TAGRISSO
ASTRAZENECA
osimertinibe
Inibidor da tirosina-quinase.
Apresentações.
Comprimidos revestidos de 40 mg em embalagens com 30 comprimidos.
Comprimidos revestidos de 80 mg em embalagens com 30 comprimidos.
VIA ORAL
USO ADULTO
Composição.
TAGRISSO 40 mg
Cada comprimido revestido contém 47,7 mg de mesilato de osimertinibe (equivalente a 40 mg de osimertinibe).
TAGRISSO 80 mg
Cada comprimido revestido contém 95,4 mg de mesilato de osimertinibe (equivalente a 80 mg de osimertinibe).
Excipientes: manitol, celulose microcristalina, hipromelose, estearil fumarato de sódio, álcool polivinílico, dióxido de titânio, macrogol, talco, óxido de ferro amarelo, óxido de ferro vermelho, óxido de ferro preto e água purificada.
Informações técnicas.
1. INDICAÇÕES
TAGRISSO (osimertinibe) é indicado para o tratamento de pacientes com câncer de pulmão de não pequenas células (CPNPC) localmente avançado ou metastático, positivo para mutação EGFR T790M, cuja doença progrediu quando em uso de, ou após a terapia com inibidores da tirosina quinase dos Receptores do Fator de Crescimento Epidérmico (EGFRs).
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Dois estudos clínicos, de braço único, abertos, AURAex [Fase II, coorte de Extensão, (n=201)] e AURA2 (n=210) foram realizados em pacientes com câncer de pulmão com mutação EGFR T790M positivo que progrediram durante a terapia sistêmica prévia, incluindo uma substância ativa inibidora da tirosina quinase do EGFR. Todos os pacientes deveriam ter CPNPC positivo para a mutação EGFR T790M identificado pelo teste cobas para mutação EGFR realizado em um laboratório central antes da administração. Todos os pacientes receberam TAGRISSO na dose de 80 mg, uma vez ao dia. A medida principal do resultado de eficácia desses dois estudos foi a Taxa de Resposta Objetiva (ORR) de acordo com o RECIST v1.1 conforme avaliação de um Comitê Independente de Revisão Central Cega (BICR). As medidas secundárias do resultado de eficácia foram a Duração da Resposta (DoR) e Taxa de Controle da Doença (DCR).
As características basais da população geral do estudo (AURAex e AURA2) foram as seguintes: mediana de idade de 63 anos, 13% dos pacientes com idade ≥75 anos, 68% mulheres, 36% caucasianos e 60% asiáticos. Todos os pacientes tinham recebido pelo menos uma linha prévia de tratamento, sendo que: 31% (N=129) receberam apenas uma linha prévia de tratamento (somente uso de EGFR-TKI, segunda linha de tratamento, virgens de tratamento com quimioterapia) e 69% (N=282) receberam duas ou mais linhas prévias de tratamento. 72% dos pacientes eram não fumantes, 99% dos pacientes tinham um Performance Status da Organização Mundial da Saúde (OMS) de 0 ou 1 e 39% dos pacientes tinham metástases cerebrais (estáveis por pelo menos 4 semanas e não precisavam de corticosteroides). 59% dos pacientes tinham metástases viscerais extra-torácicas. A mediana da duração do acompanhamento para o AURAex foi de 8,3 meses e 7,0 meses para o AURA2.
O AURA (Fase I) foi um estudo de braço único, aberto, de escalonamento de dose e expansão, que incluiu 271 pacientes pré-tratados, com CPNPC localmente avançado ou metastático durante múltiplas coortes de expansão de dose. A segurança e a eficácia de TAGRISSO 80 mg, uma vez ao dia, foi explorada em uma coorte de expansão de 63 pacientes previamente tratados com CPNPC positivo para mutação T790M confirmada centralmente. Os tratamentos prévios incluem EGFR TKI e quimioterapia. As características demográficas da população do estudo positiva para T790M (n=63) eram idade mediana de 60 anos, mulheres (62%), caucasianos (35%), asiáticos (59%), status de desempenho OMS (Organização Mundial da Saúde) de 0 ou 1 (100%) e que nunca fumaram (67%). O número de linhas de tratamento anteriores variou de 1 a 9. A duração mediana de acompanhamento foi de 8,2 meses. Os resultados de eficácia dos estudos AURA, assim como a análise agrupada (AURAex e AURA2) são resumidas na Tabela 1.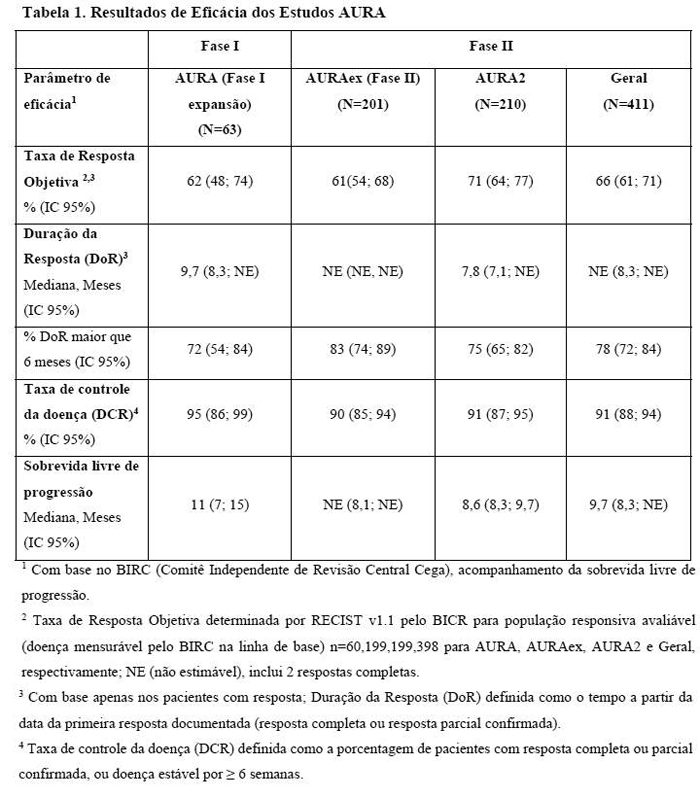
Taxas de resposta objetiva acima de 50% foram observadas em todos os subgrupos pré-definidos analisados, incluindo a linha de terapia, raça, idade e região, para ambos, investigador e avaliação independente.
Dentre os pacientes da população geral com resposta objetiva, 86% (227/263) tiveram resposta documentada no momento do primeiro exame (6 semanas); 96% (253/263) tiveram resposta documentada no momento do segundo exame (12 semanas).
Referências bibliográficas
Jänne P, Yang J, Kim D-W, Planchard D, Ohe Y, Ramalingam S, Ahn M-J, Kim S-W, Su W-C, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown K, Dickinson P, Ghiorghiu S, Ranson M. AZD9291 in EGFR inhibitor resistant Non-Small Cell Lung Cancer. N Engl J Med 2015; 37(2918):1689-99
Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, Yang JC-H, Barrett JC, Janne PA. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced NSCLC. J Clin Oncol 2016 JCO; 34(28): 3375-3382. doi: 10.1200/JCO.2016.66.7162
Cross D, Ashton S, Ghiorghiu S, Eberlein C, Nebhan C, Spitzler P, Orme J, Finlay M, Ward R, Mellor M, Hughes G, Rahi A, Jacobs V, Red Brewer M, Sun J, Jin H, Al-Kadhimi K, Klinowska T, Richmond G, Cantarini M, Kim D-W, Ranson M, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790Mmediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014; 4(9): 1046-1061. doi: 10.1158/2159-8290
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
TAGRISSO é um Inibidor da Tirosina Quinase (TKI). É um potente e seletivo inibidor oral dos Receptores do Fator de Crescimento Epidérmico (EGFRs) que abrigam mutações sensíveis (EGFRm) e mutação T790M TKI-resistente.
Estudos in vitro demonstraram que TAGRISSO possui alta potência e atividade inibitória contra o EGFR na gama de todas as linhagens de células clinicamente relevantes de câncer de pulmão de não pequenas células (CPNPC), mutadas EGFR sensíveis e mutadas T790M (IC50s aparente de 6 nM a 54 nM contra fosfo-EGFR). Isto leva à inibição do crescimento celular, embora mostre atividade menos significante contra o EGFR nas linhagens celulares do tipo selvagem (IC50s aparente de 480 nM a 1,8 mM contra fosfo-EGFR). A administração oral in vivo de TAGRISSO leva à redução do tumor em ambos os modelos de tumor de pulmão em camundongos transgênico e xenoenxerto de CPNPC, EGFRm e T790M.
Eletrofisiologia cardíaca
O potencial de prolongamento do intervalo QT de TAGRISSO foi avaliado em 210 pacientes que receberam osimertinibe 80 mg ao dia, no AURA2. ECGs seriados foram coletados após uma dose única e no estado de equilíbrio para avaliar o efeito de osimertinibe sobre os intervalos QTc. A análise farmacocinética com TAGRISSO foi preditiva de um prolongamento do intervalo QTc relacionado ao medicamento com a dose de 80 mg de 14 mseg com um limite superior de 16 mseg (IC 90%).
Propriedades Farmacocinéticas
Os parâmetros farmacocinéticos do osimertinibe foram caracterizados em voluntários sadios e em pacientes com CPNPC. Com base na análise da farmacocinética na população, a depuração plasmática aparente de osimertinibe é de 14,2 L/h, o volume de distribuição aparente é de 986 L e a meia-vida terminal de aproximadamente é de 48 horas. A AUC e a Cmax aumentaram proporcionalmente à dose ao longo da faixa de dose de 20 a 240 mg. A administração de TAGRISSO uma vez ao dia resulta em um acúmulo de aproximadamente três vezes com exposições no estado de equilíbrio atingidas em 15 dias da administração. No estado de equilíbrio, as concentrações plasmáticas circulantes são mantidas tipicamente dentro de uma faixa de 1,6 vezes ao longo do intervalo de dose de 24 horas.
Absorção:
Após administração oral de TAGRISSO, o pico da concentração plasmática de osimertinibe foi atingido com uma mediana (min-max) tmax de 6 (3 -24) horas, com diversos picos observados ao longo das primeiras 24 horas em alguns pacientes. A biodisponibilidade absoluta de TAGRISSO não foi determinada. Com base em um estudo de farmacocinética clínica em pacientes que receberam 80 mg, a alimentação não alterou a biodisponibilidade de osimertinibe em uma extensão clínica significativa [AUC aumentou 6% (IC 90% -5, 19) e Cmax diminuiu 7% (IC 90% -19, 6)]. Em voluntários sadios que receberam o comprimido de 80 mg, no qual o pH gástrico estava elevado pela administração de omeprazol por 5 dias, a exposição ao osimertinibe não foi afetada (AUC e Cmax aumentaram 7% e 2%, respectivamente) com IC de 90% para a razão da exposição contida dentro do limite de 80-125%.
Distribuição:
A média do volume de distribuição estimado para a população, no estado de equilíbrio (Vss/F) de osimertinibe é de 986 L indicando ampla distribuição no tecido. A ligação à proteína plasmática não pode ser medida devido à instabilidade, mas com base nas propriedades físico-químicas da ligação a proteína do plasma de osimertinibe, esta provavelmente é elevada. O osimertinibe demonstrou também ligar-se covalentemente às proteínas plasmáticas humanas e de ratos, albumina sérica humana e hepatócitos humanos e de ratos.
Biotransformação:
Estudos in vitro indicam que osimertinibe é metabolizado predominantemente pelas CYP3A4 e CYP3A5. O metabolismo mediado por CYP3A4 parece ser uma via metabólica menos importante. Vias metabólicas alternativas que ainda não foram totalmente caracterizadas podem existir. Com base nos estudos in vitro, dois metabólitos farmacologicamente ativos (AZ7550 e AZ5104) foram subsequentemente identificados no plasma de espécies pré-clinicas e humano após administração oral de TAGRISSO; AZ7550 mostrou um perfil farmacológico semelhante ao TAGRISSO enquanto o AZ5104 mostrou maior potência em ambos EGFRs, mutante e tipo selvagem. Os dois metabólitos apareceram lentamente no plasma após a administração de TAGRISSO aos pacientes, com uma mediana (min-max) de tmax de 24 (4-72) e 24 (6-72) horas, respectivamente. No plasma humano, osimertinibe inalterado foi responsável por 0,8%, com os dois metabólitos contribuindo para 0,08% e 0,07% da radioatividade total com a maior parte da radioatividade sendo ligada de forma covalente às proteínas no plasma. A média geométrica da exposição de ambos, AZ5104 e AZ7550, com base na AUC, foi aproximadamente de 10 % cada para a exposição ao osimertinibe no estado de equilíbrio.
A principal via metabólica do osimertinibe foi oxidação e dealquilação. Pelo menos 12 componentes foram observados nas amostras agrupadas de urina e fezes em humanos com cinco componentes representando > 1% da dose, dos quais osimertinibe inalterado, AZ5104 e AZ7550 foram responsáveis por aproximadamente 1,9, 6,6 e 2,7% da dose enquanto que o aduto cisteinil (M21), e um metabólito desconhecido (M25) foram responsáveis por 1,5% e 1,9% da dose, respectivamente.
Eliminação:
Após uma dose oral única de 20 mg, 67,8 % da dose foi recuperada nas fezes (1,2% como forma inalterada) enquanto que 14,2% da dose administrada (0,8% como forma inalterada) foi encontrada na urina em 84 dias da coleta da amostra. O osimertinibe inalterado representou aproximadamente 2% da eliminação com 0,8% na urina e 1,2% nas fezes.
Interações Medicamentosas:
Efeito de outras drogas sobre osimertinibe
Indutores CYP3A: indutores fortes CYP3A podem diminuir a exposição de osimertinibe de modo clinicamente significativo (vide item 4. Contraindicações e item 6. Interações Medicamentosas).
Inibidores fortes CYP3A: Em um estudo de farmacocinética clínica em pacientes, TAGRISSO coadministrado com 200 mg de itraconazol (um inibidor potente da CYP3A4) duas vezes ao dia, não teve efeito clinicamente significante na exposição do osimertinibe [área sob a curva (AUC) aumentada em 24% (IC 90% 15, 35) e Cmax reduzida em -20% (IC 90% -27, -13). Portanto, é provável que inibidores da CYP3A4 não afetem a exposição de osimertinibe. Outras enzimas catalizadoras não foram identificadas.
Agentes redutores da acidez estomacal: A exposição ao osimertinibe não foi afetada pela administração concomitante de dose única de 80mg de TAGRISSO após a administração de 40mg de omeprazol por 5 dias. Os agentes modificadores do pH gástrico podem ser usados concomitantemente com TAGRISSO sem quaisquer restrições.
Efeito de osimertinibe sobre outras drogas
Interação com proteínas transportadoras: Estudo in vitro mostraram que osimertinibe não é um substrato de OATP1B1 e OATP1B3. In vitro, osimertinibe não inibe OAT1, OAT3, OATP1B1, OATP1B3 e MATE2K em concentrações clinicamente relevantes. No entanto, interações com substratos MATE1 e OCT2 não podem ser excluídas.
Vias metabólicas CYP450: Com base em estudos in vitro, osimertinibe é um inibidor competitivo de CYP3A 4/5, mas não CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 e 2E1 em concentrações clinicamente relevantes. Osimertinibe induziu as enzimas CYP3A4 (indução dependente da ativação do Receptor Pregnano X - PXR) e CYP1A2. Não foram estudadas outras interações enzimáticas reguladas pelo Receptor Pregnano X (PXR) além da interação com CYP3A4 (vide item 6. Interações Medicamentosas).
Vias metabólicas UGT: Com base nos estudos in vitro, osimertinibe não é um inibidor da UGT1A1 e UGT2B7 em concentrações hepáticas clinicamente relevantes. A inibição intestinal de UGT1A1 é possível, mas o impacto clínico é desconhecido.
Interações com a glicoproteína P e BCRP: Com base nos estudos in vitro, osimertinibe é um substrato da glicoproteína P e da proteína de resistência ao câncer de mama (BCRP), mas é pouco provável que osimertinibe nas doses clínicas resulte em interações clinicamente relevantes com as substâncias ativas. Com base nos dados in vitro, osimertinibe é um inibidor da BCRP (vide item 6. Interações Medicamentosas) e Pgp. Não foram estudadas outras interações enzimáticas reguladas pelo Receptor Pregnano X (PXR) além da interação com CYP3A4.
Populações especiais:
Em análises farmacocinéticas baseada na população (n=778), não foram identificadas relações clinicamente significantes entre a exposição prevista no estado de equilíbrio (AUCss) e a idade (variação: 21 a 89 anos), gênero, etnia (incluindo caucasianos, asiáticos, japoneses, chineses e paciente não-asiáticos e nãocaucasianos) e condição de tabagismo do paciente (fumantes ativos n=24, ex-fumantes n=232). A análise da farmacocinética na população indicou que o peso corporal foi uma covariável significante com uma alteração de -20% a +30% na AUCss esperada na variação de peso corporal de 90 kg a 43 kg, respectivamente (95% a 5% quantis), quando comparado à AUCss para o peso corporal mediano de 62 kg. Levando-se em consideração os pesos corporais extremos, de < 43 kg a > 90 kg, as proporções do metabólito AZ5104 variaram de 11,8% a 9,6% enquanto para o AZ7550, variaram de 12,8% a 9,9%, respectivamente. Estas variações na exposição decorrentes das diferenças no peso corporal não são consideradas clinicamente relevantes.
Disfunção hepática
O osimertinibe é eliminado principalmente através do fígado, e assim, pacientes com disfunção hepática podem apresentar maior exposição. Estudo de farmacocinética em pacientes com disfunção hepática não foi realizado. Com base na análise da farmacocinética na população, não houve relação entre os marcadores de função hepática (ALT, AST, bilirrubina) e a exposição ao osimertinibe. O marcador albumina sérica para disfunção hepática mostrou um efeito na farmacocinética de osimertinibe. Os estudos clínicos que foram conduzidos excluíram pacientes com AST ou ALT > 2,5 x o limite superior da normalidade (LSN), ou se decorrente da malignidade de base, > 5,0 x LSN ou com bilirrubina total > 1,5 x LSN. Com base na análise farmacocinética de 44 pacientes com disfunção hepática leve (bilirrubina total ≤ LSN e AST > LSN ou bilirrubina total entre 1,0 a 1,5 vezes LSN e qualquer AST), e 330 pacientes com função hepática normal (bilirrubina total < LSN e AST < LSN), as exposições ao osimertinibe foram semelhantes. Há dados limitados em pacientes com disfunção hepática moderada ou grave (ver seção 8. Posologia e Modo de Usar).
Disfunção renal
Um estudo de farmacocinética em pacientes com disfunção renal não foi realizado. Com base em uma análise da farmacocinética na população de 330 pacientes com disfunção renal leve (CLcr 60 a menos de 90 mL/min), 149 pacientes com disfunção renal moderada (CLcr 30 a < 60 mL/min), 3 pacientes com disfunção renal grave (CLcr 15 a < 30 mL/min) e 295 pacientes com função renal normal (≥90 mL/min), as exposições a osimertinibe foram similares. Disfunção renal grave pode influenciar a eliminação de fármacos eliminados hepaticamente. Pacientes com CLcr inferior a 15 mL/min não foram incluídos nos estudos clínicos
Dados de segurança pré-clínicos
Toxicidade em doses repetidas
Os principais achados observados em estudos de toxicidade de dose repetida em ratos e cães abrangem alterações atróficas, inflamatórias e/ou degenerativas que afetam o epitélio da córnea (acompanhados por translucidez e opacidade da córnea em cães observada em exame oftalmológico), trato gastrointestinal (incluindo a língua), pele e tratos reprodutivos masculino e feminino com alterações secundárias no baço. Estes achados ocorreram em concentrações plasmáticas menores àquelas vistas em pacientes que receberam a dose terapêutica de 80 mg. Os achados apresentados após um mês de administração foram largamente reversíveis após um mês da interrupção do tratamento, com exceção de algumas alterações da córnea que foram parcialmente reversíveis.
Dados pré-clínicos indicam que osimertinibe e seu metabólito (AZ5104) inibem o canal h-ERG e não se pode excluir um efeito de prolongamento do intervalo QTc (vide item 3. Eletrofisiologia Cardíaca e item 5. Prolongamento do Intervalo QT).
Carcinogênese e Mutagênese
Estudos de carcinogenicidade não foram realizados com TAGRISSO. TAGRISSO não causou lesão genética nos ensaios in vitro e in vivo.
Toxicologia reprodutiva
Com base nos estudos em animais, a fertilidade dos machos pode estar prejudicada pelo tratamento com TAGRISSO. Alterações degenerativas estavam presentes nos testículos em ratos e cães expostos a TAGRISSO por ≥ 1 mês e houve redução na fertilidade de ratos machos após a exposição ao TAGRISSO por três meses. Esses achados foram observados em concentrações plasmáticas clinicamente relevantes. Os achados à patologia observados nos testículos, após um mês de administração foram reversíveis em ratos; no entanto, uma confirmação definitiva sobre a reversibilidades dessas lesões em cães não pode ser feita.
Com base em estudos em animais, a fertilidade em fêmeas pode ser prejudicada pelo tratamento com TAGRISSO. Nos estudos de toxicidade de dose repetida, foi observada uma incidência aumentada de anoestro, degeneração do corpo lúteo nos ovários e adelgaçamento epitelial no útero e vagina em ratas expostas a TAGRISSO por ≥ 1 mês em concentrações plasmáticas clinicamente relevantes. Os achados nos ovários vistos após um mês de administração foram reversíveis. Em um estudo de fertilidade em ratas, a administração de TAGRISSO 20mg/kg/dia (aproximadamente igual à dose clínica diária recomendada de 80 mg) não causou efeitos no ciclo estral ou no número de fêmeas que ficaram prenhas, no entanto causou morte embrionária precoce. Estes achados demonstraram evidência de reversibilidade após 1 mês sem o medicamento.
Em um estudo de desenvolvimento embrio-fetal modificado em ratos, osimertinibe causou letalidade ao embrião quando administrado a ratas prenhes antes da implantação do embrião. Esses efeitos foram observados na dose tolerada pela fêmea de 20 mg/kg/dia, na qual a exposição foi equivalente à exposição humana na dose recomendada de 80 mg ao dia (com base na AUC total). A exposição a doses de 20 mg/kg e acima, durante a organogênese causou redução no peso fetal, mas nenhum efeito adverso na morfologia externa ou visceral do feto. Quando osimertinibe foi administrado a fêmeas de ratos prenhes ao longo de toda a gestação e então até o início da lactação, houve exposição demonstrável a osimertinibe e seus metabólitos nas crias em amamentação além de uma redução na sobrevida e crescimento deficiente da prole (em doses de 20 mg/kg e acima).
4. CONTRAINDICAÇÕES
Hipersensibilidade conhecida ao osimertinibe ou a qualquer outro excipiente contido na fórmula do medicamento.
TAGRISSO não deve ser coadministrado com Erva de São João (Hypericum perforatum) (vide item 6. Interações Medicamentosas).
5. ADVERTÊNCIAS E PRECAUÇÕES
Ao se considerar o uso de TAGRISSO como tratamento para o CPNPC localmente avançado ou metastático, é importante que o status da mutação EGFR T790M seja determinado por um teste validado que deve ser realizado em laboratório clínico, usando o DNA do tecido tumoral ou o DNA tumoral circulante (ctDNA) obtido de uma amostra de plasma.
Somente teste(s) robusto(s), confiável(eis) e sensível(eis) com utilidade demonstrada para a determinação do status de mutação do EGFR deve(m) ser utilizado(s).
A determinação positiva do status da mutação T790M utilizando tanto o teste com base no tecido como no plasma indica elegibilidade para o tratamento com TAGRISSO. No entanto, se o teste no plasma (ctDNA) for utilizado e o resultado for negativo, recomenda-se que seja repetido o teste com o tecido, sempre que possível, devido à possibilidade de resultados falso-negativos do teste com base no plasma.
Doença Pulmonar Intersticial (DPI)
Reações adversas de doença intersticial pulmonar (DPI) grave, de ameaça à vida ou fatal (por exemplo, pneumonite) foram observadas em estudos clínicos em pacientes tratados com TAGRISSO. Na maioria dos casos houve melhora ou resolução com a interrupção do tratamento. Pacientes com histórico médico de DPI, DPI induzida por medicamento, pneumonite por radiação que precisou de tratamento com esteroides ou qualquer evidência de DPI clinicamente ativa foram excluídos dos estudos clínicos.
Durante todos os estudos clínicos, doença pulmonar intersticial (DPI) ou reações adversas semelhantes a DPI (por exemplo, pneumonite) foram relatadas em 2,9% e foram fatais em 0,3% (n=4) dos 1221 pacientes que receberam TAGRISSO.
Nos estudos de Fase II, a incidência de DPI foi de 6,2% nos pacientes da etnia japonesa, 1,2% nos pacientes de etnia asiática não-japoneses e 2,4% nos pacientes não-asiáticos. A mediana de tempo para início da DPI ou reações adversas semelhantes a DPI foi de 2,7 meses.
Suspenda o TAGRISSO e investigue imediatamente para DPI em todo paciente que apresentar início repentino e/ou piora não explicada de sintomas respiratórios que possam ser indicativos de DPI (por exemplo, dispneia, tosse e febre). Descontinue permanentemente TAGRISSO caso a DPI seja confirmada.
Prolongamento do Intervalo QTc
Dos 411 pacientes no AURAex e AURA2, foi observado que um paciente (menos de 1%) apresentou um QTc maior do que 500 mseg, e 11 pacientes (2,7%) tiveram um aumento em relação ao QTc basal superior a 60 mseg. A análise farmacocinética com TAGRISSO previu um aumento no prolongamento do intervalo QTc dependente da concentração. Nenhum evento de arritmia foi relatado no AURAex ou no AURA2 (ver seção Propriedades Farmacodinâmicas).
Pacientes com anormalidades clinicamente importantes no ritmo e condução de acordo com a medição do eletrocardiograma de repouso (por exemplo, intervalo QTc maior que 470 mseg) foram excluídos dos estudos clínicos.
Quando possível, evite o uso de TAGRISSO nos pacientes com síndrome de QT longo congênita. Considere o monitoramento periódico com eletrocardiogramas (ECGs) e eletrólitos nos pacientes com insuficiência cardíaca congestiva, distúrbios eletrolíticos, ou naqueles que estão em uso de medicamentos que são conhecidos por prolongar o QTc. Suspenda TAGRISSO nos pacientes que desenvolverem um intervalo QTc maior do que 500 mseg em pelo menos dois ECGs separados até que o intervalo QTc seja menor do que 481 mseg ou recuperado até o basal se o intervalo QTc for maior ou igual a 481 mseg, e então, reinicie TAGRISSO em uma dose reduzida conforme descrito na Tabela 2 da seção Posologia e Modo de Usar. Descontinue permanentemente TAGRISSO nos pacientes que desenvolverem prolongamento do intervalo QTc em combinação com qualquer um dos seguintes sintomas: Torsade de pointes, taquicardia ventricular polimórfica, sinais/sintomas de arritmia grave.
Alterações na contratilidade cardíaca
Dos 411 pacientes dos estudos AURA, diminuição igual ou superior a 10% e uma queda inferior a 50% da Fração de Ejeção do Ventrículo Esquerdo (FEVE) ocorreu em 2,4% (9/375) dos pacientes tratados com TAGRISSO que tiveram avaliação da FEVE na linha de base e pelo menos uma avaliação de acompanhamento. Com base nos dados disponíveis dos estudos clínicos, uma relação causal entre os efeitos nas alterações da contratilidade cardíaca e TAGRISSO não foi estabelecida. Nos pacientes com fatores de risco cardíaco e naqueles com condições que possam afetar a FEVE, o monitoramento cardíaco, incluindo uma avaliação da FEVE no início e durante o tratamento, deve ser considerado. Nos pacientes que desenvolverem sinais ou sintomas cardíacos relevantes durante o tratamento, o monitoramento cardíaco, incluindo avaliação da FEVE também deve ser considerado.
Ceratite
Ceratite foi relatada em 0,7% (n = 3) dos 411 pacientes tratados com TAGRISSO nos estudos AURA. Os pacientes que apresentarem sinais e sintomas agudos sugestivos de ceratite, como inflamação ocular, lacrimejamento, sensibilidade à luz, visão turva, dor nos olhos e/ou olhos vermelhos, ou piora desses sinais, devem ser imediatamente encaminhados a um oftalmologista (ver Tabela 2, seção 9. Reações Adversas).
Efeito sobre a capacidade de dirigir veículos e operar máquinas: Nenhum estudo sobre os efeitos na capacidade de dirigir e usar máquinas foi realizado. Se os pacientes apresentarem sintomas que afetam sua capacidade de se concentrar e reagir, é recomendado que eles não dirijam ou utilizem máquinas até que o efeito desapareça.
Uso durante a gravidez e lactação
Categoria de risco na gravidez: D
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Contracepção em homens e mulheres
Mulheres em idade fértil devem ser orientadas a evitar a gravidez enquanto estiverem recebendo TAGRISSO. Os pacientes devem ser orientados a continuar o uso de contracepção efetiva pelos seguintes períodos de tempo após o término do tratamento com o TAGRISSO: pelo menos 6 semanas para as mulheres e 4 meses para homens. Atualmente não se sabe se o osimertinibe pode reduzir a efetividade de contraceptivos hormonais e, portanto, mulheres utilizando contraceptivos hormonais orais devem usar também um método contraceptivo de barreira.
Gravidez
Não há dados ou há uma quantidade limitada de dados sobre a utilização de TAGRISSO em mulheres grávidas. Estudos em animais demonstraram toxicidade reprodutiva (ver seção Dados de segurança préclínicos). Com base em seu mecanismo de ação e nos dados pré-clínicos, TAGRISSO pode causar danos ao feto quando administrados a mulheres grávidas. A administração de osimertinibe a ratas prenhes foi associada com embrioletalidade, crescimento fetal reduzido e morte neonatal em exposições semelhantes às que são esperadas em humanos (ver seção Dados de segurança pré-clínicos). O uso de TAGRISSO não é recomendado durante a gravidez e por mulheres com potencial de engravidar que não estejam utilizando contraceptivos.
Amamentação
Não se sabe se TAGRISSO ou seus metabólitos são excretados no leite humano. Não há informações suficientes sobre a excreção de osimertinibe e seus metabólitos no leite de animais. No entanto, osimertinibe e seus metabólitos foram detectados em filhotes lactentes e foi associado a eventos adversos no crescimento e sobrevivência dos filhotes. O risco para crianças lactentes não pode ser excluído. A amamentação deve ser descontinuada durante o tratamento com TAGRISSO.
Fertilidade
Não existem dados sobre o efeito de TAGRISSO na fertilidade humana. Resultados de estudos animais mostraram que TAGRISSO tem efeitos nos órgão reprodutivos de machos e fêmeas e poderia diminuir a fertilidade (ver seção Dados de segurança pré-clínicos).
Atenção: este medicamento contém açúcar (147 mg/comprimidos de 40 mg e 295 mg/comprimidos de 80 mg), portanto, deve ser usado com cautela e a critério médico em pacientes portadores de diabetes.
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de outras drogas sobre osimertinibe
Indutores CYP3A: Em um estudo de farmacocinética clínica em pacientes, a AUC no estado de equilíbrio de osimertinibe foi reduzido em -78% (IC 90% -81, -76) quando coadministrado com rifampicina (600 mg diariamente por 21 dias). Do mesmo modo, a exposição do metabólito AZ5104 foi reduzida em -82% para a AUC e em -78% para Cmax. Recomenda-se que o uso concomitante de indutores potentes da CYP3A (por exemplo: fenitoína, rifampicina, carbamazepina) com TAGRISSO seja evitado. Indutores CYP3A4 moderados (por exemplo: bosentana, efavirenz, etravirina, modafinil) também podem reduzir a exposição ao osimertinibe e devem ser usados com cautela, ou evitados quando possível. Não há dados clínicos disponíveis para recomendar um ajuste de dose de TAGRISSO. O uso concomitante com Erva de São João é contraindicado (veja item 4. Contraindicações).
Efeito de osimertinibe sobre outras drogas
Substratos CYP3A: Em um estudo de farmacocinética clínica, a coadministração de TAGRISSO com sinvastatina (substrato sensível da CYP3A4) diminuiu a AUC e Cmax da sinvastatina em -9% (IC 90% -23, 8) e -23% (IC 90% -37, -6), respectivamente. Essas alterações são pequenas e provavelmente não serão de relevância clínica. Interações de farmacocinética clínica com substratos da CYP3A4 são improváveis. Não foram estudadas outras interações enzimáticas reguladas pelo Receptor Pregnano X (PXR) além da interação com CYP3A4. Um risco de exposição reduzida a contraceptivos hormonais não pode ser excluído.
Substratos BCRP: Com base nos estudos in vitro, osimertinibe é um inibidor competitivo de transportadores BCRP. Em um estudo de farmacocinética clínica, a coadministração com rosuvastatina (substrato sensível de BCRP) aumentou a AUC e a Cmax da rosuvastatina em 35% (IC 90% 15, 57) e em 72% (IC 90% 46, 103), respectivamente. Pacientes que tomam medicações concomitantes, com disposição dependente de BCRP e com índice terapêutico estreito, devem ser cuidadosamente acompanhados para sinais de mudança na tolerabilidade como resultado da maior exposição da medicação concomitante enquanto recebem TAGRISSO.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
TAGRISSO deve ser conservado em temperatura ambiente (temperatura entre 15°C a 30°C).
TAGRISSO tem validade de 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
TAGRISSO 40 mg é apresentado como um comprimido bege de 9 mm, redondo, biconvexo, gravado com "AZ" e "40" em um lado e sem gravação no lado oposto.
TAGRISSO 80 mg é apresentado como um comprimido bege, com dimensões de 7,25 x 14,5 mm, oval, biconvexo, gravado com "AZ" e "80" em um lado e sem gravação no lado oposto.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O tratamento com TAGRISSO deve ser iniciado por um médico experiente no uso de terapias anticâncer.
Ao se considerar o uso de TAGRISSO como um tratamento para o CPNPC localmente avançado ou metastático, é necessário a determinação do status da mutação EGFR T790M em amostra tumoral (DNA do tecido tumoral ou DNA tumoral circulante (ctDNA) obtido de uma amostra de plasma). O status da mutação EGFR T790M deve ser determinado mediante utilização de um método de teste validado (ver seção 5. Advertências e Precauções).
Posologia
A dose recomendada de TAGRISSO é de 80 mg de osimertinibe, uma vez ao dia, até progressão da doença ou toxicidade inaceitável.
Dose esquecida
Caso uma dose de TAGRISSO seja esquecida, esta deve ser tomada assim que o paciente se lembrar. No entanto, se faltar menos de 12 horas para a próxima dose, a dose esquecida não deve ser tomada e o paciente deve tomar a próxima dose no horário habitual.
Ajuste de dose
A interrupção da dose e/ou a redução da dose podem ser necessárias com base na segurança e tolerabilidade individuais. Se for necessária a redução da dose, então a dose de TAGRISSO deve ser reduzida para 40 mg, uma vez ao dia. As orientações para redução da dose devido à toxicidade das reações adversas são fornecidas na Tabela 2.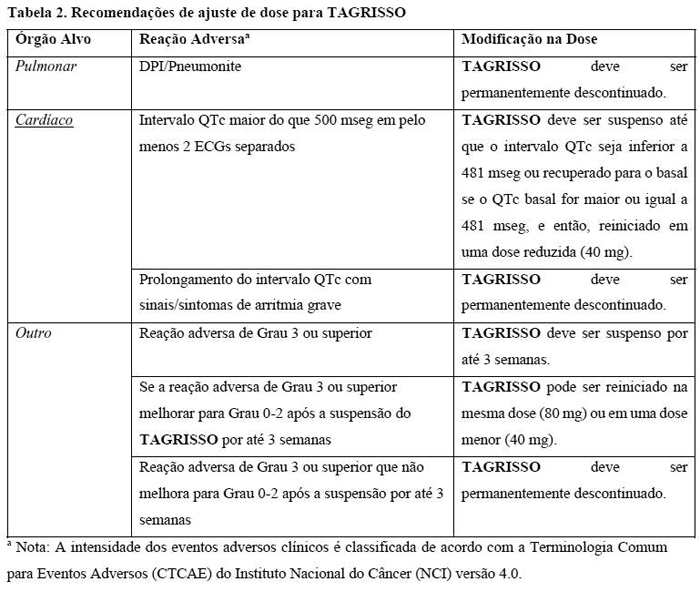
a Nota: A intensidade dos eventos adversos clínicos é classificada de acordo com a Terminologia Comum para Eventos Adversos (CTCAE) do Instituto Nacional do Câncer (NCI) versão 4.0.
Populações especiais:
Nenhum ajuste de dose é necessário devido à idade, peso corporal, sexo, etnia e condição do tabagismo do paciente (ver seção Propriedades farmacocinéticas).
Pacientes pediátricos e adolescentes:
A segurança e a eficácia de TAGRISSO em crianças ou adolescentes com idade inferior a 18 anos não foram estabelecidas. Não há dados disponíveis.
Idosos ( > 65 anos):
A análise da farmacocinética na população indicou que a idade não teve impacto na exposição ao osimertinibe e dessa forma, pode ser usado em adultos independentemente da idade.
Disfunção hepática:
Nenhum estudo clínico foi realizado para avaliar especificamente o efeito da disfunção hepática sobre a farmacocinética de osimertinibe. Com base na análise da farmacocinética na população, nenhum ajuste de dose é recomendado em pacientes com disfunção hepática leve (bilirrubina total ≤ LSN e AST > LSN ou bilirrubina total entre 1,0 a 1,5 x LSN e qualquer valor de AST). A dose apropriada de TAGRISSO não foi estabelecida em pacientes com disfunção hepática moderada ou grave (ver seção Propriedades farmacocinéticas).
Disfunção renal:
Nenhum ajuste de dose é recomendado em paciente com disfunção renal leve e moderada. Dados limitados estão disponíveis em pacientes com disfunção renal grave. A segurança e eficácia de TAGRISSO não foram estabelecidas em pacientes com doença renal em estágio terminal [depuração de creatinina (CLcr) < 15 mL/min, calculado pela equação de Cockcroft e Gault] ou em diálise. Cuidados devem ser tomados quando pacientes com disfunção renal grave ou em estágio terminal são tratados (ver seção Propriedades farmacocinéticas).
Modo de usar
TAGRISSO pode ser tomado com ou sem alimentos, no mesmo horário todos os dias.
Este medicamento destina-se ao uso oral. O comprimido deve ser engolido inteiro com água.
Este medicamento não deve ser partido, aberto ou mastigado.
Se o paciente não for capaz de engolir o comprimido, ele deve primeiro ser dissolvido em 50 mL de água não gaseificada. O comprimido deve ser colocado na água, sem esmagar, agitado até a dispersão, e imediatamente ingerido. Um volume adicional de meio copo de água deve ser adicionado para garantir que nenhum resíduo permaneça no recipiente e então ingerido imediatamente. Nenhum outro líquido deve ser adicionado.
Caso seja necessária administração por sonda nasogástrica, o mesmo processo descrito acima deve ser seguido, mas com a utilização de volumes de 15 mL para a dispersão inicial e de 15 mL para enxágue dos resíduos. O volume resultante de 30 mL de líquido deve ser administrado de acordo com as instruções do fabricante da sonda naso-gástrica com enxágues apropriados com água. A dispersão e os resíduos devem ser administrados dentro de 30 minutos da adição dos comprimidos na água.
9. REAÇÕES ADVERSAS
Resumo geral do perfil de segurança
Os dados de segurança do TAGRISSO refletem a exposição de 411 pacientes com CPNPC positivos para mutação T790M, previamente tratados, que receberam TAGRISSO em dose diária de 80 mg por dia. Dados comparativos de segurança provenientes de estudos clínicos randomizados ainda não estão disponíveis. A maior parte das reações adversas foi de grau 1 ou grau 2 em intensidade. As reações adversas ao medicamento (RAM) mais comumente relatadas foram diarreia (42%) e erupção cutânea (24%). Eventos adversos de grau 3 e grau 4 com TAGRISSO entre os dois estudos foram 26% e 1,2%, respectivamente. Nos pacientes tratados com TAGRISSO 80 mg, uma vez ao dia, reduções na dose devido a RAMs ocorreram em 3,4% dos pacientes. A descontinuação devido a eventos adversos ou parâmetros laboratoriais anormais foi de 5,6%.
Lista tabulada de reações adversas
A Tabela 3 lista as incidências das reações adversas comumente relatadas em pacientes que receberam TAGRISSO. As reações adversas ao medicamento (RAMs) a seguir foram identificadas nos estudos clínicos com pacientes que receberam TAGRISSO como tratamento para CPNPC. A avaliação de segurança relatada abaixo é baseada em 411 pacientes positivos para a mutação EGFR que receberam TAGRISSO em uma dose de 80 mg ao dia. As reações adversas ao medicamento estão listadas de acordo com a classes de sistemas de órgãos MedDRA (CSO MedDRA). Dentro de cada classe de sistema de órgão, as reações adversas ao medicamento são classificadas por frequência, com as reações mais frequentes primeiro. Dentro de cada grupo de frequência, as reações adversas ao medicamento são apresentadas em ordem decrescente de gravidade. Além disso, a categoria de frequência correspondente para cada RAM é baseada na convenção CIOMS III e é definida como: muito comum (≥1/10); comum ( > 1/100 a < 1/10); incomum (≥1/1.000 a < 1/100); rara (≥1/10.000 a < 1/1.000); muito rara ( < 1/10.000); não conhecida (não pode ser estimada a partir dos dados disponíveis). Esta seção inclui apenas dados derivados dos estudos concluídos onde a exposição do paciente é conhecida. Os dados na Tabela 3 são cumulativos e provenientes dos estudos AURA extensão (Fase II) e AURA 2; somente os eventos dos pacientes que receberam pelo menos uma dose de TAGRISSO estão sumarizados.
a Dados são cumulativos dos estudos AURA extensão (Fase II) e AURA 2; apenas os eventos dos pacientes que receberam pelo menos uma dose de TAGRISSO estão sumarizados.
b Terminologia Comum para Eventos Adversos do Instituto Nacional do Câncer, versão 4.0.
c Inclui os casos relatados dentro dos termos agrupados: doença pulmonar intersticial e pneumonite.
d 4 eventos grau 5 CTCAE (fatais) foram relatados.
e Inclui casos relatados dentro dos ter