TAKHZYRO
TAKEDA
lanadelumabe
Tratamento do angioedema hereditário (AEH).
Apresentações.
TAKHZYRO 150 mg/mL, solução injetável é fornecida na seguinte apresentação:
1 frasco-ampola de 2 mL de solução injetável (300 mg/2 mL) e conjunto de infusão.
VIA SUBCUTÂNEA
USO ADULTO E PEDIÁTRICO ACIMA DE 12 ANOS
Composição.
Cada frasco-ampola com 2 mL contém 300 mg de lanadelumabe. Excipientes: fosfato de sódio dibásico di-hidratado; ácido cítrico monoidratado; histidina; cloreto de sódio; polissorbato 80; água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
TAKHZYRO é indicado para a prevenção de rotina de crises recorrentes de angioedema hereditário (AEH) em pacientes com 12 anos de idade ou mais.
2. RESULTADOS DE EFICÁCIA
Estudo HELP
O estudo HELP foi um estudo multicêntrico, randomizado, duplo-cego, controlado por placebo, de grupos paralelos em 125 indivíduos (115 adultos e 10 adolescentes) com AEH sintomático tipo I ou II. Os indivíduos foram randomizados em 1 de 4 grupos de tratamento paralelos, estratificados pela taxa basal de crises, na proporção 3:2:2:2 (placebo, lanadelumabe 150 mg a cada 4 semanas, lanadelumabe 300 mg a cada 4 semanas, ou lanadelumabe 300 mg a cada 2 semanas por injeção SC) para o período de tratamento de 26 semanas.
A mediana (intervalo) de idade da população do estudo foi de 42 (12 a 73) anos, com 88 mulheres (70%). Foi reportada uma história de crises de angioedema na laringe em 65% (81/125) dos indivíduos e 56% (70/125) dos pacientes estavam recebendo profilaxia prévia de longo prazo (LTP). Durante o período de execução do estudo, a taxa média de crise foi de 3,7 crises / mês, com 52% (65/125) dos participantes com ≥ 3 crises / mês.
Todos os grupos de tratamento com TAKHZYRO apresentaram reduções estatisticamente significativas na taxa média de crise de AEH em comparação com o placebo em todos os desfechos primários e secundários na população de intenção de tratamento (ITT) (Tabela 1).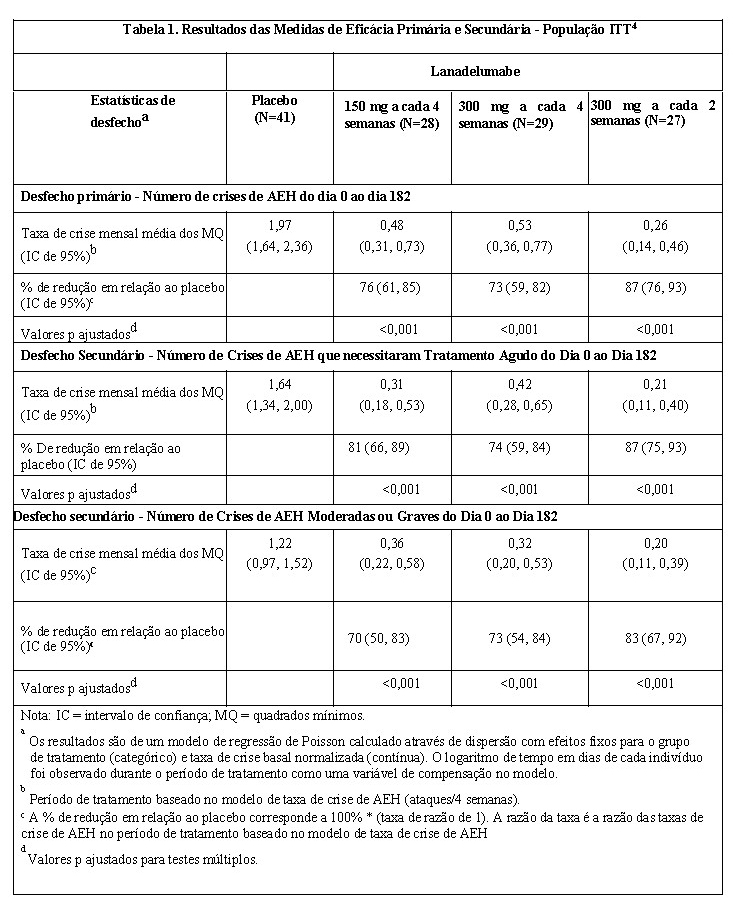
A redução média na taxa de crise de AEH foi consistentemente mais alta entre os grupos de tratamento com TAKHZYRO em comparação com placebo, independentemente do histórico basal de LTP, ataques laríngeos ou taxa de crise durante o período do estudo. A porcentagem de indivíduos sem crises é fornecida na Tabela 2.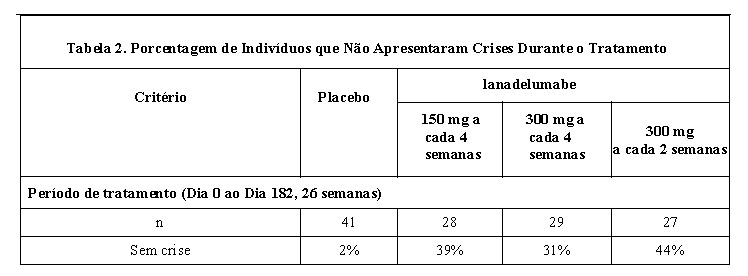
A porcentagem de pacientes que estavam sem crises nas últimas 16 semanas (Dia 70 ao Dia 182) do estudo foi de 77% no grupo de 300 mg a cada 2 semanas, em comparação com 3% dos pacientes no grupo placebo.
100% dos indivíduos no grupo 300 mg a cada 2 semanas ou a cada 4 semanas e 89% no grupo 150 mg a cada 4 semanas alcançaram uma redução de pelo menos 50% na taxa de crise de AEH em comparação com o período de recrutamento.
Qualidade de vida relacionada com saúde
Todos os grupos de tratamento TAKHZYRO mostraram uma melhoria nas pontuações total e de domínio (funcionamento, fadiga/humor, medo/vergonha e nutrição) do Questionário de Qualidade de Vida no Angioedema (AE-QoL) em comparação com o grupo placebo; a maior melhoria foi observada na pontuação de funcionamento, como mostrado na Tabela 3. Uma redução de 6 pontos é considerada uma melhoria clinicamente significativa. A percentagem de indivíduos que alcançaram uma melhoria clinicamente significativa na pontuação total do AE-QoL foi de 65% (Razão de chances vs placebo, [IC de 95%] = 3,2 [1,1, 9,2]), 63% (2,9 [1,1, 8,1]), e 81% (7,2 [2,2, 23,4]), nos grupos TAKHZYRO 150 mg a cada 4 semanas, 300 mg a cada 4 semanas e 300 mg a cada 2 semanas, respectivamente, em comparação com 37% dos pacientes no grupo placebo.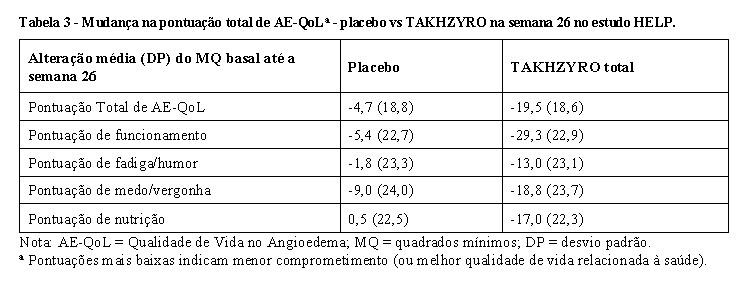
Extensão do estudo HELP
Um total de 212 indivíduos adultos e adolescentes com AEH sintomática tipo I ou II recebeu pelo menos uma dose de lanadelumabe neste estudo, incluindo 109 indivíduos que entraram como prolongamento do estudo HELP e 103 participantes novos ou que não eram parte do prolongamento do estudo (incluindo 19 indivíduos do estudo Fase 1b) que tinham um índice de histórico de crises na situação basal de ≥1 ataque em 12 semanas. Os indivíduos foram autorizados a iniciar a autoadministração depois de receber as 2 primeiras doses de um profissional de saúde em clínica e completar o treinamento apropriado. Análises parciais indicam que o efeito foi mantido por até um ano de tratamento.
Referências bibliográficas: BANERJI, P. Busse, M. Shennak, et. Inibição da Calicreína Plasmática para Profilaxia do Angioedema Hereditário. The New England journal of medicine. 2017; 376 (8): 717-728 Marc A. Riedl, Jonathan A. Bernstein, Timothy Craig, et. al. Estudo em aberto para avaliar a longo prazo a segurança e eficácia de lanadelumab para prevenção dos ataques de angioedema hereditário: desenho da extensão do estudo HELP. Clinical and Translational Allergy. 2017 7:36 Yung Chyung MD; Bradley Vince, DO; Ryan Iarrobino, BA, et. al. Um estudo de fase 1 investigando DX-2930 em pacientes saudáveis. Ann Allergy Asthma Immunol. 2014 1-7.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O lanadelumabe é um anticorpo monoclonal totalmente humano (IgG1 / cadeia leve k). O lanadelumabe inibe a atividade proteolítica da calicreína plasmática ativa. O aumento da atividade da calicreína plasmática resulta em crises de angioedema em pacientes com AEH através da proteólise do cininogênio de alto peso molecular (HMWK) para gerar HMWK clivado (cHMWK) e bradicinina. O lanadelumabe fornece controle sustentado da atividade da calicreína plasmática e, portanto, limita a geração de bradicinina em pacientes com AEH.
Efeitos farmacodinâmicos
A inibição da calicreína plasmática dependente da concentração, medida como redução dos níveis de cHMWK, foi demonstrada após administração subcutânea de TAKHZYRO 150 mg a cada 4 semanas, 300 mg a cada 4 semanas ou 300 mg a cada 2 semanas em indivíduos com AEH. A relação farmacocinética-farmacodinâmica entre TAKHZYRO e cHMWK é descrita por um modelo farmacológico de exposição-resposta indireta. A formação de cHMWK foi reduzida ao máximo em 53,7% com um IC50 de 5705 ng/mL.
Farmacocinética
A farmacocinética de dose única e múltipla de lanadelumabe foi estudada em pacientes com AEH. A farmacocinética de lanadelumabe mostrou uma resposta linear à dose-exposição com doses até 400 mg e exposição reprodutível após administração subcutânea até 12 meses. A biodisponibilidade absoluta do lanadelumabe após administração subcutânea não foi determinada. No estudo HELP, os indivíduos tratados com 300 mg a cada 2 semanas apresentaram média (DP) da área sob a curva ao longo do intervalo de dose no estado estacionário (AUCtau, ss), concentração máxima no estado estacionário (Cmax, ss) e concentração mínima no estado estacionário (Cmín, ss) de 408 mg*dia/mL (138), 34,4 mg/mL (11,2) e 25,4 mg/mL (9,18), respectivamente. O tempo previsto da população para atingir a concentração no estado estacionário foi de aproximadamente 70 dias.
Absorção
Após a administração SC, o tempo para a concentração máxima é de aproximadamente 5 dias. O local de injeção SC (coxa, braço ou abdômen) e a autoadministração não afetaram a absorção de lanadelumabe.
Distribuição
O volume médio (DP) de distribuição de lanadelumabe em pacientes com AEH é de 14,5 litros (4,53). Lanadelumabe é um anticorpo monoclonal terapêutico e não se espera que se ligue às proteínas plasmáticas.
Eliminação
Lanadelumabe tem um clearance corporal total médio (DP) de 0,0297 L/h (0,0124) e uma meia-vida de eliminação terminal de aproximadamente 14 dias.
Populações Especiais
Não foram realizados estudos específicos para avaliar a farmacocinética de lanadelumabe em populações especiais de pacientes, incluindo sexo, idade, gestantes ou a presença de comprometimento renal ou hepático. Numa análise farmacocinética populacional, após a correção do peso corporal, não se verificou influência do sexo ou da idade (12 a 75 anos) no clearance ou volume de distribuição do lanadelumabe. Embora o peso corporal tenha sido identificado como uma covariável importante descrevendo a variabilidade do clearance, um regime de dose de 300 mg a cada 2 semanas forneceu exposição suficiente para a indicação.
Insuficiência renal e hepática
Como os anticorpos monoclonais IgG são eliminados principalmente através do catabolismo intracelular, não se espera que a insuficiência renal ou a insuficiência hepática influenciem no clearance do lanadelumabe. Por conseguinte, numa análise farmacocinética populacional, a insuficiência renal (TFG estimada: 60 a 89 mL/min/1,73 m2 [leve, N = 98] e 30 a 59 mL/min/1,73 m2 [moderada, N = 9]) não teve efeito sobre o clearance ou volume de distribuição de lanadelumabe.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos componentes do produto.
Este medicamento é contraindicado para menores de 12 anos.
5. ADVERTÊNCIAS E PRECAUÇÕES Rastreabilidade
A fim de melhorar a rastreabilidade dos medicamentos biológicos, o nome e o número do lote do produto administrado devem ser claramente registrados.
Reações de hipersensibilidade
Reações de hipersensibilidade foram observadas. No caso de uma reação de hipersensibilidade grave, a administração de TAKHZYRO deve ser interrompida imediatamente e o tratamento apropriado deve ser iniciado.
Geral
TAKHZYRO não se destina ao tratamento de crises agudas de AEH. Em caso de uma crise de AEH, o tratamento individualizado deve ser iniciado com uma medicação de resgate aprovada. Não há dados clínicos disponíveis sobre o uso de lanadelumabe em pacientes com AEH com atividade normal de C1-INH.
Interferência com o teste de coagulação
O lanadelumabe pode aumentar o tempo de tromboplastina parcial ativada (aPTT) devido a uma interação de lanadelumabe com o teste de aPTT. Os reagentes utilizados no teste laboratorial do aPTT iniciam a coagulação intrínseca através da ativação da calicreína plasmática no sistema de contato. A inibição da calicreína plasmática por lanadelumabe pode aumentar o aPTT neste ensaio. Nenhum dos aumentos no aPTT em pacientes tratados com TAKHZYRO foi associado a eventos adversos hemorrágicos anormais. Não houve diferenças na razão normalizada internacional (INR) entre os grupos de tratamento.
Teor de sódio
Este medicamento contém menos de 1 mmol de sódio (23 mg) por frasco, ou seja, é essencialmente "isento de sódio".
Fertilidade, gravidez e lactação
Gravidez
Não existem dados, ou eles são limitados, sobre o uso de lanadelumabe em gestantes. Os estudos em animais não indicam efeitos nocivos diretos ou indiretos no que diz respeito à toxicidade reprodutiva ou de desenvolvimento. Como medida de precaução, é preferível evitar o uso de lanadelumabe durante a gravidez.
Gravidez -Categoria C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgiãodentista.
Lactação
Não se sabe se o lanadelumabe é excretado no leite humano. Sabe-se que as IgG humanas são excretadas no leite materno durante os primeiros dias após o nascimento, e que diminuem para baixas concentrações logo em seguida; consequentemente, um risco para a criança amamentada não pode ser excluído durante este curto período. O desenvolvimento e os benefícios da amamentação devem ser considerados juntamente com a necessidade clínica de lanadelumabe da mãe e quaisquer potenciais efeitos adversos no lactente.
Fertilidade
O efeito de lanadelumabe na fertilidade não foi avaliado em humanos. O lanadelumabe não teve efeito sobre a fertilidade masculina ou feminina em macacos cynomolgus.
Populações Especiais
Idosos
A segurança e eficácia de TAKHZYRO foram avaliadas em um subgrupo de pacientes (N=5) com idade ≥ 65 anos no estudo HELP. Os resultados das análises do subgrupo por idade foram consistentes com os resultados gerais do estudo. Não se espera que a idade afete a exposição ao lanadelumabe. Não é necessário ajuste de dose em pacientes com idade superior a 65 anos (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência renal
Nenhum estudo foi realizado em pacientes com insuficiência renal grave. Não se espera que a insuficiência renal afete a exposição ao lanadelumabe ou o perfil de segurança. Não é necessário ajuste de dose em pacientes com insuficiência renal (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Pacientes pediátricos
A segurança e eficácia de TAKHZYRO foram avaliadas em um subgrupo de pacientes (N=10) com idade entre 12 e 18 anos no estudo HELP. Os resultados da análise do subgrupo por idade foram consistentes com os resultados gerais do estudo A segurança e eficácia de TAKHZYRO em crianças com menos de 12 anos não foram estabelecidas. Nenhum dado está disponível.
Insuficiência hepática
Não foram realizados estudos em pacientes com insuficiência hepática. Não se espera que a insuficiência hepática afete a exposição ao lanadelumabe. Não é necessário ajuste de dose em pacientes com insuficiência hepática (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Efeitos sobre a capacidade de dirigir e usar máquinas
TAKHZYRO tem influência insignificante na capacidade de dirigir ou usar máquinas
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo dedicado à interação medicamentosa foi realizado. Com base nas características de lanadelumabe, não são esperadas interações farmacocinéticas com medicamentos administrados concomitantemente.
Como esperado, o uso concomitante de inibidor da C1 esterase como medicação de resgate resulta em um efeito aditivo na resposta lanadelumabe-cHMWK com base no mecanismo de ação do lanadelumabe e do inibidor da C1 esterase (ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar sob refrigeração entre 2°C e 8°C. Manter o frasco dentro do cartucho para proteger da luz. Não congelar. Prazo de validade: TAKHZYRO, solução injetável, tem validade de 24 meses a partir da data de sua fabricação.
Depois que a seringa é preparada, ela pode ser refrigerada entre 2°C e 8°C e deve ser usada dentro de 8 horas. Do ponto de vista microbiológico, a dose preparada na seringa deve ser administrada imediatamente.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
TAKHZYRO é uma solução estéril, sem conservantes, incolor a ligeiramente amarelada, apresentando-se clara ou ligeiramente opalescente.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O uso deste medicamento deve ser iniciado sob a supervisão de um médico com experiência no tratamento de pacientes com angioedema hereditário (AEH).
Posologia
A dose inicial recomendada é de 300 mg a cada 2 semanas. Em pacientes em tratamento que estão estáveis, sem crises por mais de 6 meses, pode-se considerar uma redução da dose para 300 mg a cada 4 semanas, especialmente em pacientes com baixo peso.
TAKHZYRO não se destina ao tratamento de crises agudas de AEH (ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Doses esquecidas
Se uma dose de TAKHZYRO for esquecida, o paciente deve ser instruído a administrar a dose o mais rapidamente possível, assegurando pelo menos 10 dias entre as doses.
Método de Administração
TAKHZYRO destina-se apenas à administração subcutânea (SC). Cada frasco de TAKHZYRO destina-se apenas a uma única utilização. A injeção deve ser restrita aos locais de injeção recomendados: o abdômen, as coxas e a parte exterior superior dos braços (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS). Recomenda-se a variação do local de injeção.
TAKHZYRO pode ser autoadministrado ou administrado por um cuidador apenas após um treinamento sobre a técnica de injeção SC por um profissional de saúde.
Descarte qualquer dose do medicamento, não utilizada, remanescente no frasco-ampola ou na seringa.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A reação adversa mais comumente observada (52,4%) associada ao TAKHZYRO foram as reações no local de injeção (ISR), incluindo dor no local da injeção, eritema no local da injeção e hematomas no local da injeção. Destas ISRs, 97% foram de intensidade leve, 90% resolvidas dentro de 1 dia após o início, com uma duração média de 6 minutos. Foi observada (1,2%) reação de hipersensibilidade (prurido leve e moderado, desconforto e formigamento da língua), ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES.
Lista tabelada de reações adversas
A Tabela 4 resume as reações adversas observadas no estudo HELP que incluiu 84 indivíduos com AEH, que receberam pelo menos uma dose de TAKHZYRO.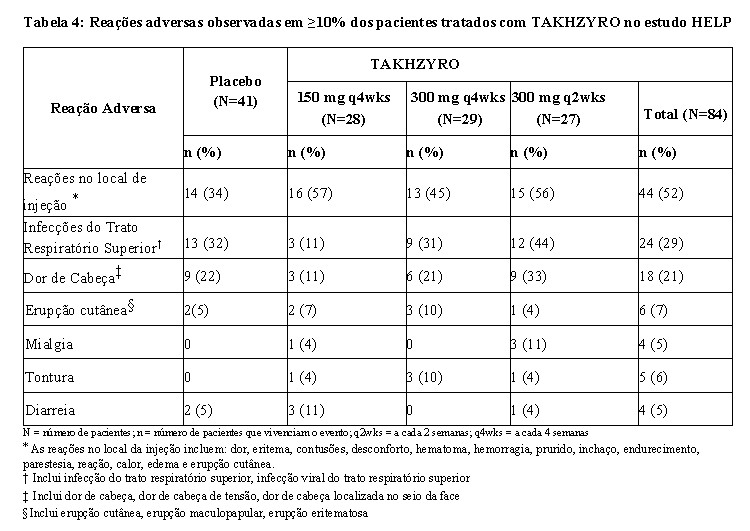
Reações adversas menos comuns
Outras reações adversas que ocorreram com maior incidência em pacientes tratados com TAKHZYRO comparado ao placebo incluem hipersensibilidade (1% vs 0%), aumento de transaminases (2% vs 0%), e aumento de alanina transaminase (2% vs 0%)
População pediátrica
A segurança de TAKHZYRO foi avaliada em um subgrupo de 23 indivíduos com idade entre 12 e < 18 anos. Os resultados da análise do subgrupo foram consistentes com os resultados gerais do estudo para todos os indivíduos.
Imunogenicidade
O tratamento com lanadelumabe foi associado ao desenvolvimento de anticorpos antimedicamento (ADA) emergentes em 11,9% (10/84) dos indivíduos. Todas as titulações de anticorpos foram baixas. A resposta de ADA foi transitória em 20% (2/10) dos indivíduos com ADA positivos. 2,4% (2/84) dos indivíduos tratados com lanadelumabe foram positivos para o teste de anticorpos neutralizantes. O desenvolvimento de ADA incluindo anticorpos neutralizantes contra TAKHZYRO não pareceu afetar adversamente a farmacocinética (PK), farmacodinâmica (PD), segurança ou resposta clínica.
Anormalidades laboratoriais
Elevação de transaminases
Durante o período de tratamento controlado por placebo no estudo HELP, o número de pacientes tratados com TAKHZYRO com nível máximo de transaminase (ALT e AST) > 8, > 5, > 3 vezes o limite normal superior (LNS) foi 1 (1,2%), 0 (0%), ou 3(3,6%) respectivamente, comparado com 0 nos pacientes tratados com placebo. Essas elevações de transaminases foram assintomáticas e transitórias. Nenhum paciente apresentou bilirrubina total elevada > 2x LNS. Um paciente tratado com TAKHZYRO descontinuou, permanentemente, o tratamento devido à elevação de transaminase (4,1 x AST). Nenhum dos pacientes foi relatado como tendo reações adversas graves de transaminases aumentadas.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Nenhum caso de superdosagem foi relatado. Não existe informação disponível para identificar potenciais sinais e sintomas de superdosagem. Se os sintomas ocorrerem, recomenda-se tratamento sintomático. Não há antídoto disponível.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS -1.0639.0290
VENDA SOB PRESCRIÇÃO MÉDICA