TECENTRIQ
ROCHE
840 mg em 14 mL
atezolizumabe
Antineoplásico. Anticorpo monoclonal.
Apresentações.
Solução para diluição para infusão.
Caixa com 1 frasco-ampola de dose única de 840 mg em 14 mL (60 mg/mL).
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada frasco-ampola de dose única com 14 mL contém: Princípio ativo: atezolizumabe 840 mg (60 mg/ mL). Excipientes: histidina, ácido acético, sacarose, polissorbato 20 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Câncer de mama triplo-negativo
Tecentriq® em combinação com nab-paclitaxel é indicado para o tratamento de pacientes adultos com câncer de mama triplo negativo localmente avançado irressecável ou metastático cujos tumores apresentam expressão de PD-L1 ≥ 1% e que não tenham recebido quimioterapia prévia para doença metastática.
2. RESULTADOS DE EFICÁCIA
Para a descrição dos estudos clínicos de Tecentriq® administrado a cada 3 semanas em dose única de 1200 mg, veja a bula de Tecentriq® solução para diluição para infusão de 1200 mg.
Câncer de mama triplo negativo
IMpassion130 (WO29522): estudo clínico de fase III, randomizado, em pacientes com câncer de mama triplo negativo localmente avançado ou metastático não tratados previamente para doença metastática
O estudo clínico de fase III, duplo-cego, com dois braços de tratamento, multicêntrico, internacional, randomizado, controlado por placebo, IMpassion130, foi conduzido para avaliar a eficácia e segurança de atezolizumabe em combinação com nab-paclitaxel em pacientes com câncer de mama triplo negativo localmente avançado irressecável ou metastático, que não receberam quimioterapia prévia para a doença metastática. Os pacientes tinham que ser elegíveis para monoterapia com taxano (isto é, ausência de progressão clínica rápida, metástases viscerais com risco de morte ou necessidade de controle rápido dos sintomas e/ou da doença) e foram excluídos se tivessem recebido quimioterapia neoadjuvante ou adjuvante prévia nos últimos 12 meses, histórico de doença autoimune, administração de vacina viva atenuada em até 4 semanas antes da randomização, administração de agentes imunoestimulantes sistêmicos em até 4 semanas ou medicações imunossupressoras sistêmicas em até 2 semanas antes da randomização ou metástases cerebrais que não haviam sido tratadas ou que fossem dependentes de corticosteroides. As avaliações tumorais foram realizadas a cada 8 semanas (± 1 semana) nos primeiros 12 meses após o Ciclo 1, dia 1 e a cada 12 semanas (± 1 semana) depois disso.
Um total de 902 pacientes foram recrutados e estratificados pela presença de metástases hepáticas, tratamento anterior com taxano e pela expressão de PD-L1 em células imunes infiltrantes de tumor (CI) (células imunes infiltrantes de tumor coradas para PD-L1 [CI] < 1% da área tumoral versus ≥ 1% da área tumoral) determinado por teste específico.
Os pacientes foram, então, randomizados para receber atezolizumabe 840 mg ou placebo por infusão intravenosa nos Dias 1 e 15 de cada ciclo de 28 dias mais nab-paclitaxel (100 mg/m2) administrado por infusão intravenosa nos Dias 1, 8 e 15 de cada ciclo de 28 dias. Os pacientes receberam tratamento até a progressão radiográfica da doença, de acordo com RECIST v1.1, ou até toxicidade inaceitável.
As características demográficas e basais da doença na população do estudo foram bem equilibradas entre os braços de tratamento. A maioria dos pacientes eram mulheres (99,6%); 67,5% eram brancos e 17,8% eram asiáticos. A idade mediana foi de 55 anos (variação: 20-86). A capacidade funcional segundo o ECOG basal foi 0 (58,4%) ou 1 (41,3%). Em geral, 41% dos pacientes recrutados apresentaram expressão de PD-L1 ≥ 1%, 27% apresentaram metástases hepáticas e 7% metástases cerebrais no perído basal. Aproximadamente metade dos pacientes havia recebido um taxano (51%) ou antraciclina (54%) no contexto (neo) adjuvante. Os dados demográficos e a doença tumoral basal dos pacientes com expressão de PD-L1 ≥ 1% foram, em geral, representativos da população mais ampla do estudo.
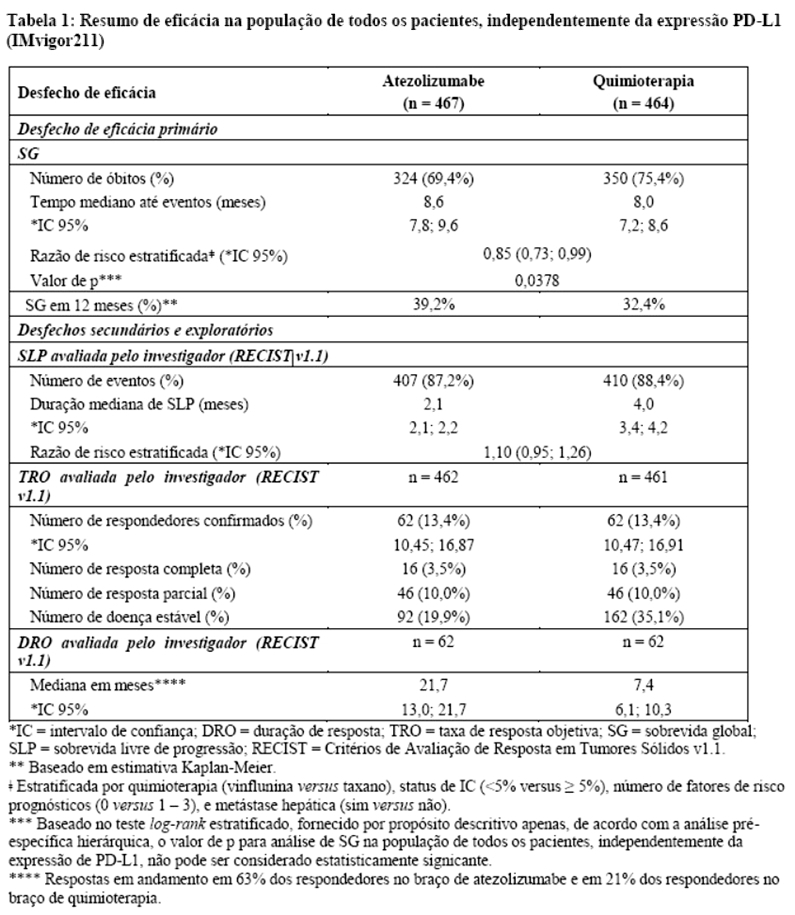
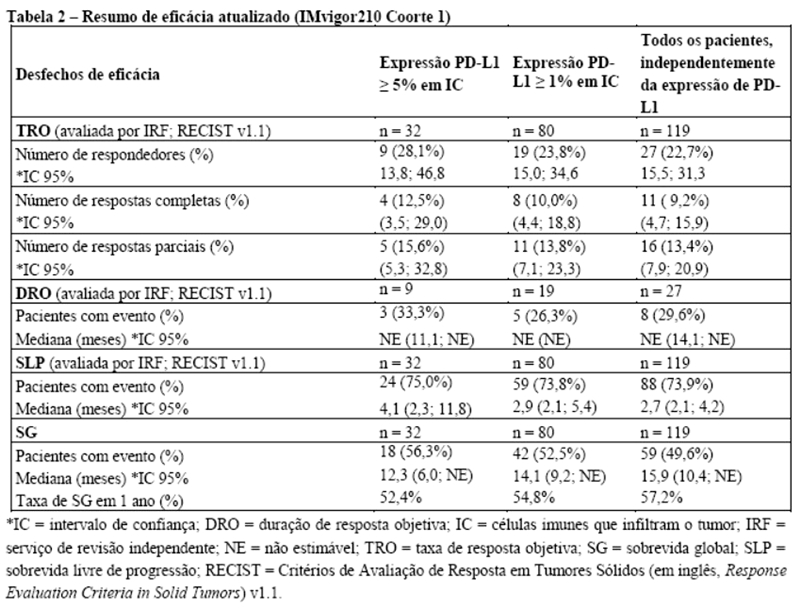
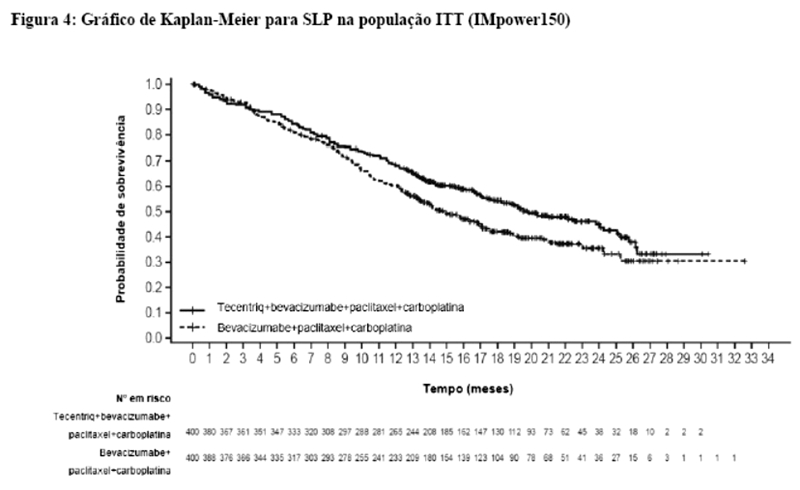
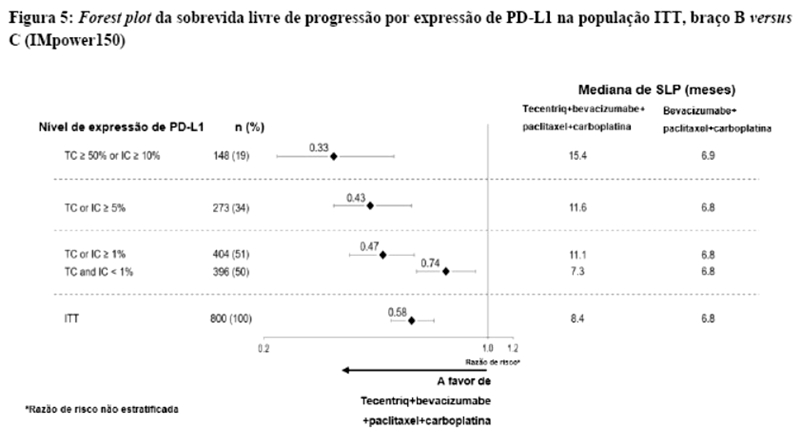
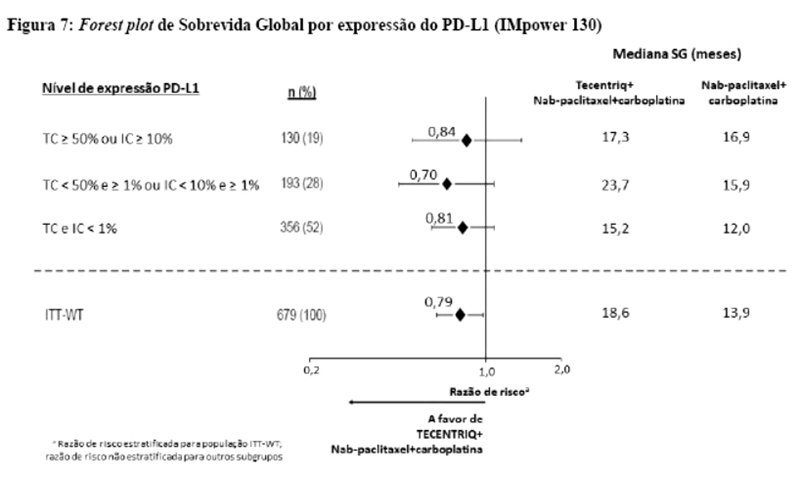
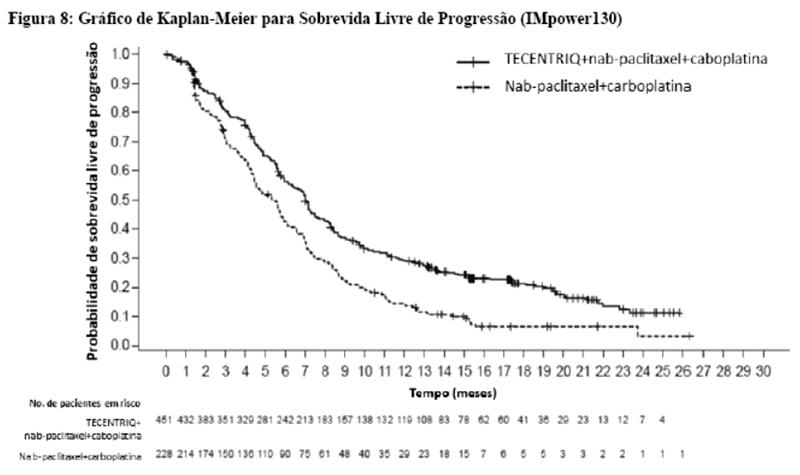
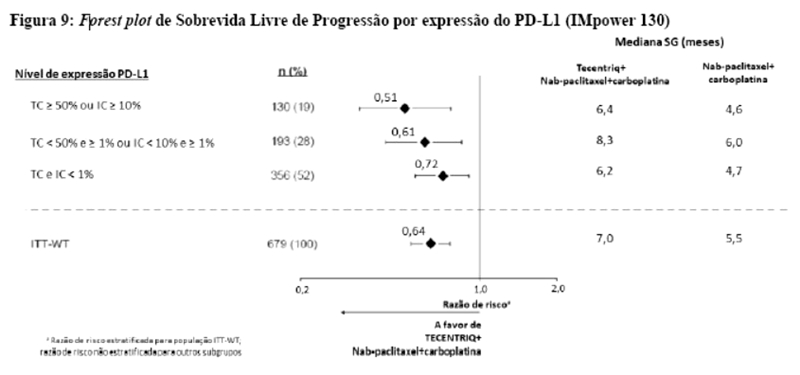
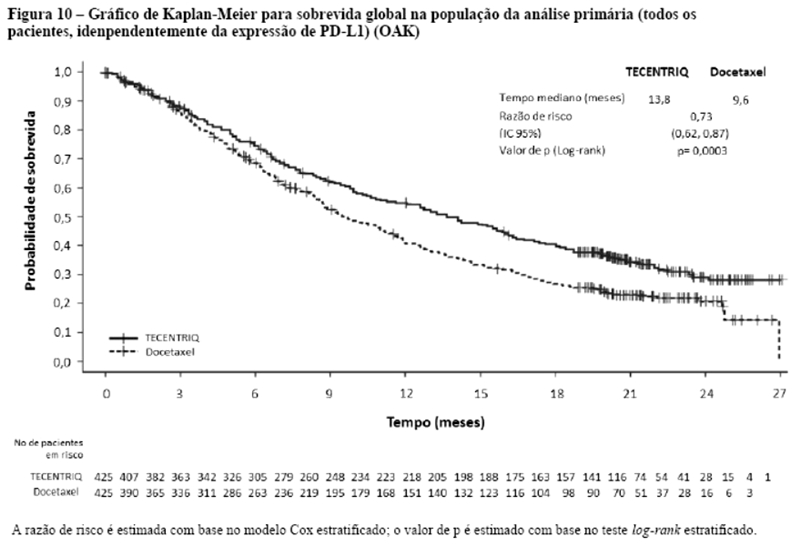
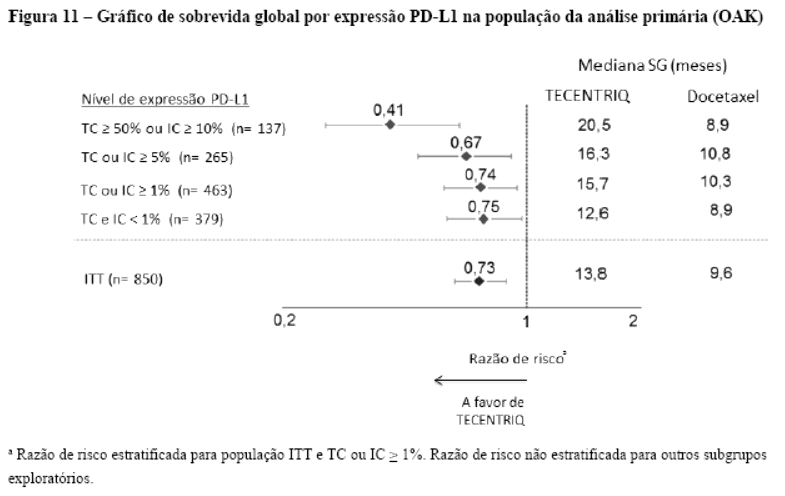
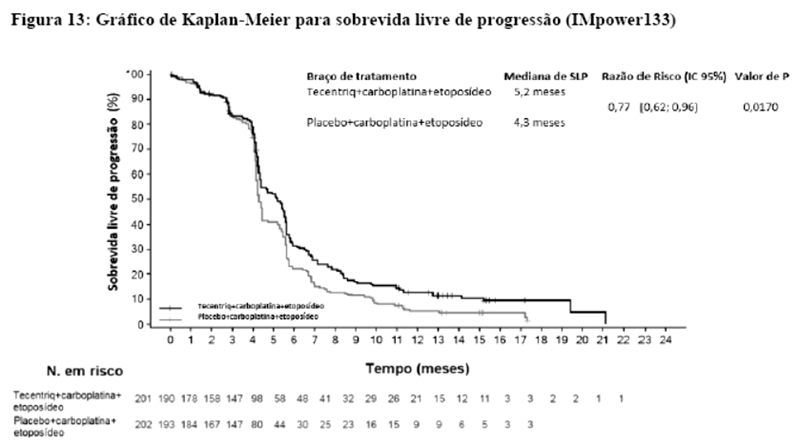
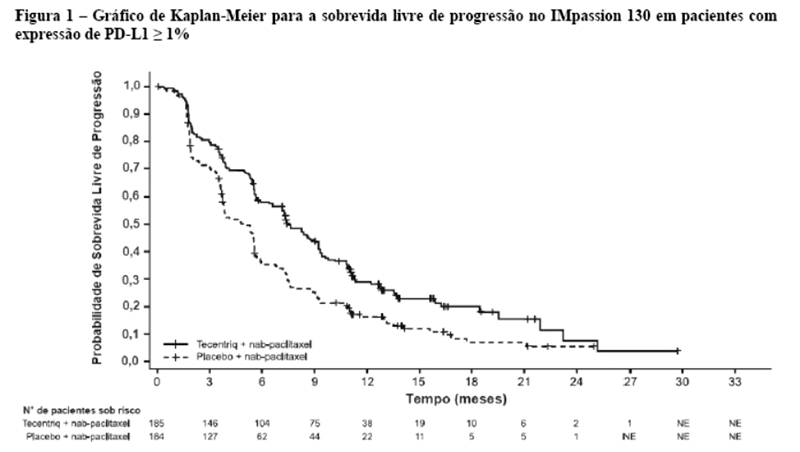
Os desfechos de eficácia co-primários incluiram sobrevida livre de progressão (SLP) avaliada pelo investigador na população de pacientes ITT (população com intenção de tratar) e nos pacientes com expressão de PD-L1 ≥1%, de acordo com RECIST v1.1, assim como sobrevida global (SG) na população ITT e nos pacientes com expressão de PD-L1 ≥1%. Os desfechos secundários de eficácia incluíram a taxa de resposta objetiva (TRO) e a duração da resposta (DOR) de acordo com o RECIST v1.1. Os resultados de SLP, TRO e DOR do IMpassion130, para os pacientes com expressão de PD-L1 ≥ 1% com acompanhamento mediano de sobrevida de 13 meses, são apresentados na tabela 1 com as curvas de Kaplan-Meier para SLP na figura 1. Pacientes com expressão de PD-L1 < 1% não demonstraram melhora em SLP quando atezolizumabe foi adicionado ao nab-paclitaxel (HR de 0,94; IC 95%: 0,78; 1,13).
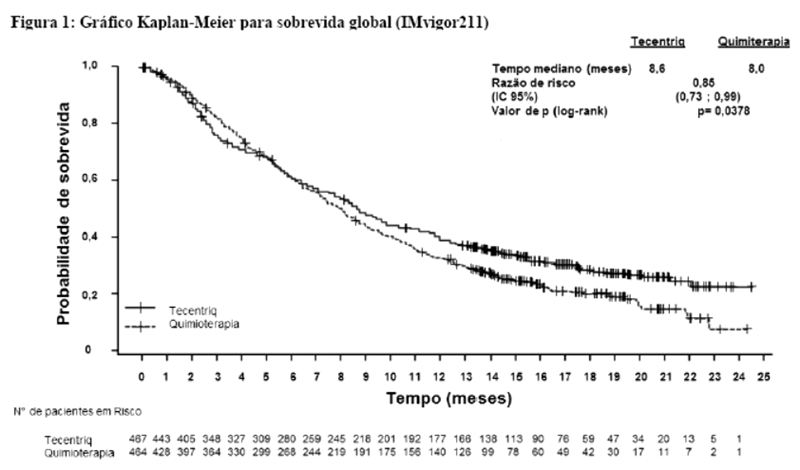
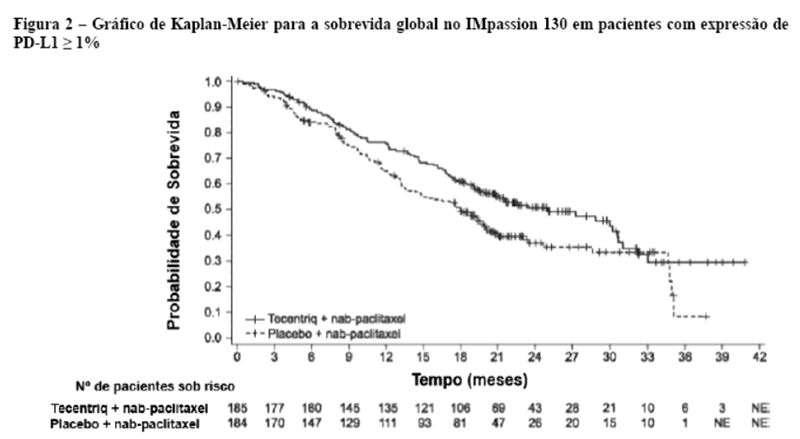
Uma análise de SG atualizada foi realizada com um acompanhamento de 18 meses, os resultados de SG são apresentados na tabela 1 e a curva de Kaplan-Meier na figura 2. Pacientes com expressão de PD-L1 < 1% não demonstraram aumento de SG quando atezolizumabe foi adicionado ao nab-paclitaxel (HR de 0,97; IC 95%: 0,78; 1,20).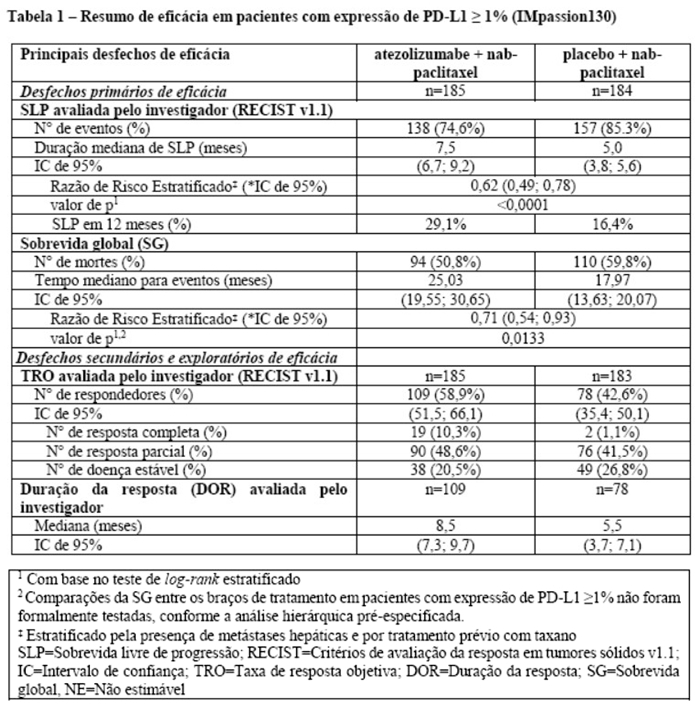
Os desfechos reportados pelos pacientes avaliados pelo EORTC QLQ-C30 sugerem que os pacientes mantiveram seu estado de saúde global e a qualidade de vida relacionada à saúde (QVRS), capacidades funcionais diárias em um período semelhante durante o tratamento. Não houve diferença no tempo para uma deterioração ≥ 10 pontos na QVRS (HR: 0,94; IC 95%: 0,69, 1,28), capacidade física (HR: 1,02; IC 95%: 0,76, 1,37) ou capacidade funcional (HR: 0,77, IC 95%: 0,57, 1,04) observados nos dois braços.
Eficácia em pacientes idosos
Não foram identificadas diferenças de eficácia em pacientes ≥ 65 anos de idade e mais jovens recebendo atezolizumabe em monoterapia. Dados para pacientes ≥75 anos de idade são muito limitados para tirar conclusões nessa população.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
O ligante de morte programada (PD-L1) pode se expressar em células tumorais e/ou células imunes que infiltram tumores e pode contribuir para a inibição de resposta imune antitumoral no microambiente tumoral. A ligação de PD-L1 aos receptores PD-1 e B7.1, encontrados nas células T e nas células apresentadoras de antígeno, suprime a atividade citotóxica das células T, a proliferação de células T e a produção de citocinas.
Atezolizumabe é um anticorpo monoclonal de imunoglobulina G1 (IgG1) humanizado com domínio Fc produzido através de engenharia genética que se liga diretamente ao PD-L1 e promove um bloqueio duplo dos receptores PD-1 e B7.1, gerando a inibição mediada pela via PD-L1/PD-1 da resposta imune, incluindo reativação de resposta imune antitumoral sem induzir citotoxicidade celular dependente de anticorpo. Atezolizumabe mantém a interação PD-L2/PD-1 intacta, permitindo que os sinais inibitórios mediados por PD-L2/PD-L1 permaneçam.
Propriedades farmacocinéticas
A exposição a atezolizumabe aumentou proporcionalmente à dose no intervalo de doses de 1 mg/kg a 20 mg/kg, incluindo a dose fixa de 1200 mg, administrada a cada 3 semanas. Uma análise populacional que incluiu 472 pacientes descreveu a farmacocinética de atezolizumabe para o intervalo de dose: 1 a 20 mg/kg com um modelo de distribuição bicompartimental linear com eliminação de primeira ordem. As propriedades farmacocinéticas de atezolizumabe 840 mg administrado a cada 2 semanas e de 1200 mg administrado a cada 3 semanas são comparáveis. Uma análise farmacocinética populacional sugere que o estado de equilíbrio dinâmico é obtido após 6 a 9 semanas após as múltiplas doses. A razão do acúmulo sistêmico máximo entre os regimes de dose é 3.3.
Absorção
Atezolizumabe é administrado em infusão intravenosa. Não foram realizados estudos com outras vias de administração.
Distribuição
Uma análise de farmacocinética populacional indica que o volume de distribuição no compartimento central (V1) é de 3,28 L e que o volume em estado de equilíbrio é de 6,91 L em um paciente típico.
Metabolismo
O metabolismo de atezolizumabe não foi estudado diretamente. Os anticorpos são eliminados principalmente por catabolismo.
Eliminação
Uma análise de farmacocinética populacional indica que a depuração (clearance) de atezolizumabe é de 0,200 L/dia e que a meia-vida de eliminação terminal típica (t1/2) é de 27 dias.
Populações especiais
Com base em análises de exposição-resposta e farmacocinética populacional, a idade (21 - 89 anos), região, etnia, insuficiência renal, insuficiência hepática leve, nível de expressão de PD-L1 ou escala de performance status de desempenho ECOG não apresentaram nenhum efeito na farmacocinética de atezolizumabe. Peso corpóreo, sexo, níveis de albumina e carga tumoral apresentaram efeito na farmacocinética de atezolizumabe estatisticamente significante, mas não clinicamente relevante. A incidência de anticorpo antiterapêutico (ADA) emergentes ao tratamento entre pacientes tratados com atezo +nP (atezolizumabe+nab-paclitaxel) correspondeu a 13,1% e 11,8% nas populações ITT e positiva para PD-L1, respectivamente. A positividade para ADA não teve um efeito clinicamente relevante sobre a farmacocinética (PK), embora a Cmín no estado de equilíbrio, em média, fosse aproximadamente 25% menor em pacientes positivos para ADA versus pacientes negativos para ADA. Independentemente do estado de ADA, a Cmín média no estado de equilíbrio permaneceu 31-42 vezes acima da concentração sérica pretendida de 6 mg/mL. Não é possível fazer uma declaração definitiva em relação ao impacto de ADA sobre a eficácia. O perfil de segurança geral foi equivalente entre pacientes positivos para ADA e negativos para ADA, com base na incidência de todos os eventos adversos (EAs) e eventos adversos de interesse especial (EAIEs) (52,6% versus 58,6%, respectivamente).
Idosos
Não foram conduzidos estudos com atezolizumabe dedicados a pacientes idosos. O efeito da idade na farmacocinética de atezolizumabe foi avaliado na análise de farmacocinética populacional. A idade não foi identificada como uma covariável significativa que influencie a farmacocinética de atezolizumabe com base em pacientes com idades que variam de 21 a 89 anos (n = 472) e mediana de 62 anos. Nenhuma diferença clinicamente importante foi observada na farmacocinética de atezolizumabe entre pacientes < 65 anos (n = 274), pacientes entre 65 - 75 anos (n = 152) e pacientes > 75 anos (n = 46) (vide item "Posologia e Modo de Usar - Populações especiais").
População pediátrica
Não foram conduzidos estudos para investigar a farmacocinética de atezolizumabe em crianças ou adolescentes.
Insuficiência renal
Não foram conduzidos estudos de atezolizumabe dedicados a pacientes com insuficiência renal. Na análise de farmacocinética populacional, não foram encontradas diferenças clinicamente importantes na depuração (clearance) de atezolizumabe em pacientes com insuficiência renal leve (taxa de filtração glomerular estimada (TFGe) de 60 a 89 mL/min/1,73 m2; n = 208) ou moderada (TFGe 30 a 59 mL/min/1,73 m2; n = 116) comparados a pacientes com função renal normal (TFGe maior ou igual a 90 mL/min/1,73 m2; n = 140). Apenas poucos pacientes apresentaram insuficiência renal severa (TFGe 15 a 29 mL/min/1,73 m2; n = 8) (vide item "Posologia e Modo de Usar - Populações especiais"). O efeito da insuficiência renal grave na farmacocinética de atezolizumabe é desconhecido.
Insuficiência hepática
Não foram conduzidos estudos de atezolizumabe dedicados a pacientes com insuficiência hepática. Na análise de farmacocinética populacional, não houve diferenças clinicamente importantes na depuração (clearance) de atezolizumabe entre pacientes com insuficiência hepática leve (bilirrubina ≤ LSN (limite superior da normalidade) e AST (aspartato aminotransferase) > LSN ou bilirrubina > 1,0 x a 1,5 x LSN e qualquer AST, n = 71) e função hepática normal (bilirrubina e AST ≤ LSN, n = 401). Não há dados disponíveis em pacientes com insuficiência hepática moderada ou grave. A insuficiência hepática foi definida pelos critérios do National Cancer Institute (NCI) para disfunção hepática (vide item "Posologia e Modo de Usar - Populações especiais"). O efeito da insuficiência hepática moderada ou grave (bilirrubina > 1,5 a 3 x LSN e qualquer AST ou bilirrubina > 3 x LSN e qualquer AST) na farmacocinética de atezolizumabe é desconhecido.
Dados de segurança pré-clínica
Carcinogenicidade
Não foram conduzidos estudos de carcinogenicidade para estabelecer o potencial carcinogênico de atezolizumabe.
Mutagenicidade
Não foram conduzidos estudos de mutagenicidade para estabelecer o potencial mutagênico de atezolizumabe. No entanto, não é esperado que anticorpos monoclonais alterem DNA ou cromossomos.
Fertilidade
Não foram conduzidos estudos de fertilidade com atezolizumabe; no entanto, a avaliação dos órgãos reprodutores de macacos cynomolgus machos e fêmeas foi incluída no estudo de toxicidade crônica. A administração semanal de atezolizumabe a macacas com área sobre a curva (ASC) estimada de, aproximadamente, 6 vezes a ASC de pacientes, recebendo a dose recomendada ocasionou um padrão irregular de ciclos menstruais e ausência de corpos lúteos recém-formados nos ovários, o qual foi reversível. Não houve nenhum efeito sobre os órgãos reprodutores dos machos.
Teratogenicidade
Não foram conduzidos estudos de teratogenicidade ou de reprodução em animais com atezolizumabe. Estudos em animais demonstraram que a inibição da via PD-L1/PD-1 pode levar à rejeição imunorrelacionada do feto em desenvolvimento e resultar em morte fetal. A administração de atezolizumabe pode causar dano fetal, incluindo letalidade embriofetal.
4. CONTRAINDICAÇÕES
Tecentriq® é contraindicado a pacientes com hipersensibilidade a atezolizumabe ou quaisquer dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Para aumentar a rastreabilidade dos medicamentos biológicos, o nome comercial e o número de lote do produto administrado devem ser claramente registrados no prontuário médico do paciente.
Reações adversas imunorrelacionadas
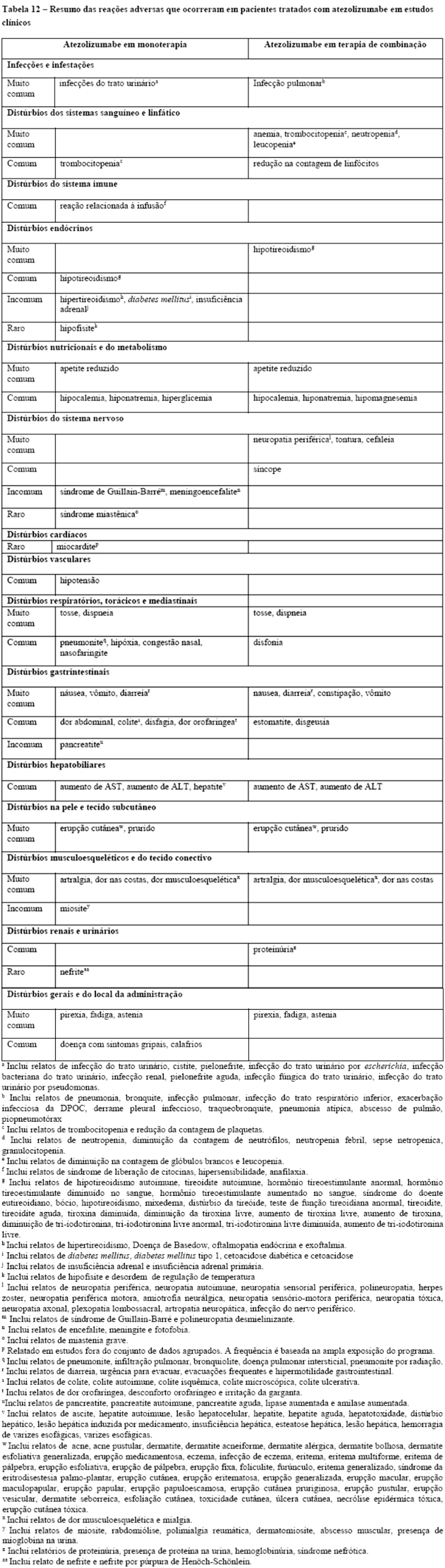
A maioria das reações adversas imunorrelacionadas que ocorreram durante o tratamento com atezolizumabe foram reversíveis com a interrupção de atezolizumabe e a introdução de corticosteroides e/ou cuidados paliativos. Foram observadas reações adversas imunorrelacionadas que afetaram mais de um sistema do corpo.
Reações adversas imunorrelacionadas a atezolizumabe podem ocorrer após a última dose de atezolizumabe.
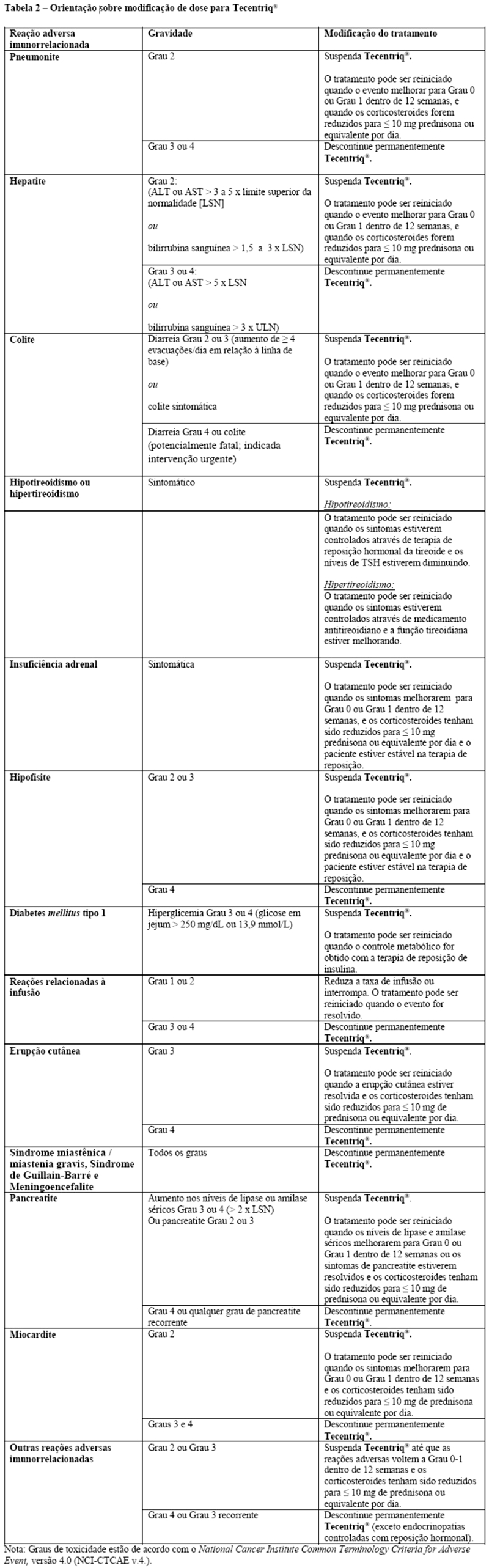
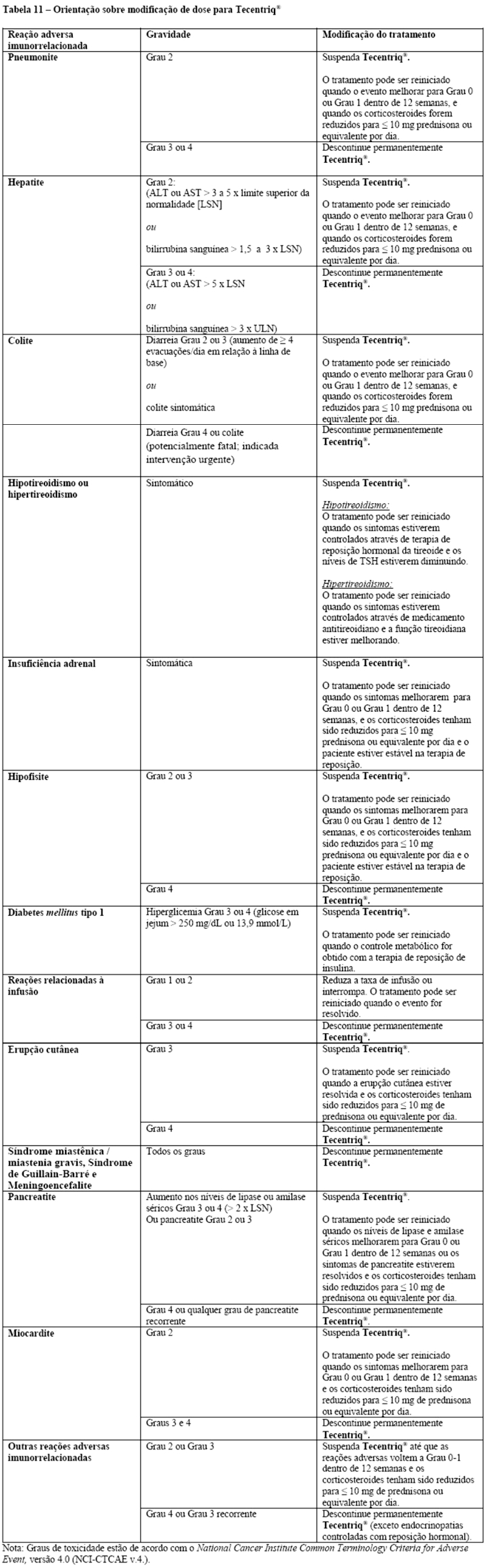
Em caso de suspeita de reações adversas imunorrelacionadas, deve-se realizar uma avaliação completa para confirmar a etiologia ou excluir outras causas. Com base na gravidade da reação adversa, atezolizumabe deve ser descontinuado e corticosteroides devem ser administrados. Após a melhoria para Grau ≤ 1, os corticosteroides devem ser reduzidos gradualmente durante ≥ 1 mês. Com base em dados limitados de estudos clínicos em pacientes, cujas reações adversas imunorrelacionadas não puderam ser controladas com o uso de corticosteroides sistêmicos, a administração de outros imunossupressores sistêmicos pode ser considerada.
Atezolizumabe deve ser permanentemente descontinuado em qualquer reação adversa imunorrelacionada de Grau 3, que se repita e em quaisquer reações adversas imunorrelacionadas de Grau 4, com exceção das endocrinopatias controladas por reposição hormonal (vide itens "Posologia e Modo de Usar" e "Reações Adversas").
Pneumonite imunorrelacionada
Casos de pneumonite, incluindo casos fatais, foram observados em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados em relação a sinais e sintomas de pneumonite.
O tratamento com atezolizumabe deve ser suspenso para pneumonite Grau 2 e deve ser introduzida prednisona 1 - 2 mg/kg/dia ou equivalente. Se os sintomas melhorarem para ≤ Grau 1, reduza os corticosteroides gradualmente durante ≥ 1 mês. O tratamento com atezolizumabe pode ser reiniciado se o evento melhorar até ≤ Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia. O tratamento com atezolizumabe deve ser permanentemente descontinuado para pneumonite nos Graus 3 ou 4.
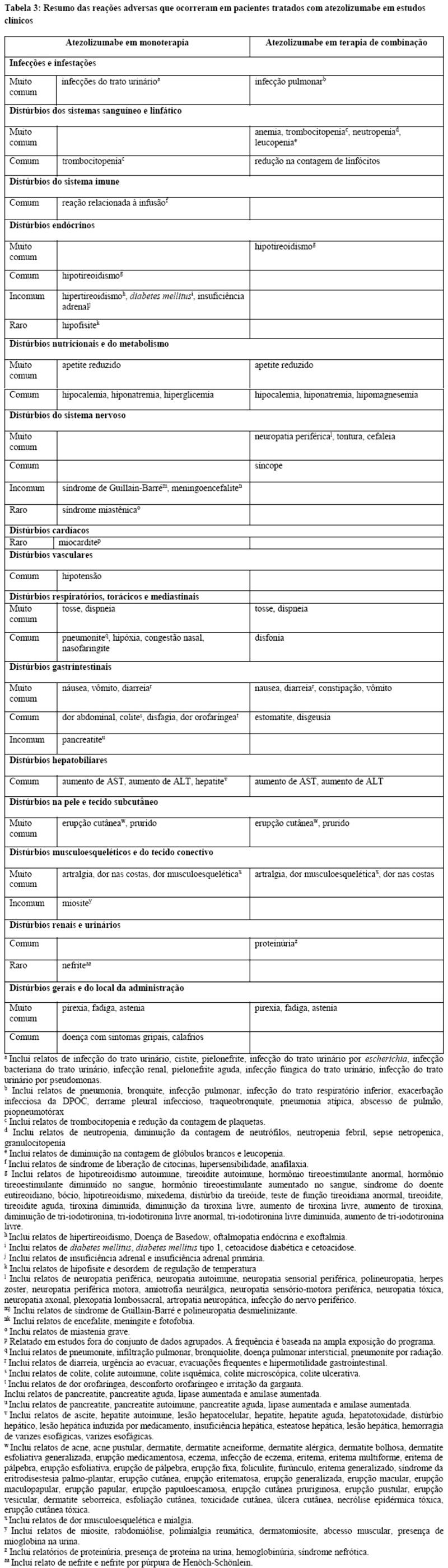
Nos estudos clínicos envolvendo 2616 pacientes com diversos tipos de câncer que receberam Tecentriq® como monoterapia (vide item "Reações Adversas"), pneumonite ocorreu em 2,5% dos pacientes, incluindo pneumonite imunorrelacionada Grau 3 (0,6%), Grau 4 (0,1%) e Grau 5 ( < 0,1%). O tempo mediano para o início da pneumonite foi 3,6 meses (3 dias a 20,5 meses) e a mediana da duração da pneumonite foi 1,4 meses (1 dia a 15,1 meses). A pneumonite foi solucionada em 67% dospacientes. Pneumonite levou à descontinuação de Tecentriq® em 0,4% dos 2616 pacientes. O uso de corticosteroides sistêmicos foi necessário em 1,5% dos pacientes, incluindo 0,8% que receberam altas doses de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) por um tempo mediano de duração de 4 dias (1 a 45 dias) seguido de redução gradual de dose de corticosteroides.
Nos estudos clínicos envolvendo 2421 pacientes com câncer de pulmão de não pequenas células e pequenas células que receberam Tecentriq® em combinação com quimioterapia a base de platina (vide item "Reações Adversas"), pneumonite imunorrelacionada ocorreu em 5,5% dos pacientes, incluindo Graus 3 - 4 em 1,4% dos pacientes. Corticosteróides sistêmicos foram necessários em 4,2% dos pacientes, incluindo 3,1% que receberam altas doses de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) por um tempo mediano de duração de 5 dias (1 a 98 dias) seguido de redução gradual de dose de corticosteroides.
Hepatite imunorrelacionada
Casos de hepatite, alguns levando a evoluções fatais, foram observados em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados para sinais e sintomas de hepatite.
Deve-se monitorar aspartato aminotransferase (AST), alanina aminotransferase (ALT) e bilirrubinas previamente, periodicamente durante o tratamento com atezolizumabe e conforme indicado na avaliação clínica.
O tratamento com atezolizumabe deve ser suspenso se eventos de Grau 2 (ALT ou AST > 3 a 5 x LSN ou bilirrubina no sangue > 1,5 a 3 x LSN) persistirem por mais do que 5 a 7 dias e devem ser introduzidos 1 - 2 mg/kg/dia de prednisona ou equivalente. Se os eventos melhorarem para ≤ Grau 1, reduza gradualmente os corticosteroides durante ≥ 1 mês.
O tratamento com atezolizumabe pode ser reiniciado se o evento melhorar para ≤ Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia. O tratamento com atezolizumabe deve ser permanentemente descontinuado para eventos Grau 3 ou Grau 4 (ALT ou AST > 5,0 x LSN ou bilirrubina no sangue > 3 x LSN).
Nos estudos clínicos envolvendo 2616 pacientes com diversos tipos de câncer que receberam Tecentriq® como monoterapia (vide item "Reações Adversas"), hepatite ocorreu em 9% dos pacientes, incluindo Grau 3 (2,3%), Grau 4 (0,6%) e Grau 5 ( < 0.1%). O tempo mediano para o início da hepatite foi 1,4 meses (1 dia a 25,8 meses) e a mediana da duração da pneumonite foi 24 dias (1 dia a 13 meses). A hepatite foi solucionada em 71% dos pacientes. Hepatite levou à descontinuação de Tecentriq® em 0,4% dos 2616 pacientes. O uso de corticosteroides sistêmicos foi necessário em 2% dos pacientes, com 1,3% requerendo altas doses de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) por um tempo mediano de duração de 3 dias (1 a 35 dias) seguido de redução gradual de dose de corticosteroides.
Nos estudos clínicos envolvendo 2421 pacientes com câncer de pulmão de não pequenas células e pequenas células que receberam Tecentriq® em combinação com quimioterapia a base de platina (vide item "Reações Adversas"), hepatite imunorrelacionada ocorreu em 14% dos pacientes, incluindo Graus 3 - 4 em 4,1% dos pacientes. Corticosteróides sistêmicos foram necessários em 4,8% dos pacientes, incluindo 3,4% que receberam altas doses de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) por um tempo mediano de duração de 6 dias (1 a 144 dias) seguido de redução gradual de dose de corticosteroides.
Colite imunorrelacionada
Casos de diarreia ou colite foram observados em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados para sinais e sintomas de colite.
O tratamento com atezolizumabe deve ser suspenso para diarreia de Graus 2 ou 3 (aumento de ≥ 4 evacuações/dia em relação ao basal) ou colite (sintomática). Para diarreia ou colite de Grau 2, se os sintomas persistirem > 5 dias ou recorrerem, inicie 1 - 2 mg/kg/dia de prednisona ou equivalente. Para diarreia ou colite Grau 3, inicie corticosteroides intravenosos (1 - 2 mg/kg/dia de metilprednisolona ou equivalente). Depois que os sintomas melhorarem, inicie 1 - 2 mg/kg/dia de prednisona ou equivalente. Se os sintomas melhorarem para ≤ Grau 1, reduza gradualmente os corticosteroides durante ≥ 1 mês. O tratamento com atezolizumabe pode ser reiniciado se o evento melhorar para ≤ Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia. O tratamento com atezolizumabe deve ser permanentemente descontinuado para diarreia ou colite Grau 4 (potencialmente fatal; intervenção urgente é indicada).
Nos estudos clínicos envolvendo 2616 pacientes com diversos tipos de câncer que receberam Tecentriq® como monoterapia (vide item "Reações Adversas"), diarreia ou colite ocorreu em 20% dos pacientes, incluindo eventos de Grau 3 (1,4%). O tempo mediano para o início da diarreia ou colite foi 1,5 meses (1 dia a 41 meses). A diarreia e a colite foram solucionadas em 85% dos pacientes. Diarreia ou colite levou à descontinuação de Tecentriq® em 0,2% dos 2616 pacientes. O uso de corticosteroides sistêmicos foi necessário em 1,1% dos pacientes e alta dose de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) foi necessária em 0,4% dos pacientes por um tempo mediano de duração de 3 dias (1 a 11 dias) seguido de redução gradual de dose de corticosteroides.
Nos estudos clínicos envolvendo 2421 pacientes com câncer de pulmão de não pequenas células e pequenas células que receberam Tecentriq® em combinação com quimioterapia a base de platina (vide item "Reações Adversas"), diarreia ou colite ocorreu em 29% dos pacientes, incluindo Graus 3 - 4 em 4,3% dos pacientes. Corticosteróides sistêmicos foram necessários em 4,7% dos pacientes, incluindo 2,9% que receberam alta dose de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente) por um tempo mediano de duração de 4 dias (1 a 170 dias) seguido de redução gradual de dose de corticosteroides.
Endocrinopatias imunorrelacionadas
Hipotireoidismo, hipertireoidismo, insuficiência adrenal, hipofisite e diabetes mellitus tipo 1, incluindo cetoacidose diabética, foram observados em estudos clínicos com atezolizumabe (vide item "Reações Adversas").
Os pacientes devem ser monitorados para sinais e sintomas clínicos de endocrinopatias. Monitore a função tireoidiana previamente e periodicamente durante o tratamento com atezolizumabe. O gerenciamento aproriado de pacientes com provas de função tireoidiana anormal no período basal deve ser considerado.
Pacientes assintomáticos com provas de função tireoidiana anormais podem receber atezolizumabe. Para hipotireoidismo sintomático, atezolizumabe deve ser suspenso e a reposição do hormônio tireoidiano deve ser iniciada se necessário. Hipotireoidismo isolado pode ser tratado com terapia de reposição e sem corticosteroides. Para hipertireoidismo sintomático, atezolizumabe deve ser suspenso e uma droga antitireoide deve ser introduzida se necessário. O tratamento com atezolizumabe pode ser reiniciado quando os sintomas estiverem controlados e a função tireoidiana estiver melhorando.
Nos estudos clínicos envolvendo 2616 pacientes que receberam Tecentriq® como monoterapia (vide item "Reações Adversas"), hipotireoidismo ocorreu em 4,6% dos pacientes e para 3,8% dos pacientes foi necessário o uso terapia de reposição hormonal. Hipertireoidismo ocorreu em 1,6% dos pacientes. Um paciente apresentou tireoidite aguda. Nos estudos clínicos envolvendo 2421 pacientes com câncer de pulmão de não pequenas células e pequenas células que receberam Tecentriq® em combinação com quimioterapia a base de platina (vide item "Reações Adversas"), hipotireoidismo ocorreu em 11% dos pacientes, incluindo Graus 3 - 4 em 0,3% dos pacientes; em 8,2% dos 2421 pacientes foi necessário o uso terapia de reposição hormonal. A frequência e a severidade do hipertireoidismo e tireoidite foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer, quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
Para insuficiência adrenal sintomática, atezolizumabe deve ser suspenso e o tratamento com corticosteroides intravenosos (1 - 2 mg/kg/dia de metilprednisolona ou equivalente) deve ser iniciado. Depois que os sintomas melhorarem, siga com 1 - 2 mg/kg/dia de prednisona ou equivalente. Se os sintomas melhorarem para ≤ Grau 1, reduza gradualmente os corticosteroides durante ≥ 1 mês. O tratamento pode ser reiniciado se o evento melhorar para ≤ Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia e o paciente estiver estável com terapia de reposição (se necessário).
Nos estudos clínicos envolvendo 2616 pacientes que receberam Tecentriq® como monoterapia, insuficiência adrenal ocorreu em 0,4% dos pacientes, incluindo insuficiência adrenal Grau 3 ( < 0.1%). O tempo mediano para o início foi 5,7 meses (3 dias a 19 meses). As informações foram insuficientes para caracterizar adequadamente o tempo mediano de duração da insuficiência adrenal. Insuficiência adrenal foi solucionada em 27% dos pacientes. O uso de corticosteroides sistêmicos foi necessário em 0,3% dos 2616 pacientes, incluindo 0,1% que necessitou alta dose de corticosteroides (predinisona ≥ 40 mg por dia ou equivalente). A frequência e a severidade da insuficiência adrenal foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
O tratamento com atezolizumabe deve ser suspenso em caso de hipofisite Grau 2 e Grau 3 e tratamento com corticosteroides intravenosos (1 - 2 mg/kg/dia de metilprednisolona ou equivalente) e a reposição hormonal devem ser iniciados, se necessário. Depois que os sintomas melhorarem, seguir com 1 - 2 mg/kg/dia de prednisona ou equivalente. Se os sintomas melhorarem para < Grau 1, reduza gradualmente os corticosteroides durante > 1 mês. O tratamento pode ser reiniciado se o evento melhorar para < Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para < 10 mg de prednisona ou equivalente por dia e o paciente estiver estável com terapia de reposição (se necessário). O tratamento com atezolizumabe deve ser permanentemente descontinuado para hipofisite Grau 4. Nos estudos clínicos envolvendo 2616 pacientes que receberam Tecentriq® como monoterapia, hipofisite Grau 2 ocorreu em < 0,1% dos pacientes. A frequência e a severidade da hipofisite foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
Monitore os pacientes quanto à hiperglicemia ou outros sinais e sintomas de diabetes. O tratamento com insulina deve ser iniciado para diabetes mellitus tipo 1. Para hiperglicemia ≥ Grau 3 (glicose em jejum > 250 mg/dL ou 13,9 mmol/L), atezolizumabe deve ser suspenso. O tratamento com atezolizumabe pode ser reiniciado se o controle metabólico for atingido com terapia de reposição de insulina. Nos estudos clínicos envolvendo 2616 pacientes que receberam Tecentriq® como monoterapia, diabetes mellitus tipo 1 ocorreu em < 0,1% dos pacientes. Insulina foi necessária para um paciente. A frequência e a severidade da diabetes mellitus foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
Meningoencefalite imunorrelacionada
Meningoencefalite foi observada em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados para sinais e sintomas clínicos de meningite ou encefalite.
O tratamento com atezolizumabe deve ser permanentemente descontinuado para qualquer grau de meningite ou encefalite. Tratamento com corticosteroides intravenosos (1 - 2 mg/kg/dia de metilprednisolona ou equivalente) deve ser iniciado. Depois que os sintomas melhorarem, seguir o tratamento com 1 - 2 mg/kg/dia de prednisona ou equivalente.
Neuropatias imunorrelacionadas
Síndrome miastênica/miastenia gravis ou síndrome de Guillain-Barré, que podem ser potencialmente fatais, foram observadas em pacientes recebendo atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados para sintomas de neuropatia motora ou sensorial.
O tratamento com atezolizumabe deve ser permanentemente descontinuado para qualquer grau de síndrome miastênica/miastenia gravis ou síndrome de Guillain-Barré. A introdução de corticosteroides sistêmicos (na dose de 1 - 2 mg/kg/dia de prednisona ou equivalente) deve ser considerada.
Pancreatite imunorrelacionada
Pancreatite, incluindo aumentos na amilase sérica e níveis de lipase, foi observada em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados de perto para sinais e sintomas sugestivos de pancreatite aguda.
O tratamento com atezolizumabe deve ser suspenso para amilase sérica ≥ Grau 3 ou níveis elevados de lipase ( > 2 x LSN), ou pancreatite de Graus 2 ou 3, e deve-se iniciar tratamento com corticosteroides intravenosos (1 - 2 mg/kg/dia de metilprednisolona ou equivalente). Após melhora dos sintomas, siga com 1 - 2 mg/kg/dia de prednisona ou equivalente. O tratamento com atezolizumabe pode ser reiniciado quando os níveis de amilase sérica e lipase melhorarem para ≤ Grau 1 dentro de 12 semanas ou os sintomas de pancreatite forem resolvidos e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia. O tratamento com atezolizumabe deve ser permanentemente descontinuado para Grau 4 ou qualquer grau de pancreatite recorrente.
Miocardite imunorrelacionada
Nos estudos clínicos com atezolizumabe foi observada miocardite (vide item "Reações Adversas"). Pacientes devem ser monitorados para sinais e sintomas de miocardite.
O tratamento com atezolizumabe deve ser suspenso para miocardite Grau 2 e deve ser iniciado tratamento com corticosteroides sistêmicos na dose de 1 a 2 mg/kg/dia de prednisona ou equivalente. O tratamento com atezolizumabe pode ser reiniciado se o evento melhorar para ≤ Grau 1 dentro de 12 semanas e os corticosteroides tiverem sido reduzidos para ≤ 10 mg de prednisona ou equivalente por dia. O tratamento com atezolizumabe deve ser permanentemente descontinuado para miocardite Grau 3 ou 4.
Nefrite imunorrelacionada
Nefrite tem sido observada nos estudos clínicos com atezolizumabe. Os pacientes devem ser monitorados quanto às alterações na função renal.
Miosite imunorrelacionada
Casos de miosite, incluindo casos fatais, têm sido observados em estudos clínicos com atezolizumabe (vide item "Reações Adversas"). Os pacientes devem ser monitorados quanto aos sinais e sintomas de miosite.
Reações relacionadas à infusão
Reações relacionadas à infusão tem sido observadas com atezolizumabe (vide item "Reações Adversas").
A taxa de infusão deve ser reduzida ou o tratamento deve ser interrompido em pacientes com reações relacionadas à infusão Grau 1 ou 2. Atezolizumabe deve ser permanentemente descontinuado em pacientes com reações relacionadas à infusão Grau 3 ou 4. Pacientes com reações relacionadas à infusão Grau 1 ou 2 podem continuar a receber atezolizumabe com monitoramento constante; premedicação com antipirético e anti-histamínicos deve ser considerada.
Nos estudos clínicos envolvendo 2616 pacientes com vários tipos de câncer que receberam Tecentriq® como monoterapia (vide item "Reações Adversas"), reações relacionadas à infusão ocorreram em 1,3% dos pacientes, incluindo Grau 3 (0,2%). A frequência e a severidade da reação relacionada à infusão foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
Outras reações adversas imunomediadas
O Tecentriq® pode causar reações adversas imunomediadas graves e fatais. Estas reações imunomediadas podem envolver qualquer sistema orgânico. Embora as reações imunomediadas usualmente se manifestem durante o tratamento com Tecentriq®, as reações adversas imunomediadas também podem se manifestar após a descontinuação de Tecentriq®.
Para suspeita de reações adversas imunomediadas mediadas por Grau 2, exclua outras causas e inicie os corticosteroides conforme clinicamente indicado. Para reações adversas graves (Grau 3 ou 4), administrar corticosteroides, prednisona 1 a 2 mg/ kg/dia ou equivalentes, seguidos de redução gradual. Interromper ou descontinuar permanentemente o Tecentriq®, com base na gravidade da reação.
Se a uveíte ocorrer em combinação com outras reações adversas imunomediadas, avalie a síndrome de Vogt-Koyanagi-Harada, que foi observada com outros produtos desta classe e pode requerer tratamento com esteroides sistêmicos para reduzir o risco de perda permanente da visão.
As seguintes reações adversas imunomediadas clinicamente significativas ocorreram com uma incidência < 1% em 2616 pacientes que receberam o Tecentriq® ou foram reportados em outros produtos desta classe:
Dermatológicas: dermatite bolhosa, penfigóide, eritema multiforme, síndrome de Stevens Johnson (SSJ) / necrólise epidérmica tóxica (NET).
Geral: síndrome de resposta inflamatória sistêmica, linfadenite necrosante histiocitária.
Hematológico: anemia hemolítica auto-imune, púrpura trombocitopênica imunológica.
Musculosqueléticas: miosite, rabdomiólise.
Neurológico: síndrome de Guillain-Barré, síndrome miastenia / miastenia gravis, desmielinização, meningoencefalite relacionada ao sistema imunológico, meningite asséptica, encefalite, paresia do nervo facial e abducente, polimialgia reumática, neuropatia autoimune e síndrome de Vogt-Koyanagi-Harada.
Oftalmológico: uveíte, irite.
Renal: síndrome nefrótica, nefrite.
Vascular: vasculite.
Infecções
O Tecentriq® pode causar infecções graves, incluindo casos fatais. Monitore os pacientes quanto a sinais e sintomas de infecção. Para infecções de Grau 3 ou superior, suspender o Tecentriq® e retomar uma vez clinicamente estável (vide itens "Posologia e Modo de Usar" e "Reações Adversas"). Em estudos clínicos envolvendo 2616 pacientes com vários tipos de tumores tratados com Tecentriq® em monoterapia, ocorreram infeções em 42% dos pacientes, incluindo infecções de Grau 3 (8,7%), Grau 4 (1,5%) e Grau 5 (1%). Em pacientes com carcinoma urotelial, as infecções mais comuns de Grau 3 ou superiores foram infecções de trato urinário, que ocorreram em 6,5% dos pacientes. Em pacientes com CPNPC a infecção mais comum Grau 3 ou superior foi pneumonia, ocorrendo em 3,8% dos pacientes. A frequência e a severidade das infecções foram similares tanto em Tecentriq® administrado em monoterapia em pacientes com vários tipos de câncer quanto quando administrado em combinação com outros medicamentos antineoplásicos em CPNPC e CPPC.
Precauções específicas da doença
Uso de atezolizumabe em combinação com nab-paclitaxel em câncer de mama triplo negativo
A neutropenia e neuropatias periféricas que ocorrem durante o tratamento com atezolizumabe e nab-paclitaxel podem ser reversíveis com interrupções do atezolizumabe e/ou nab-paclitaxel. Os médicos prescritores devem consultar a bula do nab-paclitaxel para precauções e advertências específicas desse medicamento.
Uso de atezolizumabe em combinação com bevacizumabe, paclitaxel e carboplatina em câncer de pulmão de não pequenas células não escamoso
Os médicos devem cuidadosamente considerar os riscos combinados do regime terapêutico com quatro drogas atezolizumabe, bevacizumabe, paclitaxel e carboplatina antes de iniciar o tratamento (vide item "9. Reações adversas").
Pacientes excluídos dos estudos clínicos
Os pacientes com as seguintes condições foram excluídos dos ensaios clínicos: história de doença autoimune, história de pneumonite, metástase cerebral ativa, HIV, infecção por hepatite B ou hepatite C, doença cardiovascular significativa, pacientes com resultados hematológicos inadequados e com função do órgão alvo inadequada. Pacientes que receberam uma vacina viva atenuada dentro de 28 dias antes da inclusão no estudo, agentes imunoestimulantes sistêmicos dentro de 4 semanas ou medicamentos imunossupressores sistêmicos dentro de 2 semanas antes da inclusão no estudo foram excluídos dos ensaios clínicos.
Pacientes com escala de performance ECOG ≥ 2 na linha de base foram excluídos (vide item "Resultados de eficácia").
Uso de atezolizumabe em combinação com bevacizumabe, paclitaxel e carboplatina
Pacientes com CPNPC com tumor claramente infiltrado nos grandes vasos toráxicos ou com cavitação clara das lesões pulmonares, conforme observação de imagem, foram excluídos do estudo clínico pivotal IMpower150 após a ocorrência de vários casos de hemorragia pulmonar fatal, que é um fator de risco conhecido do tratamento com bevacizumabe.
Na ausência de dados, atezolizumabe deve ser administrado com cautela nessas populações após uma avaliação cuidadosa do balanço risco-benefíci