-1 (ver item 2. Resultados de Eficácia), que incluiu pacientes adultos com mieloma múltiplo recidivado ou refratário. Os pacientes receberam doses para escalonamento de 0,06 mg/kg e 0,3 mg/kg de TecvayliTM seguido de TecvayliTM 1,5 mg/kg, por via subcutânea uma vez por semana (N=165). Entre os pacientes que receberam TecvayliTM, 47% foram expostos ao medicamento por 6 meses ou mais e 7% foram expostos ao medicamento por um ano ou mais. A idade mediana dos pacientes que receberam TecvayliTM foi de 64 anos (intervalo: 33 a 84 anos); 58% eram do sexo masculino; 81% eram brancos, 13% eram negros ou afro-americanos e 2% eram asiáticos. Reações adversas graves ocorreram em 54% dos pacientes que receberam TecvayliTM. As reações adversas graves que ocorreram em uma frequencia maior que 2% dos pacientes incluíram