TALZENNA
PFIZER
tosilato de talazoparibe
Inibidor das enzimas PARP, PARP1 e PARP2.
Apresentações.
Talzenna® 0,25 mg em frascos contendo 30 cápsulas duras.
Talzenna® 1 mg em frascos contendo 30 cápsulas duras.
VIA DE ADMINISTRAÇÃO: USO ORAL
USO ADULTO
Composição.
Cada cápsula de Talzenna® 0,25 mg contém 0,363 mg de tosilato de talazoparibe que equivale a 0,25 mg de base livre de talazoparibe.
Cada cápsula de Talzenna® 1 mg contém 1,453 mg de tosilato de talazoparibe que equivale a 1 mg de base livre de talazoparibe.
Excipientes:
0,25 mg: celulose microcristalina silicificada, hipromelose, dióxido de titânio, hipromelose, óxido de ferro amarelo, dióxido de titânio.
1 mg: celulose microcristalina silicificada, hipromelose, dióxido de titânio, hipromelose, óxido de ferro amarelo, óxido de ferro vermelho, dióxido de titânio.
Informações técnicas.
1. INDICAÇÕES
Talzenna® é indicado para o tratamento de pacientes adultos com câncer de mama metastático ou localmente avançado negativo para receptor de fator de crescimento epidérmico humano 2 (HER2), não suscetível a radiação curativa ou cirurgia, com uma mutação do gene de suscetibilidade ao câncer de mama de linha germinativa (BRCA1/2), deletéria ou suspeitamente deletéria, que foram previamente tratados com quimioterapia em ambiente neoadjuvante, adjuvante, localmente avançado ou metastático, a menos que sejam considerados inadequados para esses tratamentos.
2. RESULTADOS DE EFICÁCIA
Estudo de Fase 3 randomizado EMBRACA
EMBRACA foi um estudo multicêntrico, aberto, randomizado, paralelo e de dois braços de Talzenna® versus quimioterapia (capecitabina, eribulina, gencitabina ou vinorelbina) em pacientes com câncer de mama localmente avançado ou metastático de linha germinativa BRCA com mutação, HER2 negativo, que receberam no máximo três regimes prévios com quimioterapia citotóxica para a doença metastática ou localmente avançada. Os pacientes deveriam ter recebido tratamento com uma antraciclina e/ou um taxano (a menos que contraindicado) no cenário neoadjuvante adjuvante e/ou metastático. Pacientes com terapia prévia com platina para doença avançada deveriam comprovar ausência de progressão da doença durante a terapia com platina. Nenhum tratamento prévio com inibidor de PARP foi permitido.
No total, 431 pacientes foram randomizados na proporção 2:1 para receber cápsulas de 1 mg de Talzenna® uma vez por dia ou quimioterapia em doses padrão até progressão ou toxicidade inaceitável. Dos 431 pacientes randomizados no EMBRACA, 287 foram randomizados para o braço de Talzenna® e 144 para o braço da quimioterapia. A randomização foi estratificada de acordo com a quimioterapia prévia para doença avançada (0 versus 1, 2 ou 3), estado triplo negativo da doença (câncer de mama triplo negativo [CMTN] versus não CMTN) e histórico de metástase do sistema nervoso central (sim versus não). A maioria dos pacientes 408/431 (95%) foi selecionada utilizando o teste BRAC Analysis e o estado de mutação BRCA (positivo para o gene 1 de susceptibilidade ao câncer de mama [BRCA1] ou positivo para o gene 2 de susceptibilidade ao câncer de mama [BRCA2]) foi semelhante em ambos os braços de tratamento.
As características demográficas e basais do paciente foram, em geral, semelhantes nos dois braços de tratamento em estudo. A idade mediana dos pacientes tratados com Talzenna® foi de 45 anos (faixa de variação de 27 a 84) e 50 anos (faixa de variação de 24 a 88) entre pacientes tratados com quimioterapia. Notavelmente, 63% versus 47% dos pacientes tinham < 50 anos nos braços de talazoparibe e quimioterapia respectivamente, 27% versus 47% tinham de 50 a < 65 anos, e 9% versus 7% tinham ≥65 anos. Entre todos os pacientes randomizados, 1% versus 2% eram do sexo masculino, 66,9% versus 75,0% eram brancos; 10,8% versus 11,1% eram asiáticos e 4,2% versus 0,7% eram negros ou afro-americanos nos braços de talazoparibe e quimioterapia, respectivamente. Quase todos os pacientes (97,7%) em ambos os braços apresentavam estado de desempenho do Eastern Cooperative Oncology Group (ECOG) de 0 ou 1. Aproximadamente 55,9% dos pacientes apresentavam receptor positivo de hormônio (receptor de estrogênio [RE] positivo ou receptor de progesterona [RP] positivo); 44,1% dos pacientes tinham doença triplo negativa e as proporções estavam equilibradas entre os braços de tratamento. O tempo mediano entre o diagnóstico inicial de câncer de mama e o diagnóstico de câncer de mama avançado foi de 1,9 ano (faixa de variação de 0 a 22) no braço do talazoparibe e 2,7 anos (faixa de variação de 0 a 24) no braço de quimioterapia. O intervalo livre de doença (ILD) foi < 12 meses em 37,6% dos pacientes no braço do talazoparibe e em 29,2% dos pacientes nos braços de quimioterapia. Entre todos os pacientes inscritos, o número mediano de esquemas citotóxicos anteriores para câncer de mama avançado foi um, em que 38,3% dos pacientes não receberam nenhum tratamento prévio para doença avançada ou metastática, 37,4% receberam um, 19,7% receberam dois e 4,6% receberam > 3 antes dos esquemas, respectivamente. Dezesseis por cento dos pacientes no braço do talazoparibe e 20,8% dos pacientes no braço de quimioterapia receberam tratamento prévio com platina.
O desfecho primário de eficácia foi a sobrevida livre de progressão (SLP) avaliada de acordo com os Critérios de Avaliação da Resposta em Tumores Sólidos (RECIST) versão 1.1, conforme avaliado por revisão central independente cega (BICR). Os objetivos secundários foram taxa de resposta objetiva (TRO), sobrevida global (SG), segurança e PK. Os objetivos exploratórios incluíram duração da resposta (DOR), taxa de benefício clínico em 24 semanas (CBR24), qualidade de vida (QoL) avaliada pelo Questionário de Qualidade de Vida da European Organisation for Research and Treatment of Cancer (EORTC) (Organização Europeia para Pesquisa e Tratamento de Câncer) - Núcleo 30 (QLQ-C30)/Questionário de Qualidade de Vida EORTC - Módulo de Câncer de Mama (QLQ-BR23) e pesquisa de biomarcador.
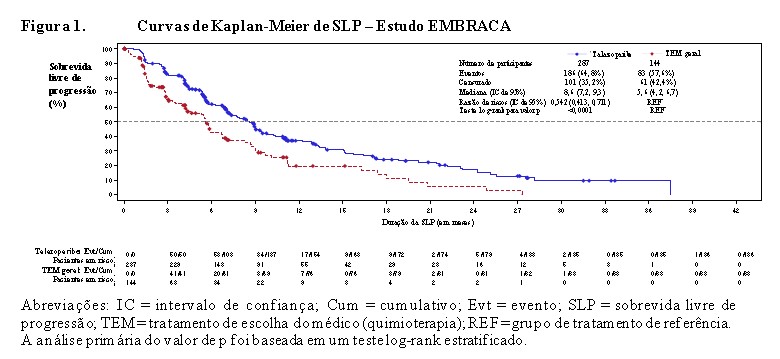
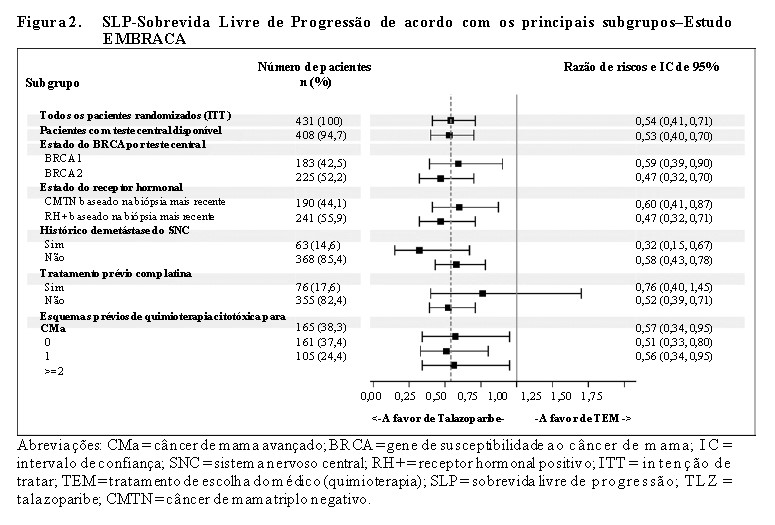
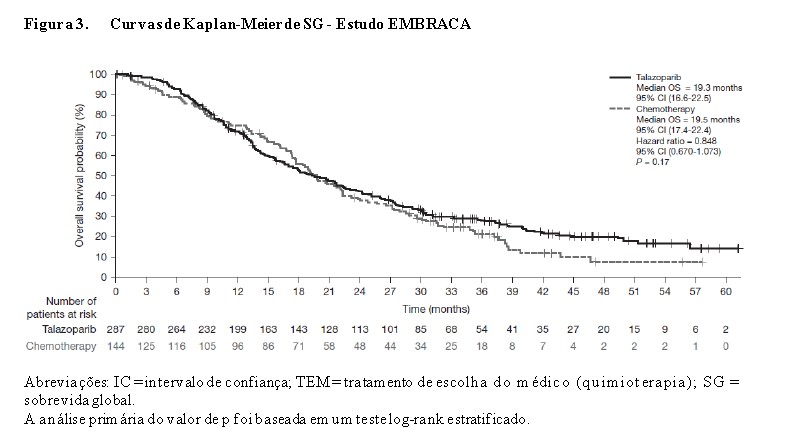
O estudo atingiu seu objetivo primário ao demonstrar melhora estatisticamente significativa na SLP com Talzenna® em comparação à quimioterapia (razão de riscos [RR] 0,54; intervalo de confiança [IC] de 95%: 0,41, 0,71; p < 0,0001). Uma análise de sensibilidade da SLP avaliada pelo investigador foi compatível com os resultados da SLP avaliados por BICR. Os dados de eficácia do EMBRACA estão resumidos na Tabela 1 e as curvas de Kaplan-Meier para SLP e SG são mostradas nas Figuras 1 e 3. Resultados consistentes foram observados nos subgrupos de pacientes pré-especificados (Figura 2).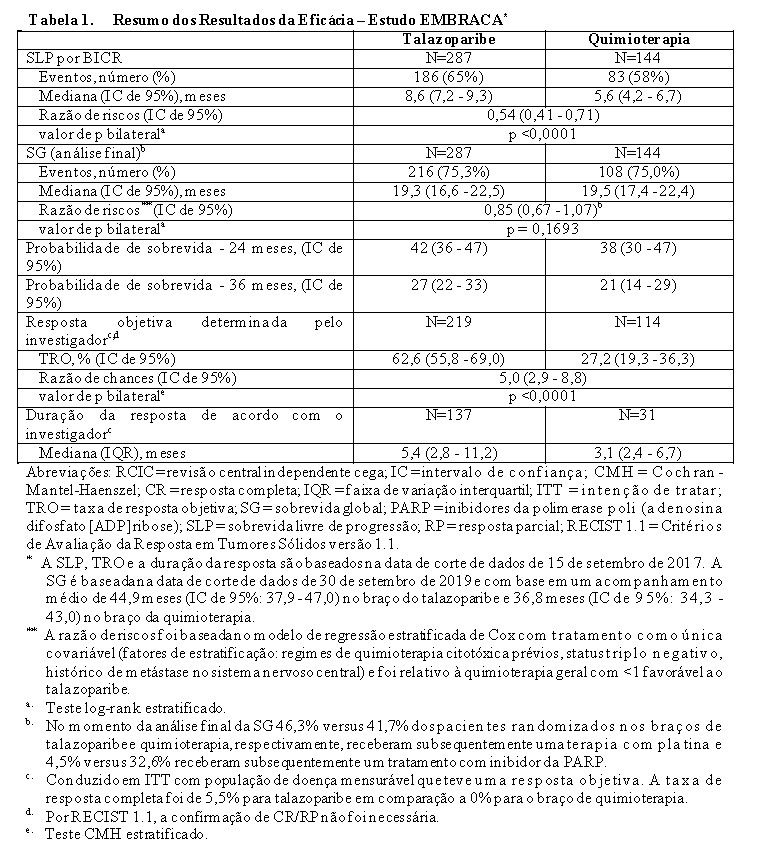
REFERÊNCIAS
Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature, 2005, Apr 14, 434(7035):913-7.
de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; Henshaw, J.W.; Herriott, A.; Patterson, M.; Curtin, N.J.; Byers, L.A.; Wainberg, Z.A. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov, 2017, Jun;7(6), 620-629.
Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; Martin, N.M.; Jackson, S.P., Smith, G.C.; Ashworth, A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 2005, Apr 14; 434(7035), 917-21.
Hoffman, J.; Chakrabarti, J.; Plotka, A.; Milillo Naraine, A.; Kanamori, D.; Moroose, R.; Nguyen, L.; Wang, D.; Wainberg, Z. A. Talazoparib has no clinically relevant effect on QTc interval in patients with advanced solid tumors. Anticancer Drugs, 2019, Jun, 30(5), 523-532.
Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer, 2011, 105, 1114-1122.
Litton, J.K.; Rugo, H.S.; Ettl J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; Roché, H.; Im, Y.H.; Quek, R.G.W.; Markova, D.; Tudor, I.C.,; Hannah, A.L.; Eiermann, W.; Blum, J.L. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med, 2018, Aug 23, 379(8), 753-763.
Murai, J; Huang, S.N.; Brata Das, B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Differential trapping of PARP1 and PARP2 by clinical PARP inhibitor. Cancer Res, 2012, Nov 1, 72(21), 5588-5599.
Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci, 2004, 95, 866-871.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação e efeitos farmacodinâmicos
Talzenna® é um potente inibidor das enzimas PARP, PARP1 e PARP2. As enzimas PARP estão envolvidas nas vias de sinalização de resposta a danos no DNA celular, tais como reparo de DNA, transcrição gênica, regulação do ciclo celular e morte celular. Os inibidores de PARP (PARPi) exercem efeitos citotóxicos nas células cancerígenas por dois mecanismos: inibição da atividade catalítica de PARP e captura de PARP, já que a proteína PARP ligada a uma PARPi não se dissocia facilmente de uma lesão de DNA, impedindo, assim, a reparação, a replicação e a transcrição do DNA e, finalmente, levando à apoptose e/ou morte celular. O tratamento de linhagens celulares cancerígenas portadoras de defeitos em genes de reparo de DNA com talazoparibe como agente único leva ao aumento do nível de cH2AX, que é um marcador da quebra da cadeia dupla de DNA, resultando em diminuição da proliferação celular e aumento da apoptose. A potente citotoxicidade observada com talazoparibe em múltiplas linhagens celulares tumorais contendo mutações nas vias de resposta a danos no DNA (RDD) pode ser atribuída à sua inibição da atividade catalítica de PARP e à captura robusta de PARP. A atividade antitumoral do talazoparibe também foi observada em câncer de mama por modelo de xenoenxerto derivado de pacientes (XDP) com mutação de BRCA previamente tratado com esquema à base de platina. No modelo XDP, talazoparibe diminuiu o crescimento do tumor e aumentou o nível de cH2AX e a apoptose nos tumores.
Detecção de mutação de BRCA
Os pacientes serão elegíveis para o tratamento com Talzenna® se tiverem uma mutação de linha germinativa BRCA deletéria confirmada ou suspeita, (ou seja, uma mutação que interrompe a função normal do gene) Esta detecção da mutação deve ser realizada por um laboratório experiente, utilizando um método validado.
Propriedades Farmacocinéticas
No geral, a exposição ao talazoparibe aumentou proporcionalmente com a dose na faixa de variação de 0,025 mg a 2 mg após administração diária de doses múltiplas. Após a dosagem diária repetida de 1 mg de talazoparibe em pacientes, a área geométrica média sob a curva de concentração plasmática-tempo (AUC) e a concentração plasmática máxima observada (Cmax) de talazoparibe no estado de equilíbrio estava na faixa de variação de 126 ng•h/mL a 208 ng•h/mL e 11,4 ng/mL a 19,1 ng/mL, respectivamente. Após a dosagem diária repetida, a concentração plasmática de talazoparibe atingiu o estado de equilíbrio entre 2 e 3 semanas. A razão de acúmulo mediana de talazoparibe após administração oral repetida de 1 mg uma vez por dia estava na faixa de variação de 2,33 a 5,15.
Absorção
Após a administração oral de talazoparibe, o tempo mediano para a Cmax (Tmax) foi geralmente entre 1 e 2 horas após a administração. O estudo de biodisponibilidade absoluta não foi realizado em humanos. No entanto, com base nos dados de excreção urinária, a biodisponibilidade absoluta é de pelo menos 54,6%, com fração absorvida de pelo menos 68,7% (ver Eliminação).
O efeito de alimentos
A ingestão de alimentos diminuiu a taxa, mas não a extensão da absorção de talazoparibe. Após uma dose oral única de talazoparibe com alimentos ricos em gorduras e altamente calóricos (aproximadamente 827 calorias, 57% de gordura), a Cmax média do talazoparibe diminuiu cerca de 46% e a Tmax mediana foi atrasada de 1 a 4 horas, ao passo que a AUCinf não foi afetada. Com base nesses resultados, Talzenna® pode ser administrado com ou sem alimentos.
Distribuição
A média populacional do volume de distribuição aparente (Vss/F) de talazoparibe foi de 420 L. In vitro, o talazoparibe é ligado aproximadamente 74% às proteínas plasmáticas, sem dependência da concentração na faixa de variação de concentração de 0,01 mM a 1 mM. O comprometimento renal ou hepático não parece impactar a ligação proteica de talazoparibe, uma vez que não havia tendências óbvias na fração média de talazoparibe do fármaco não ligado (fu) no plasma humano in vivo com piora da função renal ou hepática.
Metabolismo
O talazoparibe passa por metabolismo hepático mínimo em humanos. Após a administração oral de uma dose única de 1 mg de talazoparibe [14C] a humanos, não foram identificados metabólitos circulantes importantes no plasma, e o talazoparibe foi a única entidade derivada do medicamento identificada. Nenhum metabólito que representasse individualmente mais de 10% da dose administrada foi recuperado na urina ou nas fezes. As vias metabólicas identificadas do talazoparibe em humanos incluem: 1) mono-oxidação; 2) desidrogenação; 3) conjugação de cisteína de mono-desfluoro-talazoparibe; e 4) conjugação com glucuronida.
In vitro, o talazoparibe não é um inibidor de citocromo (CYP)1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4/5 ou indutor de CYP1A2, CYP2B6 e CYP3A4 em concentrações clinicamente relevantes.
In vitro, o talazoparibe não inibiu nenhum dos principais transportadores de membrana intestinal, hepática ou renal (P-gp, PRCM, polipeptídeo transportador de ânion orgânico [OATP]1B1, OATP1B3, transportador catiônico orgânico [OCT]1 OCT2, transportador de ânion orgânico [OAT]1, OAT3, bomba de exportação de sais biliares [BSEP] e extrusão de toxinas e vários medicamentos [MATE] 1 e MATE2-K) em concentrações clinicamente relevantes.
In vitro, o talazoparibe não inibiu nenhuma das principais isoformas de uridina-difosfato glucuronosiltransferase (UGT) (1A1, 1A4, 1A6, 1A9, 2B7 e 2B15) em concentrações clinicamente relevantes.
Eliminação
A meia-vida plasmática terminal média de talazoparibe foi de 89,8 horas e o clearance oral aparente médio da população (CL/F) foi de 6,45 L/h em pacientes com câncer. Em seis pacientes do sexo feminino com tumores sólidos avançados que receberam uma dose oral única de talazoparibe[14C], uma média de 68,7% e 19,7% da dose radioativa total administrada foi recuperada na urina e nas fezes, respectivamente. A excreção de talazoparibe inalterado na urina foi a principal via de eliminação responsável por 54,6% da dose administrada, enquanto o talazoparibe inalterado recuperado nas fezes foi responsável por 13,6%.
Idade, sexo, raça e peso corporal
A análise PK populacional foi realizada usando dados de 490 pacientes com câncer para avaliar o impacto da idade (variando de 18 a 88 anos), sexo (53 homens e 437 mulheres), raça (361 brancos, 41 asiáticos, 16 negros, 9 outros e 63 não relatada) e peso corporal (variando de 35,7 kg a 162 kg) na PK de talazoparibe. Os resultados mostraram que idade, sexo, raça e peso corporal não tiveram efeito clinicamente relevante na PK de talazoparibe.
População pediátrica
A farmacocinética do talazoparibe não foi avaliada em pacientes < 18 anos.
População idosa
Foi feita uma análise combinada de 494 pacientes que receberam Talzenna®, 85 pacientes tinham ≥65 anos de idade. Não foi observada nenhuma diferença global na segurança ou efetividade de Talzenna® entre esses pacientes e os pacientes mais jovens, mas uma maior sensibilidade em alguns indivíduos mais velhos não pode ser descartada.
Insuficiência renal
Dados de um estudo de farmacocinética em pacientes com câncer avançado com graus variáveis de insuficiência renal indicam que a exposição total ao talazoparibe (AUC0-24), após doses múltiplas de talazoparibe uma vez ao dia, aumentou 12%, 43% e 163% em pacientes com insuficiência renal leve (TFG [Taxa de Filtração Glomerular] 60 - 89 mL/min/1,73 m2), moderada (TFG 30 - 59 mL/min/1,73 m2) e severa (TFG 15 - 29 mL/min/1,73 m2), respectivamente, em relação aos pacientes com função renal normal (TFG ≥90 mL/min/1,73 m2). A Cmáx do talazoparibe aumentou 11%, 32% e 89% em pacientes com insuficiência renal leve, moderada e severa, respectivamente, em relação aos pacientes com função renal normal. Consistente com essas descobertas, uma análise de PK populacional que incluiu 490 pacientes, em que 132 pacientes apresentavam insuficiência renal leve (60 mL/min ≤ CCR [Clearance de Creatinina] < 90 mL/min), 33 pacientes apresentavam insuficiência renal moderada (30 mL/min ≤ CCR < 60 mL/min), e um paciente apresentava insuficiência renal grave (CCR < 30 mL/min), demonstrou que a CL/F de talazoparibe diminuiu 14,4% e 37,1% em pacientes com insuficiência renal leve e moderada, correspondente a 17% e 59% de aumento na ASC, respectivamente, quando comparados com pacientes com função renal normal (CCR ≥90 mL/min). A PK do talazoparibe não foi estudada em pacientes que necessitam de hemodiálise.
Insuficiência hepática
Com base em análise de PK populacional com 490 pacientes, em que 118 pacientes tiveram insuficiência hepática leve (
bilirrubina total ≤1
,0 x LSN e AST > LSN ou bilirrubina total > 1,0 a 1,5 x LSN e qualquer AST),
a insuficiência hepática leve não teve efeito na PK de talazoparibe. A PK do talazoparibe em pacientes com função hepática normal, insuficiência hepática leve, insuficiência hepática moderada (bilirrubina total > 1,5 a 3,0 × LSN e qualquer AST) ou insuficiência hepática grave (bilirrubina total > 3,0 × LSN e qualquer AST) foi estudada em um teste de PK. A análise PK populacional usando dados deste teste de PK indicou que o comprometimento hepático leve, moderado ou grave não teve impacto significativo na PK do talazoparibe.
Eletrofisiologia cardíaca
O efeito de talazoparibe na repolarização cardíaca foi avaliado por eletrocardiogramas (ECGs) de tempo para avaliar a relação entre a mudança do intervalo QT corrigido para frequência cardíaca (QTc) de valor basal e as correspondentes concentrações plasmáticas de talazoparibe em 37 pacientes com tumores sólidos avançados. O talazoparibe não teve um efeito clinicamente relevante no prolongamento do intervalo QTc na dose máxima clinicamente recomendada de 1 mg uma vez por dia.
Dados de Segurança Pré-Clínicos
Carcinogenicidade
Estudos de carcinogenicidade não foram conduzidos com talazoparibe.
Genotoxicidade
O talazoparibe não foi mutagênico no teste de mutação bacteriana reversa (Ames). O talazoparibe foi clastogênico em um ensaio de aberração cromossômica in vitro em linfócitos do sangue periférico humano e em ensaio de micronúcleos in vivo em ratos, com exposições semelhantes às doses clinicamente relevantes. Essa clastogenicidade é compatível com a instabilidade genômica resultante da farmacologia primária de talazoparibe, indicando o potencial de genotoxicidade em humanos.
Toxicidade de dose repetida
Em estudos de toxicidade repetida de até 13 semanas, talazoparibe foi clinicamente tolerado em ratos a 0,04 mg/kg/dia e em cães a 0,01 mg/kg/dia, e as margens de exposição da AUC24 ao nível de ausência de efeitos adversos são 0,2 vez a exposição humana relevante. Os principais achados em exposições subterapêuticas incluíram hipocelularidade da medula óssea com diminuição da dose dependente de células hematopoiéticas, depleção de tecido linfoide em múltiplos órgãos e atrofia e/ou alterações degenerativas nos testículos, epidídimo e túbulos seminíferos. Achados adicionais em exposições mais altas incluíram aumento dependente da dose em apoptose/necrose no trato gastrointestinal (GI), fígado e ovário. A maioria dos achados histopatológicos foi geralmente reversível, enquanto os achados dos testículos foram parcialmente reversíveis após quatro semanas de interrupção da dosagem. Esses achados de toxicidade são compatíveis com a farmacologia do talazoparibe e seu padrão de distribuição tecidual.
Toxicologia reprodutiva
Em um estudo de desenvolvimento embriofetal em ratos, o talazoparibe resultou em morte embrionária, malformação fetal (depressão do bulbo ocular, olhos pequenos, estérnebra dividida, arco vertebral cervical fundido) e variações estruturais nos ossos na exposição materna sistêmica à AUC24 de aproximadamente 0,09 vezes a exposição humana relevante na dose recomendada.
4. CONTRAINDICAÇÕES
Talzenna® é contraindicado em pacientes com hipersensibilidades ao talazoparibe ou a qualquer componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Mielossupressão
Foi relatada mielossupressão, que consiste em anemia, leucopenia/neutropenia e/ou trombocitopenia, em pacientes tratados com talazoparibe (vide item 9. Reações Adversas). Não inicie talazoparibe até que os pacientes tenham se recuperado da toxicidade hematológica causada pela terapia anterior (≤ Grau 1).
Devem ser tomadas precauções para monitorar rotineiramente os parâmetros hematológicos e os sinais e sintomas associados à anemia, leucopenia/neutropenia e/ou trombocitopenia em pacientes que recebem talazoparibe. Na ocorrência de tais eventos, modificações de dose (redução ou interrupção) são recomendadas (vide item 8. Posologia e Modo de Usar). Tratamento de suporte com ou sem transfusões de sangue e/ou plaquetas e/ou administração de fatores estimulantes de colônias podem ser usados conforme apropriado.
Síndrome mielodisplásica/Leucemia mieloide aguda
Foi relatada síndrome mielodisplásica/leucemia mieloide aguda (SMD/LMA) em pacientes que receberam inibidores da polimerase poli (adenosina difosfato [ADP] ribose) (PARP). No geral, foi relatada SMD/LMA em < 1% dos pacientes com tumores sólidos tratados com talazoparibe em estudos clínicos. Os potenciais fatores que contribuem para o desenvolvimento de SMD/LMA incluem quimioterapia anterior contendo platina, outros agentes prejudiciais ao DNA ou radioterapia. Deve ser realizado um hemograma completo na avaliação inicial e monitorado mensalmente quanto a sinais de toxicidade hematológica durante o tratamento. Se SMD/LMA forem confirmadas, o talazoparibe deverá ser descontinuado.
Toxicidade embriofetal
Estudos em animais mostraram toxicidade embriofetal, e talazoparibe foi clastogênico em ensaios in vitro e in vivo (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos). O talazoparibe não deve ser administrado a pacientes grávidas ou que pretendam engravidar durante o tratamento. Mulheres em idade reprodutiva devem ser orientadas a evitar a gravidez durante o uso de Talzenna®. Talzenna® pode prejudicar o feto quando administrado a uma mulher grávida. Oriente pacientes grávidas sobre o risco potencial para o feto (vide item 5. Advertências e Precauções - Fertilidade, gravidez e lactação).
Um método altamente efetivo de contracepção é necessário para pacientes do sexo feminino durante o tratamento com Talzenna® e por, pelo menos, sete meses após o término da terapia. Oriente pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo e parceiras grávidas a usar contracepção efetiva (mesmo após vasectomia) durante o tratamento com Talzenna® por pelo menos 4 meses após a dose final.
Fertilidade, gravidez e lactação
Mulheres em idade fértil/contracepção em homens e mulheres
Mulheres em idade fértil não devem engravidar enquanto recebem Talzenna® e não devem estar grávidas no início do tratamento. Um teste de gravidez deve ser realizado em todas as mulheres em idade fértil antes do tratamento (vide item 5. Advertência e Precauções).
Um método altamente efetivo de contracepção é necessário para pacientes do sexo feminino durante o tratamento com Talzenna® e por, pelo menos, sete meses após o término da terapia. Oriente pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo e parceiras grávidas a usar preservativo (mesmo após vasectomia) durante o tratamento com Talzenna® por pelo menos 4 meses após a dose final (vide item 5. Advertência e Precauções).
Gravidez
Não há dados sobre o uso da Talzenna® em mulheres grávidas. Estudos em animais mostraram toxicidade embriofetal (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos). Talzenna® pode prejudicar o feto quando administrado a uma mulher grávida. Talzenna® não é recomendado durante a gravidez ou para mulheres em idade fértil que não estejam usando métodos contraceptivos (vide item 5. Advertência e Precauções).
Talzenna® é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Amamentação
Não se sabe se Talzenna® é excretado no leite humano. Não pode ser excluído um risco para recém-nascidos/lactentes e, portanto, a amamentação não é recomendada durante o tratamento com Talzenna® e por pelo menos 1 mês após a dose final.
Fertilidade
Não há informações sobre a fertilidade em pacientes. Com base em achados não clínicos em testículos e ovários, a fertilidade masculina e feminina pode ser comprometida pelo tratamento com Talzenna® (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos).
Efeitos na Habilidade de Dirigir e Operar Máquinas
Não foram conduzidos estudos sobre o efeito do talazoparibe na habilidade para dirigir e operar máquinas. Entretanto, os pacientes que apresentarem fadiga/astenia ou tontura com o uso de Talzenna® deverão ter cautela ao dirigir ou operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
O talazoparibe é um substrato para os transportadores de medicamento P-gp e PRCM e é principalmente eliminado por depuração renal como composto inalterado.
Agentes que podem afetar a concentração plasmática de talazoparibe
Efeito dos inibidores de P-gp
Dados de um estudo de interação medicamentosa realizado em pacientes com tumores sólidos avançados, indicaram que a coadministração de doses diárias múltiplas de um inibidor de P-gp, itraconazol 100 mg duas vezes ao dia com uma dose única de talazoparibe 0,5 mg aumentou a exposição total do talazoparibe (AUCinf) e o pico de concentração (Cmáx) em aproximadamente 56% e 40%, respectivamente, em relação a uma dose única de 0,5 mg de talazoparibe administrada isoladamente.
A análise farmacocinética (PK) populacional demonstrou que o uso concomitante de inibidores potentes da P-gp com o Talzenna® aumentou a exposição de talazoparibe em 44,7% em comparação com o Talzenna® administrado isoladamente. Se os pacientes precisarem de administração concomitante com um inibidor potente de P-gp, aqueles que resultam em aumento de ≥2 vezes na exposição de um substrato da amostra P-gp in vivo, (incluindo, entre outros, amiodarona, carvedilol, claritromicina, cobicistate, darunavir, dronedarona, eritromicina, indinavir, itraconazol, cetoconazol, lapatinibe, lopinavir, propafenona, quinidina, ranolazina, ritonavir, saquinavir, telaprevir, tipranavir, valspodar e verapamil), reduza a dose de Talzenna® para 0,75 mg uma vez por dia (vide item 8. Posologia e Modo de Usar).
A análise farmacocinética (PK) populacional em estudos clínicos demonstrou que a co-administração com inibidores relativamente fracos da P-gp (incluindo azitromicina, atorvastatina, diltiazem, felodipina, fluvoxamina e quercetina), não tiveram efeitos significativos na exposição ao talazoparibe.
Efeito dos indutores de P-gp
Dados de um estudo de interação medicamentosa em pacientes com tumores sólidos avançados indicaram que a coadministração de um indutor P-gp (rifampicina 600 mg uma vez ao dia), com uma dose única de talazoparibe 1 mg, aumentou a Cmáx do talazoparibe em 37%, sem afetar a exposição ao talazoparibe.
Efeito dos inibidores de PRCM
O efeito dos inibidores de PRCM na PK do talazoparibe não foi estudado. O uso concomitante de inibidores potentes de PRCM (incluindo, entre outros, curcumina, ciclosporina e elacridar [GF120918]) deve ser evitado (vide item 8. Posologia e Modo de Usar).
Efeito de agentes redutores de ácido
A análise de PK populacional indica que a administração concomitante de agentes redutores de ácido, incluindo inibidores da bomba de prótons (IBP), antagonistas do receptor 2 de histamina (H2RA) ou outros agentes redutores de ácido não tiveram impacto significativo na absorção do talazoparibe.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Talzenna® deve ser conservado em temperatura ambiente (entre 15 °C e 30 °C) e pode ser utilizado por 48 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas:
Talzenna® 0,25 mg: cápsulas de tamanho 4, com tampa de cor marfim opaca com inscrição "Pfizer" em preto e corpo branco com inscrição "TLZ 0,25" em preto.
Talzenna® 1 mg: cápsulas de tamanho 4, com tampa de cor vermelho clara com inscrição "Pfizer" em preto e corpo branco com inscrição "TLZ 1" em preto.
8. POSOLOGIA E MODO DE USAR
O tratamento com Talzenna® deve ser iniciado e supervisionado por um médico com experiência no uso de medicamentos antineoplásicos.
A detecção de mutações nos genes BRCA1 e BRCA2 relacionados ao câncer de mama hereditário deve ser realizada por um laboratório experiente, utilizando um método de teste validado (vide item 3. Propriedades Farmacológicas - Propriedades Farmacodinâmicas).
Posologia
A dose recomendada de Talzenna® é uma cápsula de 1 mg uma vez por dia, via oral. Os pacientes devem ser tratados até que ocorra progressão da doença ou toxicidade inaceitável.
Dose omitida
Caso o paciente apresente vômito ou deixe de tomar uma dose, ele não deve tomar uma dose adicional. A dose prescrita seguinte deve ser tomada no horário habitual.
Modificações de dose
Para tratar reações adversas ao medicamento, considere a interrupção do tratamento ou a redução da dose com base na gravidade e na apresentação clínica. As reduções de dose recomendadas estão indicadas na Tabela 2.
Monitorar o hemograma antes do início da terapia com Talzenna® e mensalmente e conforme indicação clínica (vide Tabela 3 e item 5. Advertências e Precauções).
Tratamento concomitante com inibidores da glicoproteína P (P-gp)
Inibidores potentes da P-gp podem levar ao aumento da exposição a talazoparibe. O uso concomitante de inibidores potentes da P-gp durante o tratamento com talazoparibe deve ser evitado. A administração concomitante somente deve ser considerada após avaliação cuidadosa dos possíveis benefícios e riscos. Se a administração concomitante com um inibidor potente da P-gp for inevitável, a dose de Talzenna® deve ser reduzida a próxima menor dose. Quando o inibidor potente da P-gp for descontinuado, a dose de Talzenna® deve ser aumentada (após 3 a 5 meias-vidas do inibidor da P-gp) até a dose utilizada antes do início do inibidor potente da P-gp (vide item 6. Interações Medicamentosas).
Tratamento concomitante com inibidores da Proteína Resistente ao Câncer de Mama (PRCM)
O efeito da administração concomitante de inibidores da PRCM com o Talzenna® não foi estudado. Sendo assim, o uso concomitante de inibidores da PRCM durante o tratamento com talazoparibe deve ser evitado (vide item 6. Interações Medicamentosas).
Populações especiais
Insuficiência hepática
Nenhum ajuste de dose é necessário para pacientes com insuficiência hepática leve (bilirrubina total ≤1× limite superior da normalidade [LSN] e aspartato aminotransferase (AST) > LSN, ou bilirrubina total > 1,0 a 1,5 × LSN e qualquer AST), insuficiência hepática moderada (bilirrubina total > 1,5 a 3,0 × LSN e qualquer AST) ou insuficiência hepática grave (bilirrubina total > 3,0 × LSN e qualquer AST) (vide item 3. Propriedades Farmacocinéticas).
Insuficiência renal
Nenhum ajuste de dose é necessário para pacientes com insuficiência renal leve (60 mL/min ≤ clearance de creatinina [CCR] < 90 mL/min). Para pacientes com insuficiência renal moderada (30 mL/min ≤ClCr < 60 mL/min), a dose recomendada de Talzenna® é de 0,75 mg uma vez por dia. Para pacientes com insuficiência renal grave (15 mL/min ≤ CCR < 30 mL/min), a dose recomendada de Talzenna® é de 0,5 mg uma vez ao dia. Talzenna® não foi estudado em pacientes que necessitam de hemodiálise (vide item 3. Propriedades Farmacocinéticas)
População idosa
Não é necessário ajuste de dose em pacientes idosos (≥65 anos de idade) (vide item 3. Propriedades Farmacocinéticas).
População pediátrica
A segurança e a eficácia de Talzenna® em crianças e adolescentes com idades < 18 anos não foram estabelecidas.
Este medicamento não deve ser partido, aberto ou mastigado.
9.REAÇÕES ADVERSAS
O perfil de segurança global de Talzenna® é baseado em dados agrupados de 494 pacientes que receberam 1 mg por dia de talazoparibe em estudos clínicos para tumores sólidos, incluindo 286 pacientes de um estudo randomizado de Fase 3 com câncer de mama localmente avançado ou metastático, de linha germinativa BRCA com mutação, negativo para HER2, e 83 pacientes de um estudo não randomizado de Fase 2 em pacientes com câncer de mama localmente avançado ou metastático, de linha germinativa BRCA com mutação (Tabela 4).
As reações adversas mais frequentes (≥25%) em pacientes tratados com talazoparibe nesses estudos clínicos foram fadiga (57,1%), anemia (49,6%), náusea (44,3%), neutropenia (30,2%), trombocitopenia (29,6%) e cefaleia (26,5%). As reações adversas de Grau ≥3 mais comuns (≥10%) de talazoparibe foram anemia (35,2%), neutropenia (17,4%) e trombocitopenia (16,8%).
Ocorreram modificações de dose (reduções de dose ou interrupções de dose) por alguma reação adversa em 62,3% dos pacientes que receberam Talzenna®. As reações adversas mais comuns que levaram às modificações de dose foram anemia (33,0%), neutropenia (15,8%) e trombocitopenia (13,4%).
A descontinuação permanente por reação adversa ocorreu em 3,6% dos pacientes que receberam Talzenna®. A anemia foi o motivo principal para a descontinuação permanente do medicamento em estudo, ocorrendo em 3 pacientes (0,6%), todos os outros eventos adversos que foram o motivo principal para a descontinuação permanente do medicamento em estudo ocorreram em 1 paciente cada. A duração mediana da exposição foi de 5,4 meses (faixa de variação de 0,03 a 61,1).
Mielossupressão
As reações adversas relacionadas à mielossupressão de anemia, neutropenia e trombocitopenia foram notificadas com muita frequência em pacientes tratados com talazoparibe 1 mg/dia. Eventos relacionados à mielossupressão de grau 3 e grau 4 foram relatados para anemia 34,8% e 0,4%, neutropenia 15,6% e 1,8% e trombocitopenia 12,8% e 4,0%. Nenhuma morte foi relatada devido a reações adversas relacionadas à mielossupressão. Eventos adversos relacionados à mielossupressão associados a modificações de dose foram relatados em até aproximadamente 30% dos pacientes na população de talazoparibe 1 mg/dia e aqueles associados à descontinuação permanente do medicamento do estudo foram relatados em menos de 1% dos pacientes.
A Tabela 4 resume as reações adversas com base em conjuntos de dados agrupados listados por classe de sistema de órgãos e categoria de frequência. As categorias de frequência são definidas como: muito comum (≥ 1/10) e comum (≥ 1/100 a < 1/10). Dentro de cada grupo de frequência, as reações adversas são apresentadas por ordem decrescente de gravidade.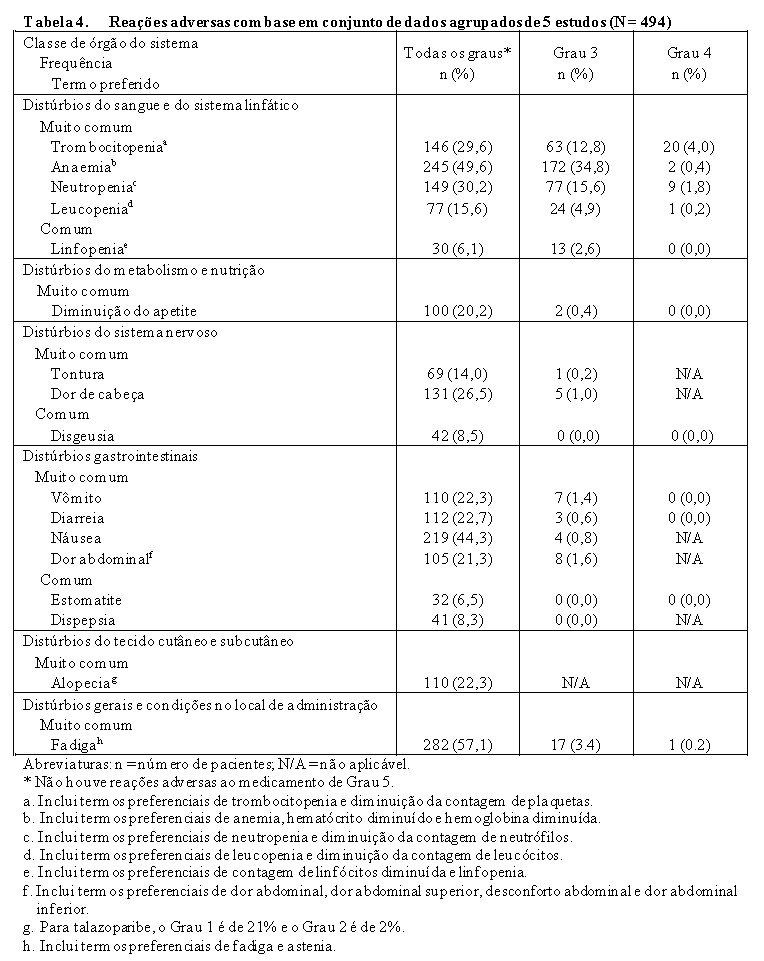
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Não existe tratamento específico em caso de superdosagem com talazoparibe e os sintomas de superdosagem não foram estabelecidos. Em caso de superdosagem, o tratamento com talazoparibe deve ser interrompido e os médicos devem considerar a descontaminação gástrica, seguir as medidas gerais de suporte e tratar os sintomas.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.2110.0482
VENDA SOB PRESCRIÇÃO MÉDICA