SUTENT®
PFIZER
sunitinibe, malato
Antineoplásico.
Apresentações.
Sutent® cápsulas de 12,5 mg em embalagens contendo 1 frasco com 28 cápsulas.
Sutent® cápsulas de 25 mg em embalagens contendo 1 frasco com 28 cápsulas.
Sutent® cápsulas de 50 mg em embalagens contendo 1 frasco com 28 cápsulas.
VIA DE ADMINISTRAÇÃO: ORAL
USO ADULTO
Composição.
Cada cápsula de Sutent® 12,5 mg, 25 mg ou 50 mg contém malato de sunitinibe equivalente a 12,5 mg, 25 mg ou 50 mg de sunitinibe base, respectivamente. Excipientes: manitol, croscarmelose sódica, povidona e estearato de magnésio.
Indicações.
Sutent® (malato de sunitinibe) cápsula é indicado para o tratamento de tumor estromal gastrintestinal (GIST) após falha do tratamento com mesilato de imatinibe em decorrência de resistência ou intolerância.
Sutent® é indicado para o tratamento de carcinoma metastático de células renais (RCCm) avançado.
Sutent® também é indicado para o tratamento de tumores neuroendócrinos pancreáticos não ressecáveis.
Resultados de eficácia.
A segurança e eficácia clínica de malato de sunitinibe foram estudadas no tratamento de pacientes com tumor estromal gastrintestinal maligno (GIST) resistentes ao imatinibe (por ex., aqueles que apresentaram progressão da doença durante ou após o tratamento com imatinibe) ou com intolerância ao imatinibe (por ex., aqueles que apresentaram toxicidade significativa durante o tratamento com imatinibe impedindo tratamento adicional), em pacientes com carcinoma metastático de células renais (RCCm) e em pacientes com tumores neuroendócrinos pancreáticos não ressecáveis.
A eficácia está baseada no Tempo até a Progressão Tumoral (TPT) e no aumento da sobrevida nos pacientes com GIST.
A eficácia em pacientes virgens de tratamento e com RCCm refratário a citocinas está baseada na sobrevida livre de progressão (SLP) e nas taxas de resposta objetivas (TRO), respectivamente, e na sobrevida livre de progressão em tumores neuroendócrinos pancreáticos.
Tumores Estromais Gastrintestinais (GIST)
Um estudo inicial aberto, de titulação de dose, foi conduzido em pacientes com GIST após falha do imatinibe (mediana de dose máxima diária de 800 mg) em decorrência de resistência ou intolerância. Noventa e sete pacientes foram incluídos para receber tratamento com diversas doses e esquemas; 55 pacientes receberam 50 mg no esquema de tratamento recomendado, ou seja, 4 semanas com tratamento e 2 semanas sem tratamento (esquema 4/2). Nesse estudo, o TPT mediano e Sobrevida Livre de Progressão (SLP) mediana foram de 34,0 semanas (IC 95% = 22,0 - 46,0 semanas).
Um estudo randomizado de fase 3, duplo-cego, placebo-controlado, de sunitinibe foi conduzido em pacientes com GIST que apresentaram intolerância ou progressão da doença durante ou após o tratamento com imatinibe (mediana de dose máxima diária de 800 mg). Neste estudo, 312 pacientes foram randomizados (2:1) para receber 50 mg de sunitinibe ou placebo, por via oral, uma vez ao dia, seguindo o esquema 4/2 até a progressão da doença ou retirada do paciente do estudo por outro motivo (207 pacientes receberam sunitinibe e 105 pacientes receberam placebo). O endpoint primário de eficácia do estudo foi o Tempo até a Progressão do Tumor (TPT), avaliado pela Revisão Independente, definido como o tempo decorrido entre a randomização e a primeira documentação de progressão tumoral objetiva. Os objetivos secundários incluíram Sobrevida Livre de Progressão (SLP), Taxa de Resposta Objetiva (TRO) e Sobrevida Global (SG).
No momento da análise interina pré-especificada, o TPT mediano de sunitinibe foi de 28,9 semanas (IC 95% = 21,3 - 34,1 semanas), conforme avaliado pelo Investigador, e 27,3 semanas (IC 95% = 16,0 - 32,1 semanas), conforme avaliado pela Revisão Independente, e foi estatística e significativamente mais longo que o TPT de 5,1 semanas (IC 95% = 4,4 - 10,1 semanas), conforme avaliado pelo Investigador, e 6,4 semanas (IC 95% = 4,4 - 10,0 semanas), conforme avaliado pela Revisão Independente. A diferença de sobrevida global foi estatisticamente favorável ao sunitinibe [taxa de risco: 0,491 (IC 95% = 0,290 - 0,0831)]; o risco de morte foi 2 vezes maior para pacientes do braço placebo em comparação ao braço sunitinibe. Informações adicionais de eficácia são apresentadas a seguir na Tabela 1.
Após a análise interina positiva da eficácia e segurança, por recomendação do DSMB Independente (Comitê de Monitoramento de Segurança Independente), o código cego do estudo foi quebrado e aos pacientes do braço placebo foi oferecido tratamento aberto com sunitinibe.
Um total de 225 pacientes receberam sunitinibe na fase aberta do estudo, incluindo 99 pacientes que foram tratados inicialmente com placebo. Nesta análise final, o braço placebo incluiu aqueles pacientes randomizados para placebo que, em seguida, receberam tratamento aberto com sunitinibe.
As análises finais dos endpoints primário e secundário do estudo reafirmaram os resultados obtidos no momento da análise intermediária, como mostrado na Tabela 1 abaixo: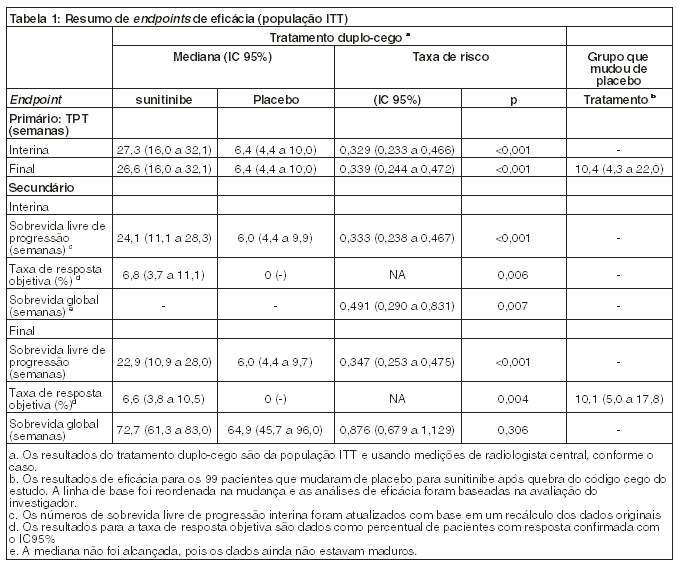
Dos pacientes randomizados para o braço de sunitinibe, 62,7% sobreviveram mais de 1 ano, 35,5% sobreviveram mais de 2 anos e 22,3% sobreviveram mais de 3 anos.
No geral, o estudo demonstrou uma melhora estatística e clinicamente significativa no TPT, o endpoint primário, para sunitinibe com os melhores cuidados de suporte comparado com placebo com os melhores cuidados de suporte.
Carcinoma de Células Renais
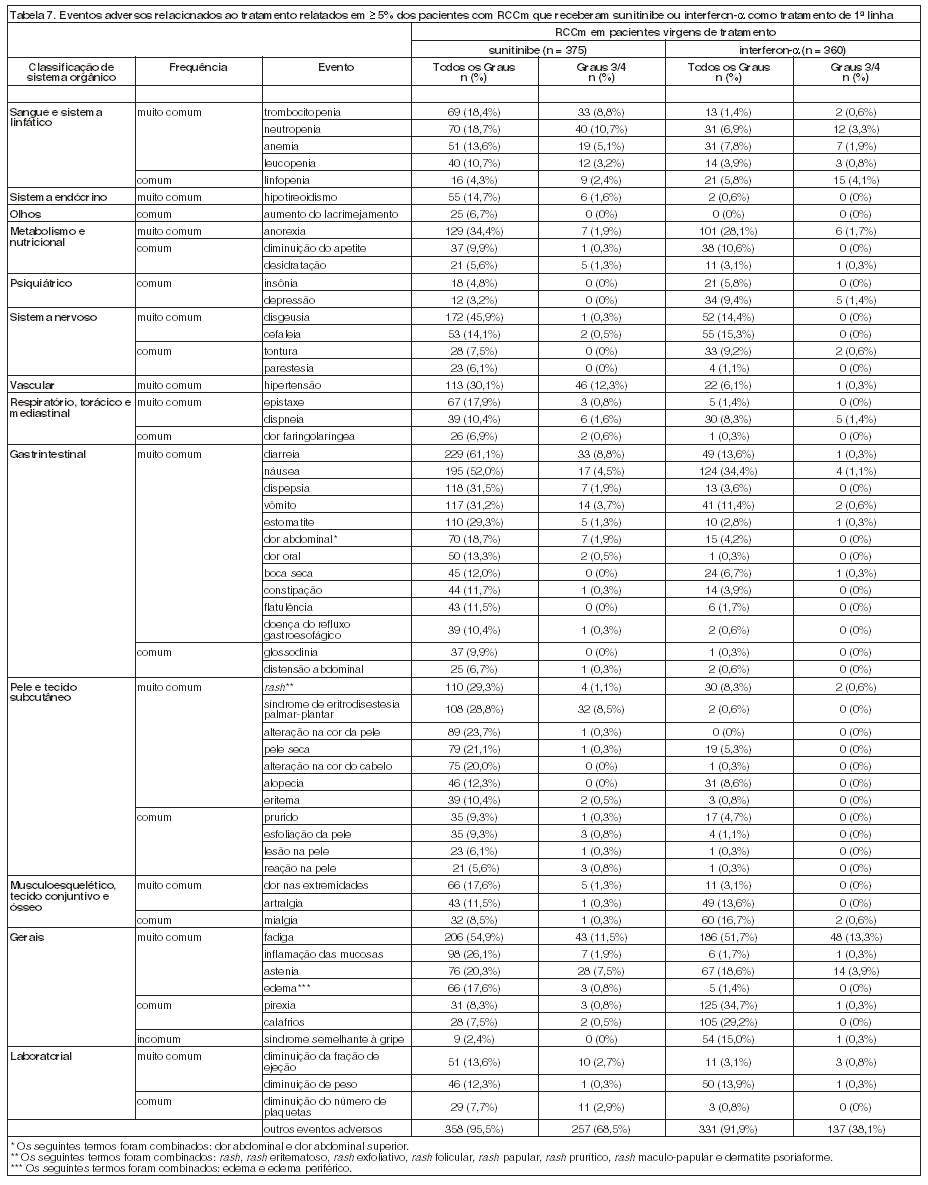
RCCm em pacientes virgens de tratamento
Um estudo randomizado de fase 3 comparando sunitinibe como agente único e interferon-a foi conduzido em pacientes com RCCm virgens de tratamento. O objetivo principal foi comparar a SLP em pacientes recebendo sunitinibe versus pacientes recebendo interferon-a. Os objetivos secundários incluíram TPT, TRO, segurança SG e o desfecho reportado pelo paciente (PRO). Setecentos e cinquenta (750) pacientes foram randomizados (1:1) para receber sunitinibe 50 mg, uma vez ao dia, no esquema 4/2, ou para receber interferon-a administrado subcutaneamente na dose de 9 milhões de unidades internacionais (MIU) três vezes na semana. Os pacientes foram tratados até a progressão da doença ou até serem retirados do estudo por outra razão.
A população ITT (Intenção de Tratamento) incluiu 750 pacientes, 375 randomizados para sunitinibe e 375 randomizados para interferon-a. A idade, o sexo, a raça e o Performance Status ECOG no pré-tratamento foram comparáveis e balanceados entre os grupos recebendo sunitinibe e interferon-a. As características demográficas dos pacientes são demonstradas na tabela a seguir (Tabela 2). O local mais comum de metástases nessa amostra foi o pulmão (78% versus 80%, respectivamente), seguido por acometimento linfonodal (58% versus 53%, respectivamente) e ósseo (30% em cada braço). A maioria dos pacientes apresentava metástases múltiplas (2 ou mais) no pré-tratamento (80% versus 77%, respectivamente).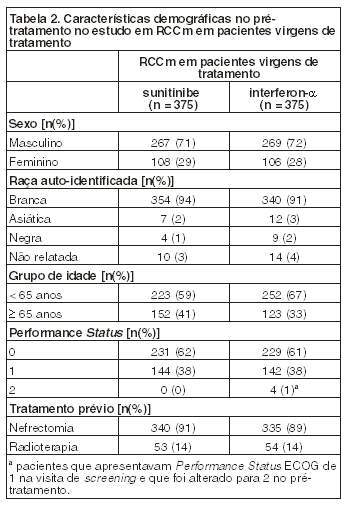
A duração mediana do tratamento foi de 11,1 meses (variação: 0,4 - 46,1) para o tratamento com sunitinibe e 4,1 meses (variação: 0,1 - 45,6) para o tratamento com interferon-a. Interrupções da administração ocorreram em 202 pacientes (54%) com sunitinibe e 141 pacientes (39%) com interferon-a. Reduções da dose ocorreram em 194 pacientes (52%) com sunitinibe e 98 pacientes (27%) com interferon-a. As taxas de descontinuação por causa de reações adversas foram de 20% para sunitinibe e 23% para interferon-a. Os pacientes foram tratados até a progressão da doença ou a retirada do estudo. O endpoint primário de eficácia foi sobrevida livre de progressão (SLP). Uma análise interina planejada demonstrou uma vantagem estatisticamente significativa para sunitinibe em relação ao interferon-a no endpoint primário de SLP, com SLP no grupo recebendo sunitinibe mais que duas vezes maior daquela no grupo recebendo interferon-a (47,3 e 22,0 semanas, respectivamente). O endpoint secundário de TRO foi mais que quatro vezes maior no grupo recebendo sunitinibe que no grupo recebendo interferon-a (27,5% e 5,3%, respectivamente). Os dados não estavam suficientemente maduros para determinar o benefício de sobrevida global; no momento desta análise interina, 374 de 750 pacientes incluídos (50%) continuavam no estudo, 248/375 (66%) no braço recebendo sunitinibe e 126/375 (34%) no braço recebendo interferon-a.
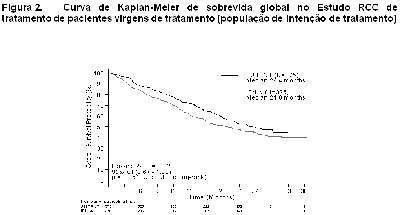
No momento da análise final, houve uma vantagem estatisticamente significativa para sunitinibe em relação ao interferon-a no endpoint de sobrevida livre de progressão (vide Tabela 3 e Figura 1). Nos fatores de estratificação pré-especificados de LDH ( > 1,5 LSN vs. ≤ 1,5 LSN), Performance Status ECOG (0 vs. 1) e nefrectomia anterior (sim vs. não), a taxa de risco favoreceu sunitinibe em relação a interferon. Avaliação de radiologia foi descontinuada depois que o endpoint primário de eficácia foi alcançado. A TRO determinada pela avaliação do investigador foi de 46% (IC95%: 41, 51) para o braço de sunitinibe e 12% (IC95%: 9, 16) para o braço de interferon-a (p < 0,001) (veja Tabela 3).
Os resultados foram similares nas análises de suporte e eles foram robustos quando controlados pelas características demográficas (idade, sexo, raça e Performance Status) e pelos fatores de risco conhecidos. Para 262 de 750 pacientes (35%) sem nenhum fator de risco (critérios do MSKCC - Memorial Sloan Kettering Cancer Center), a SLP mediana foi de 64,1 semanas no braço recebendo sunitinibe e foi de 34,1 semanas no braço recebendo interferon-a (taxa de risco: 0,447, IC 95% [0,313 - 0,640]); para os 424 pacientes (56%) com 1 ou 2 fatores de risco, a SLP mediana foi de 46,6 semanas no braço recebendo sunitinibe e 16,1 semanas no braço recebendo interferon-a (taxa de risco: 0,547, IC 95% [0,423 - 0,707]); e para os 47 pacientes (6%) com 3 ou mais fatores de risco, a SLP mediana foi de 12,1 semanas no braço recebendo sunitinibe e 5,7 semanas no braço recebendo interferon-a (taxa de risco: 0,679 IC 95% [0,330 - 1,398]).
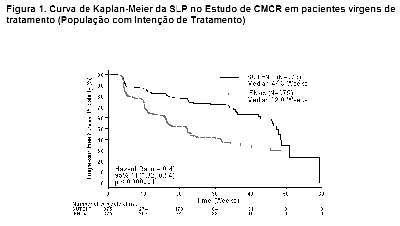
Como mostrado na Figura 2, o tratamento com sunitinibe foi associado com uma sobrevida mais longa comparado com interferon-a. A sobrevida global mediana foi de 114.6 semanas para o braço de sunitinibe (IC 95%: 100,1 - 142,9) e 94,9 semanas para o braço de interferon-a (IC 95%: 77,7 - 117,0) [taxa de risco: 0,821 (IC 95%: 0,673 - 1,001); p = 0,0510 pelo teste de log rank, p = 0,013 pelo teste de Wilcoxon]. Na análise estratificada (LDH > vs. ≤ 1,5 x LSN, Performance Status ECOG 0 vs. ≥ 1 e ausência ou presença de nefrectomia prévia) a razão de risco foi de 0,818 (IC 95%: 0,669 a 0,999; p = 0,049 pelo teste log rank). A sobrevida global mediana para o braço de interferon-a inclui 25 pacientes que descontinuaram o tratamento com interferon-a por causa da progressão da doença e passaram para o tratamento com sunitinibe. Após a descontinuação do estudo, 213 pacientes do braço de interferon-a receberam tratamento pós-estudo contra o câncer, incluindo 32% que receberam sunitinibe; 182 pacientes no braço de sunitinibe receberam tratamento pós-estudo contra o câncer, incluindo 11% que receberam sunitinibe. Em análises post-hoc de censo dos pacientes que passaram do tratamento com interferon-a para o tratamento com sunitinibe no momento da passagem, a sobrevida global foi de 114,6 vs. 86,7 semanas (taxa de risco não estratificado: 0,808; p=0,0361 por teste de log rank; p=0,0081 pelo teste de Wilcoxon). Ao excluir pacientes que receberam tratamento pós-estudo contra o câncer, a sobrevida global mediana foi de 121,9 v.s 61,3 semanas com sunitinibe vs. interferon-a, respectivamente (taxa de risco: 0,647; IC 95%: 0,482 a 0,867; p = 0,0033 pelo teste de log rank; p = 0,0013 pelo teste de Wilcoxon).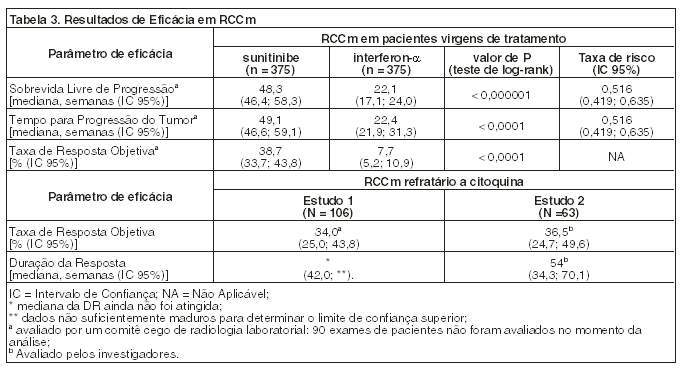
Os desfechos relatados pelos pacientes foram mensurados utilizando a Avaliação Funcional da Terapia do Câncer - Índice de Sintomas do Câncer Renal em Estágio Avançado (FKSI) e da Avaliação Funcional da Terapia do Câncer - Geral (FACT-G). Os endpoints da PRO incluem o escore FKSI, a sua sub-escala de Sintomas Relacionados à Doença (FKSI-DRS), o escore total do FACT-G e suas quatro sub-escalas de escore (bem-estar físico (PWB), bem-estar social/familiar (SWB), bem-estar emocional (EWB) e bem-estar funcional (FWB). O FKSI-DRS foi pré-especificado como o endpoint primário de PRO e utilizado para avaliar os sintomas relacionados ao câncer renal reportado pelos pacientes (falta de energia/fadiga, dor/dor nos ossos, perda de peso, falta de ar, tosse, febre e hematúria) em 719 pacientes. Os pacientes tratados com sunitinibe reportaram índices melhores estatisticamente significativos no escore FKSI-DRS (p≤0,0071), no escore FKSI (p≤0,0133), no escore total FACT-G (p≤0,0244), nos escores de bem-estar físico (p≤0,0208) e de bem-estar funcional (p≤0,0044) quando comparados com pacientes tratados com interferon-a em todos os pontos de tempo avaliados após o prétratamento até o vigésimo ciclo de tratamento. Para o bem-estar físico, bem-estar social/familiar e o bem-estar emocional, o nível estatisticamente significativo aumentou acima do nível 0,05 após o ciclo 13, o ciclo 15 dia 1, e o ciclo 10, respectivamente. Comparadas com as diferenças mínimas clinicamente importantes pré-estabelecidas para estes endpoints, as diferenças entre o tratamento dos sintomas relacionados ao câncer renal (FKSI em todos os pontos de tempo após a linha de base e FKSI-DRS após o ciclo 3, dia 1) e à qualidade de vida geral (FACT-G), em todos os pontos de tempo pós linha de base, foram consideradas clinicamente significativas.
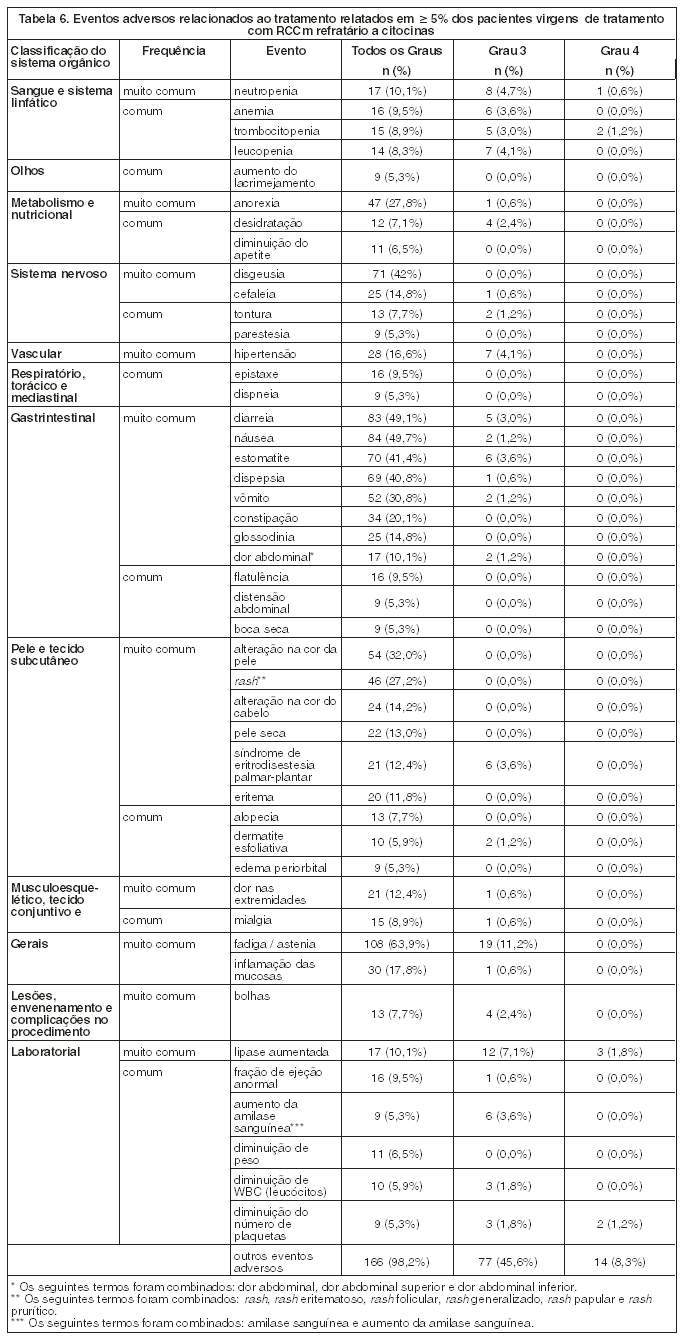
RCCm Refratário a citocinas
Um estudo de fase 2 de sunitinibe foi conduzido em pacientes refratários a uma terapia anterior baseada em citoquina, como interleucina-2 ou interferon-a. Sessenta e três pacientes receberam uma dose inicial de 50 mg de sunitinibe por via oral, uma vez ao dia, no Esquema 4/2 (n = 63). O endpoint primário de eficácia foi a Taxa de Resposta Objetiva (TRO) com base nos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST). Os endpoints secundários incluíam avaliação de TPT, SLP, Duração da Resposta (DR) e SG.
Neste estudo, a taxa de resposta objetiva foi de 36,5% (IC 95% = 24,7% - 49,6%) e o TPT e SLP de 37,7 semanas (IC 95% = 24,0 - 46,4 semanas).
Um estudo confirmatório aberto, multicêntrico e de braço único avaliou a eficácia e segurança de sunitinibe e foi conduzido em pacientes com carcinoma metastático de células renais (RCCm), refratários a uma terapia anterior baseada em citoquina. Cento e seis pacientes receberam no mínimo uma dose de 50 mg de sunitinibe no Esquema 4/2 (n = 106). O endpoint primário de eficácia deste estudo era a TRO. Os endpoints secundários incluíam o TPT, SLP, DR e SG.
Neste estudo, a TRO foi de 34,0 % (IC 95% = 25,0% - 43,8%). As medianas do TPT, da SLP, DR e da SG ainda não foram atingidas.
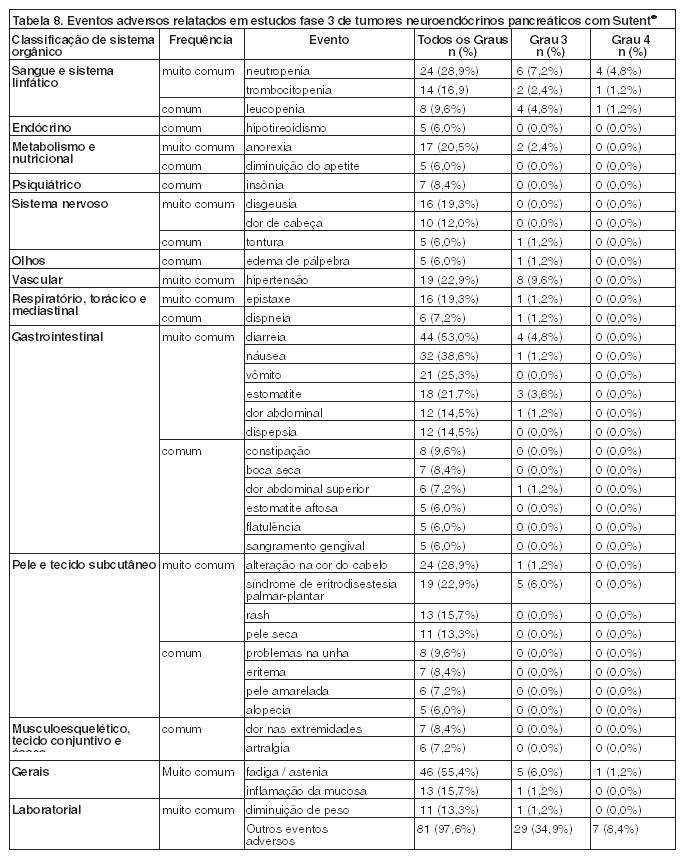
Tumores neuroendócrinos pancreáticos
Um estudo Fase 2, aberto, multicêntico avaliou a eficácia e a segurança do agente único Sutent® 50 mg por dia no Esquema 4/2 (4 semanas de tratamento, 2 semanas de descanso) em pacientes com tumores neuroendócrinos pancreáticos avançados, não ressecáveis. No coorte de 66 pacientes com tumor das ilhotas pancreáticas, foi observada uma Taxa de Resposta Objetiva (TRO) de 17%.
Um estudo pivotal de Fase 3, multicêntrico, internacional, randomizado, duplo-cego, placebo-controlado de sunitinibe como agente único foi conduzido em pacientes com tumores neuroendócrinos pancreáticos não ressecáveis. Os pacientes tinham que ter progressão documentada, baseada nos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST), dentro dos 12 meses anteriores e foram randomizados (1:1) para receber 37,5 mg de sunitinibe uma vez ao dia sem um período de descanso programado (n=86) ou placebo (n=85). O objetivo primário foi comparar a Sobrevida Livre de Progressão (SLP) em pacientes recebendo sunitinibe versus pacientes recebendo placebo. Outros endpoints incluíram a Sobrevida Global (SG), TRO, Desfechos Relatados por Pacientes (DRP) e segurança.
Os dados demográficos foram comparáveis entre os grupos de sunitinibe e de placebo. Além disto, 49% dos pacientes de sunitinibe tiveram tumores não-funcionantes versus 52% dos pacientes placebo e 92% dos pacientes em ambos os braços tiveram metástases hepáticas. O uso de análogos da somatostatina foi permitido no estudo. Um total de 66% dos pacientes de sunitinibe recebeu tratamento sistêmico prévio comparado com 72% dos pacientes placebo. Além disto, 24% dos pacientes de sunitinibe tinha recebido análogos da somatostatina comparado com 22% dos pacientes placebo.
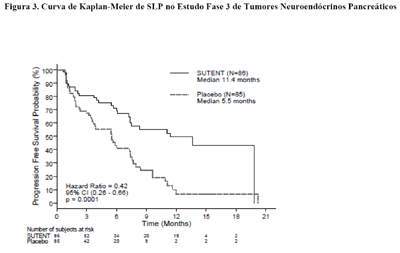
Foi observada uma vantagem clinicamente significativa na SLP para sunitinibe em relação ao placebo. A SLP mediana foi de 11,4 meses para o braço de sunitinibe comparado com 5,5 meses para o braço placebo [taxa de risco: 0,418 (IC95% 0,263; 0,662) valor de p = 0,0001]. Resultados semelhantes foram observados quando as avaliações das respostas do tumor pelo critério de RECIST para medição do tumor pelo investigador foram usadas para determinar a progressão da doença, como mostra a Tabela 4. A taxa de risco favorecendo sunitinibe foi observada em todos os subgrupos de características basais avaliadas, incluindo uma análise por número de terapias sistêmicas prévias. Um total de 29 pacientes no braço do sunitinibe e 24 no braço do placebo não receberam tratamento sistêmico prévio. Entre estes pacientes, a taxa de risco para SLP foi de 0,365 (IC 95% 0,156, 0,857), p = 0,0156. Da mesma forma, entre 57 pacientes no braço de sunitinibe (incluindo 28 com uma terapia sistêmica prévia e 29 com 2 ou mais terapias sistêmicas prévias) e 61 pacientes no braço do placebo (incluindo 25 com uma terapia sistêmica prévia e 36 com 2 ou mais terapias sistêmicas prévias) que tinham recebido terapia sistêmica prévia, a taxa de risco para SLP foi 0,456 (IC 95% 0,264, 0,787), p = 0,0036.
Na análise de sensibilidade da SLP a progressão foi baseada em medições de tumor relatadas pelo investigador e na qual todos os dados censurados por motivos que não o encerramento do estudo foram tratados como se fossem eventos SLP. Esta análise apresentou uma estimativa conservadora do efeito do tratamento com sunitinibe e deu suporte à análise primária, demonstrando uma taxa de risco de 0,507 (IC 95% 0,350, 0,733) e p = 0,000193. O estudo pivotal de tumores neuroendócrinos pancreáticos foi encerrado prematuramente por recomendação de uma Comissão Independente de Monitorização de Medicamentos, e o endpoint primário foi baseado na avaliação do investigador, que podem ter afetado as estimativas do efeito do tratamento.
A fim de excluir viés da avaliação da SLP do pesquisador, uma revisão central, cega e independente foi realizada e deu suporte a avaliação do investigador, como mostra a Tabela 4.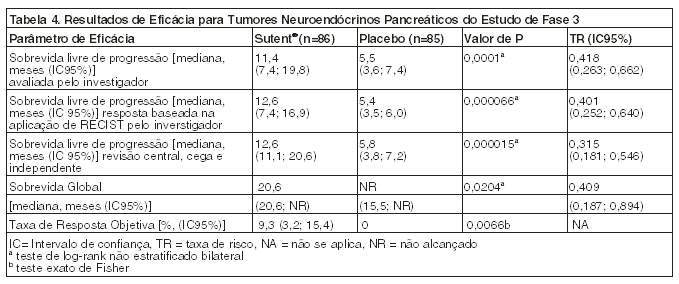
Os dados de Sobrevida Global (SG) não estavam maduros na época da análise. Houve 9 óbitos no braço de sunitinibe e 21 óbitos no braço de placebo. Foi observada uma diferença estatisticamente significativa na Taxa de Resposta Objetiva (TRO) em favor de sunitinibe em relação ao placebo.
Após a progressão da doença, pacientes teriam o medicamento aberto e para os pacientes do braço placebo seria oferecido tratamento aberto com sunitinibe através de um estudo de extensão separado. Como resultado do encerramento antecipado do estudo, os pacientes que permaneceram tiveram o medicamento aberto e foi oferecido acesso ao sunitinibe aberto, em um estudo de extensão. Um total de 59 pacientes do braço placebo recebeu Sutent® em um estudo de extensão.
Os resultados do Questionário de Qualidade de Vida de Câncer da Organização Europeia para Pesquisa e Tratamento (EORTC QLQC-30) demonstrou que a qualidade de vida relacionada à saúde global as cinco esferas de função (física, funcional, cognitiva, emocional e social) foram mantidas para pacientes em tratamento com sunitinibe quando comparado com placebo com efeitos sintomáticos adversos limitados.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
O sunitinibe inibe múltiplos receptores de tirosina-quinase (TKRs) que implicam no crescimento tumoral, na angiogênese patológica e na progressão metastática do câncer. O sunitinibe foi identificado como um inibidor dos receptores do fator de crescimento derivado de plaquetas (PDGFRa e PDGFRb), dos receptores do fator de crescimento vascular endotelial (VEGFR1, VEGFR2 e VEGFR3), do receptor do fator de células tronco (KIT), da tirosina quinase-3 similar a Fms (FLT3), do receptor do fator de estimulação de colônias Tipo 1 (CSF-1R) e do receptor do fator neurotrófico derivado de linhagem celular glial (RET). A inibição da atividade de tirosinaquinase destes receptores pelo sunitinibe foi demonstrada em ensaios bioquímicos e celulares, e a inibição da função foi demonstrada em ensaios de proliferação celular. O principal metabólito exibe uma potência similar ao sunitinibe nos ensaios bioquímicos e celulares.
O sunitinibe inibiu a fosforilação de múltiplos TKRs (PDGFRa, VEGFR2, KIT) em tumores xenográficos com expressão de TKR-alvo in vivo e demonstrou inibição do crescimento tumoral ou regressão tumoral, e/ou inibiu metástases em alguns modelos experimentais de câncer. O sunitinibe demonstrou capacidade de inibir o crescimento de células tumorais com expressão de TKRs-alvo desregulados (PDGFR, RET ou KIT) in vitro e inibir a angiogênese de tumores PDGFRa e VEGFR2-dependentes in vivo.
Propriedades Farmacocinéticas
A farmacocinética do sunitinibe e do malato de sunitinibe foi avaliada em 135 voluntários sadios e em 266 pacientes com tumores sólidos.
Absorção
As concentrações plasmáticas máximas (Cmáx) são observadas geralmente entre 6 - 12 horas (Tmáx) após administração oral. Os alimentos não afetam a biodisponibilidade do sunitinibe.
Distribuição
A ligação de sunitinibe e seu principal metabólito ativo às proteínas plasmáticas humanas in vitro foi de 95% e 90%, respectivamente, sem dependência aparente da concentração na faixa de 100 - 4000 ng/mL. O volume aparente de distribuição (Vd/F) do sunitinibe foi grande (2230 L), indicando distribuição nos tecidos. Nas faixas de dose de 25 a 100 mg, a área sob a curva de concentração-tempo plasmática (AUC) e a Cmáx aumentam de forma proporcional com a dose.
Metabolismo
Os valores de Ki calculados in vitro para todas as isoformas da CYP testadas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 e CYP4A9/11) indicaram que é improvável que o sunitinibe e seu principal metabólito ativo tenham quaisquer interações medicamentosas de relevância clínica com medicamentos que possam ser metabolizados por essas enzimas.
Os estudos in vitro indicam que o sunitinibe não induz e não inibe as principais enzimas da CYP, incluindo CYP3A4 (vide item 6. Interações Medicamentosas).
O sunitinibe é metabolizado principalmente pela CYP3A4, a enzima do citocromo P450, que produz seu principal metabólito ativo, o qual é, então, metabolizado pela CYP3A4. O principal metabólito ativo representa 23 a 37% da exposição total.
Eliminação
A excreção é feita principalmente através das fezes (61%), sendo que 16% da dose administrada e os metabólitos são eliminados por via renal. O sunitinibe e seu principal metabólito ativo (ambos marcados) foram os principais compostos relacionados à droga, identificados no plasma, urina e fezes, representando respectivamente 91,5%, 86,4% e 73,8% da radioatividade nas amostras. Metabólitos secundários foram identificados na urina e fezes, mas geralmente não foram encontrados no plasma. O clearance oral total (CL/F) foi de 34-62 L/h com a variabilidade de 40% entre pacientes.
Após a administração de uma dose oral única a voluntários sadios, as meias-vidas terminais de sunitinibe e de seu principal metabólito ativo desetil foram de aproximadamente 40 - 60 horas e 80 - 110 horas, respectivamente.
Farmacocinética em Populações Especiais de Pacientes
Insuficiência Hepática
O sunitinibe e seu principal metabólito são principalmente metabolizados pelo fígado. Exposições sistêmicas após dose única de sunitinibe são similares em indivíduos com insuficiência hepática leve (Classe A de Child-Pugh) ou moderada (Classe B de Child-Pugh) quando comparadas a indivíduos com função hepática normal. O sunitinibe não foi estudado em indivíduos com insuficiência hepática grave (Classe C de Child-Pugh).
Insuficiência Renal
As análises farmacocinéticas da população mostraram que a farmacocinética de sunitinibe foi inalterada em pacientes com clearance de creatinina calculados na faixa de 42 - 347 mL/min. Exposições sistêmicas após uma dose única de Sutent® foram similares em indivíduos com insuficiência renal grave (CLcr < 30 mL/min) em comparação com indivíduos com função renal normal (CLcr > 80 mL/min). Embora o sunitinibe e o seu metabólito primário não tenham sido eliminados por hemodiálise, em pacientes com doença renal terminal (ESRD), a exposição sistêmica total foi inferior em 47% para sunitinibe e 31% para seu metabólito primário em comparação com indivíduos com função renal normal.
Eletrofisiologia Cardíaca
O prolongamento do intervalo QT foi investigado em um estudo clínico fase 1 com 24 pacientes avaliáveis, com idades entre 20 e 87 anos, com neoplasias malignas avançadas. Nas concentrações plasmáticas terapêuticas a alteração média máxima do QTcF em relação ao pré-tratamento foi de 9,6 ms (IC 90% 15,1 ms). Em concentrações com o dobro das concentrações plasmáticas terapêuticas, a alteração média máxima do QTcF em relação ao pré-tratamento foi 15,4 ms (IC 90% 22,4 ms). O moxifloxacino (400 mg), usado como controle positivo, mostrou alteração média máxima do QTcF em relação ao pré-tratamento de 5,6 ms. Nenhum indivíduo apresentou efeito no intervalo QTc maior que Grau 2 (CTCAE v. 3.0). Nenhum paciente apresentou arritmia cardíaca (vide item 5. Advertências e Precauções).
Farmacocinética Plasmática
Após administração oral de uma dose única em voluntários sadios, a meia-vida de eliminação do sunitinibe e de seu principal metabólito ativo é de aproximadamente 40 - 60 horas e 80 - 110 horas, respectivamente. Com administração diária repetida, ocorre acúmulo de 3 a 4 vezes de sunitinibe, enquanto seu principal metabólito acumula de 7 a 10 vezes. As concentrações de steady state de sunitinibe e de seu principal metabólito são atingidas dentro de 10 a 14 dias. No dia 14, as concentrações plasmáticas combinadas de sunitinibe e de seu metabólito ativo são de 62,9 - 101 ng/mL, as quais são concentrações-alvo previstas a partir dos dados pré-clínicos para inibir a fosforilação in vitro do receptor e resultar em estase tumoral/redução do crescimento tumoral in vivo. Não foram observadas alterações significativas na farmacocinética do sunitinibe ou do seu principal metabólito ativo com administração diária repetida, nem com os ciclos repetidos nos regimes de doses testados.
A farmacocinética foi similar em todas as populações com tumores sólidos testadas e em voluntários sadios.
Farmacocinética Populacional
As análises de farmacocinética populacional dos dados demográficos indicaram que não há efeitos clinicamente relevantes para idade, peso corpóreo, clearance de creatinina, sexo, raça ou pontuação ECOG (Eastern Cooperative Oncology Group) na farmacocinética de sunitinibe ou de seu principal metabólito ativo.
Peso, Performance Status: análises farmacocinéticas populacionais dos dados demográficos indicam que não são necessários ajustes iniciais da dose para o peso ou o performance status medido pela escala do ECOG.
Sexo: os dados disponíveis indicam que mulheres podem ter um clearance aparente (CL/F) de sunitinibe cerca de 30% menor do que os homens: esta diferença, entretanto, não necessita de ajustes da dose inicial.
Dados de Segurança Pré-clínica
Em ratos e macacos, nos estudos de toxicidade de doses repetidas por até 9 meses de duração, os efeitos primários nos órgãos-alvo foram identificados no trato gastrintestinal (emese e diarreia em macacos), glândula suprarrenal (congestão cortical e/ou hemorragia em ratos e macacos, com necrose seguida de fibrose em ratos), sistema hemolinfopoiético (hipocelularidade da medula óssea, e depleção linfoide do timo, baço, e linfonodos), pâncreas exócrino (desgranulação de célula acinar com necrose celular), glândulas salivares (hipertrofia acinar), articulações ósseas (espessamento da placa de crescimento), útero (atrofia) e ovários (desenvolvimento folicular diminuído). Todos os achados ocorreram em níveis de exposição plasmática de sunitinibe clinicamente relevantes. Os efeitos adicionais, observados em outros estudos, incluíram prolongamento de intervalo QTc, reduções na FEVE, hipertrofia hipofisária e atrofia tubular dos testículos, aumento da matriz mesangial nos rins, hemorragia do trato gastrintestinal e da mucosa oral e hipertrofia das células hipofisárias anteriores. Acredita-se que as alterações no útero (atrofia endometrial) e na placa de crescimento ósseo (espessamento epifisário ou displasia da cartilagem) estejam relacionadas à ação farmacológica do sunitinibe. A maioria destes achados foi reversível após 2 a 6 semanas sem tratamento.
Genotoxicidade
O potencial genotóxico de sunitinibe foi avaliado in vitro e in vivo. O sunitinibe não foi mutagênico em bactérias utilizando ativação metabólica proporcionada por fígado de ratos. O sunitinibe não induziu in vitro aberrações cromossômicas estruturais em células linfocíticas de sangue periférico humano. Poliploidia (aberrações cromossômicas numéricas) foi observada in vitro em linfócitos de sangue periférico humano, tanto na presença como na ausência de ativação metabólica. O sunitinibe não foi clastogênico in vivo em medula óssea de ratos. O principal metabólito ativo não foi avaliado quanto ao potencial de toxicidade genética.
Carcinogenicidade
Em um estudo de 1 mês de definição de faixa de dose por gavagem oral (0, 10, 25, 75 ou 200 mg/kg/dia) com administração diária contínua em camundongos transgênicos H2ras foram observados carcinoma e hiperplasia de glândulas de Brunner do duodeno na dose mais elevada testada (200 mg/kg/dia).
Um estudo de 6 meses de carcinogenicidade por gavagem oral (0, 8, 25, 75 [reduzida para 50] mg/kg/dia) com administração diária foi conduzido em camundongos transgênicos H2ras. Carcinomas gastroduodenais, incidência aumentada de hemangiosarcomas de fundo e/ou hiperplasia da mucosa gástrica foram observados em doses ≥ 25 mg/kg/dia após duração de 1 ou 6 meses (≥ 7,3 vezes a AUC em pacientes que recebem a dose recomendada diária).
Em um estudo de 2 anos de carcinogenicidade em ratos (0, 0,33, 1 ou 3 mg/kg/dia), a administração de sunitinibe em ciclos de 28 dias seguidos por períodos de 7 dias livres de medicação resultaram em aumentos da incidência de feocromocitomas e hiperplasia da medula suprarrenal em ratos machos que receberam 3 mg/kg/dia após > 1 ano de administração (≥ 7,8 vezes a AUC em pacientes recebendo a dose diária recomendada). Carcinoma de glândulas de Brunner ocorreu no duodeno com ≥ 1 mg/kg/dia em fêmeas e com 3 mg/kg/dia em machos, e hiperplasia de células mucosas foi evidente no estômago glandular com 3 mg/kg/dia em machos, tendo ocorrido com ≥ 0,9, 7,8 e 7,8 vezes a AUC em pacientes que receberam a dose diária recomendada, respectivamente. A relevância para humanos dos achados neoplásicos observados em estudos de carcinogenicidade em camundongos (transgênicos H2ras) e em ratos com o tratamento com sunitinibe não está claro.
Reprodução e Toxicidade
Não foram observados efeitos na fertilidade dos ratos recebendo doses durante 58 dias antes do acasalamento com fêmeas não tratadas. Não foram observados efeitos reprodutivos nas ratas tratadas durante 14 dias antes do acasalamento com machos não tratados, com doses resultando em exposição sistêmica de aproximadamente 5 vezes a exposição sistêmica em pacientes. Entretanto, nos estudos de toxicidade de doses repetidas realizados em ratos e macacos, foram observados efeitos na fertilidade em fêmeas na forma de atresia folicular, degeneração do corpo lúteo, alterações endometriais no útero e diminuição dos pesos uterino e ovariano com níveis de exposições sistêmicas clinicamente relevantes. Além disso, em estudos de toxicidade de doses repetidas conduzidos em ratos foram observados efeitos na fertilidade em machos na forma de atrofia tubular dos testículos, redução de espermatozoides nos epidídimos e depleção coloide na próstata e vesículas seminais com níveis de exposição plasmática 18 vezes maior do que é observado na prática clínica. Nem todos os efeitos observados em ratos machos foi reversível no fim do período da recuperação (6 semanas).
Em ratos, a mortalidade embrio-fetal relacionada ao tratamento foi evidente por reduções significativas no número de fetos vivos, número aumentado de reabsorções (precoce e total); aumento das perdas pós-implantação e perda total da ninhada em 8 de 28 fêmeas grávidas com níveis de exposição plasmática de 5,5 vezes maior do que observado na prática clínica. Em coelhos, reduções do peso do útero gravídico e do número de fetos vivos foram devidos ao aumento no número de reabsorções (precoce e total), aumentos da perda pós-implantação e perda completa da ninhada em 4 de 6 fêmeas grávidas com níveis de exposição plasmática 3 vezes maior do que observado na prática clínica.
O tratamento com sunitinibe em ratos durante a organogênese resultou em efeitos sobre o desenvolvimento com doses ≥ 5 mg/kg/dia e consistiu em um aumento na incidência de malformações esqueléticas fetais, predominantemente caracterizadas como ossificação retardada das vértebras torácica/lombar. Os efeitos sobre o desenvolvimento em ratos ocorreram em níveis de exposição plasmática 6 vezes maiores do que observado na prática clínica. Em coelhos, os efeitos sobre o desenvolvimento consistiram no aumento da incidência da fissura do lábio em níveis de exposição plasmática, aproximadamente iguais aos observados na prática clínica, e fissuras do lábio e do palato em níveis de exposição plasmática 2,7 vezes maiores do que o observado na prática clínica.
Não foi conduzido um estudo definitivo de toxicidade sobre o desenvolvimento embrio-fetal em coelhos, visto que os efeitos embrio-fetais foram demonstrados claramente em ratos e relatados no estudo preliminar conduzido em coelhos.
Contraindicações.
Sutent® é contraindicado em pacientes com hipersensibilidade ao malato de sunitinibe ou a qualquer componente da fórmula.
Sutent® é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Advertências e precauções.
Pele e Tecidos
A alteração da cor da pele, possivelmente devido à presença da substância ativa colorida (amarelo), é um evento adverso muito comum relacionado ao tratamento, ocorrendo em cerca de 30% dos pacientes. Os pacientes devem ser informados que pode ocorrer despigmentação dos cabelos ou coloração da pele com o uso de Sutent®. Outros efeitos dermatológicos possíveis de ocorrer incluem secura, espessamento ou rachadura da pele, bolhas ou rash ocasional na palma das mãos e na planta dos pés.
Esses eventos não são cumulativos, foram tipicamente reversíveis e geralmente não necessitam de descontinuação do tratamento.
Dor/irritação da boca foi relatada em aproximadamente 14% dos pacientes. Disgeusia foi relatada em aproximadamente 28% dos pacientes.
Eventos Hemorrágicos
Eventos hemorrágicos relatados através da experiência pós-comercialização, alguns dos quais foram fatais, incluíram hemorragias gastrintestinais, respiratórias, tumorais, do trato urinário e cerebrais. Em estudos clínicos, hemorragia tumoral relacionada ao tratamento ocorreu em aproximadamente 2% dos pacientes com GIST. Esses eventos podem ocorrer repentinamente, e no caso de tumores de pulmão, podem apresentar-se como hemoptise grave e de risco de morte ou hemorragia pulmonar. Hemorragia pulmonar fatal ocorreu em 2 pacientes que receberam sunitinibe em um estudo clínico de pacientes com câncer de pulmão metastático de células não pequenas (NSCLC); e foram também relatados, alguns com desfecho fatal, em estudos clínicos e experiência pós-comercialização em pacientes em tratamento com sunitinibe para RCCm, GIST e NSCLC. Ambos os pacientes tiveram histologia de células escamosas. Sutent® não é indicado para pacientes com câncer pulmonar metastático de células não pequenas.
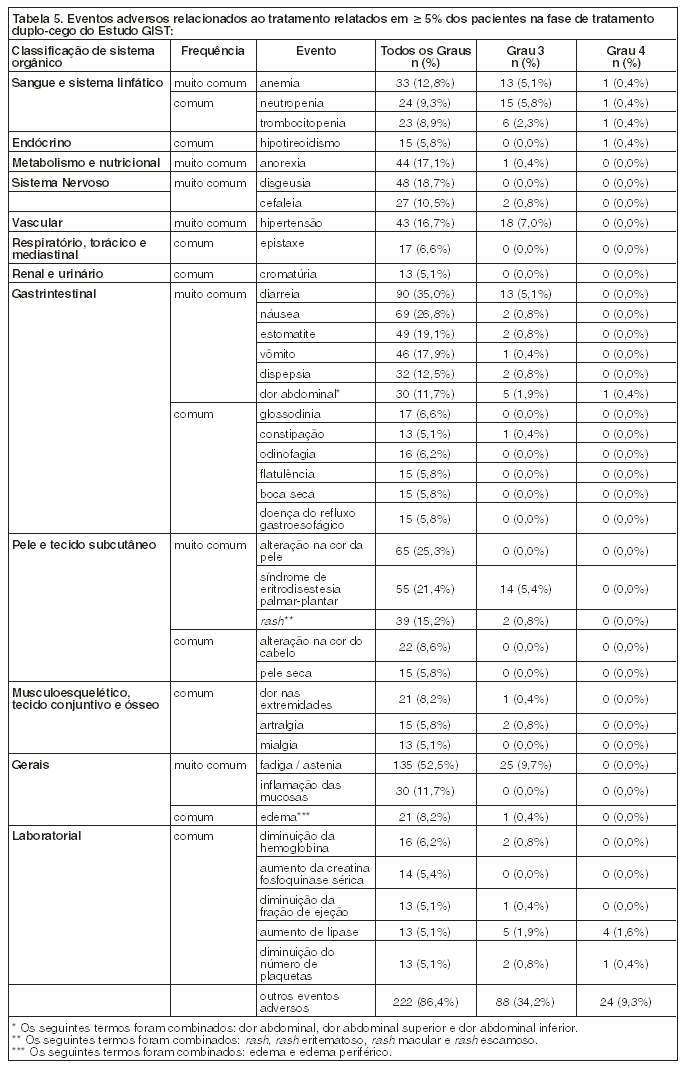
Eventos de sangramento emergentes do tratamento ocorreram em 18% de pacientes recebendo sunitinibe na fase de tratamento duplo-cego do Estudo GIST e em 17% de pacientes recebendo placebo. Em pacientes recebendo sunitinibe para RCCm em pacientes virgens de tratamento, 39% dos pacientes apresentaram eventos de sangramento, comparado com 11% de pacientes recebendo interferon-a. Onze pacientes (3,1%) em tratamento com sunitinibe apresentaram eventos de sangramento relacionados ao tratamento de Grau 3 ou maior versus 1 (0,3%) de pacientes em tratamento com interferon-a. Dos pacientes recebendo sunitinibe para RCCm refratário a citoquina, 26% apresentaram sangramento. Eventos de sangramento, com exceção de epistaxe, ocorreram em 19% dos pacientes que receberam sunitinibe em um estudo de tumores neuroendócrinos pancreáticos de Fase 3 comparado a 4% dos pacientes que receberam placebo. A avaliação de rotina deste evento deve incluir hematimetria completa e exame físico.
Epistaxe relacionada ao tratamento foi relatada em 8% dos pacientes com tumores sólidos. A epistaxe foi o evento adverso hemorrágico relacionado ao tratamento mais comum, relatado por aproximadamente metade dos pacientes com tumores sólidos que apresentaram eventos hemorrágicos.
Trato Gastrintestinal
Complicações gastrintestinais graves, algumas vezes fatais, incluindo perfuração g