SPRAVATO
JANSSEN-CILAG
escetamina
Antidepressivo.
Apresentações.
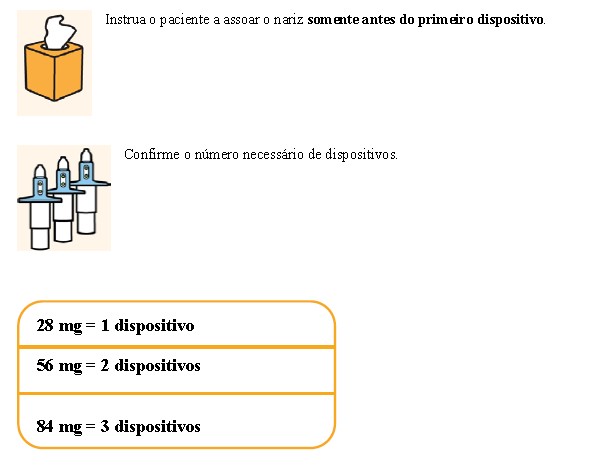
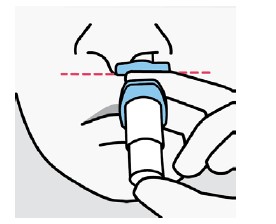
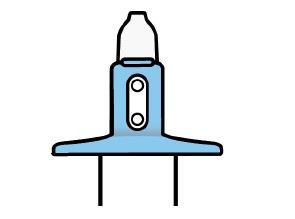
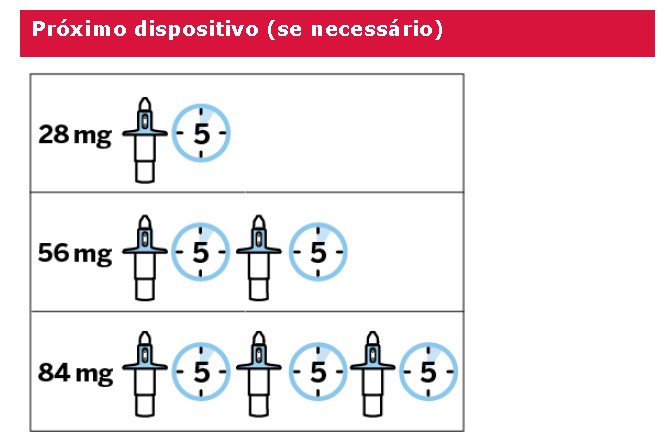
Solução spray nasal em frasco de uso único com 28 mg de escetamina em embalagens com 1 dispositivo de 0,2 mL.
USO INTRANASAL
USO ADULTO
Composição.
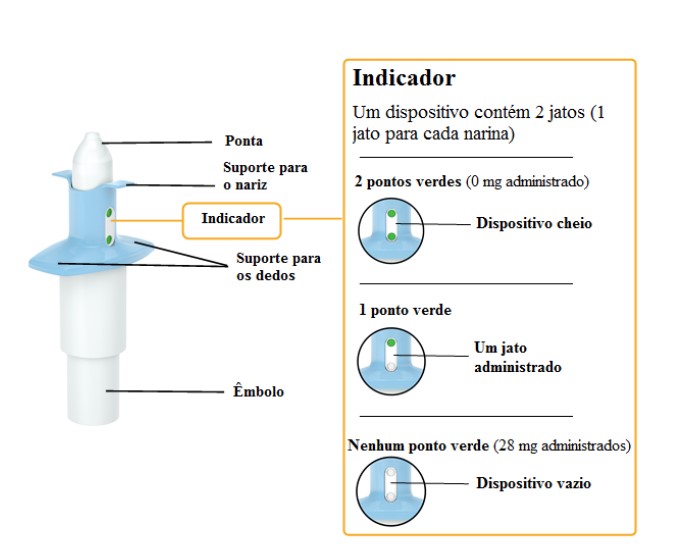
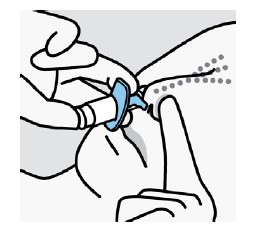
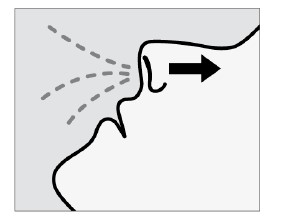
Cada dispositivo de frasco spray nasal individual de uso único contém 32,3 mg de cloridrato de escetamina, equivalente a 28 mg de escetamina, e libera dois jatos, um jato para cada narina.
Excipientes: ácido cítrico monoidratado, edetato dissódico (EDTA), hidróxido de sódio e água para injeção.
Informações técnicas.
1. INDICAÇÕES
Spravato® é indicado para Transtorno Depressivo Maior em adultos que não tenham respondido adequadamente a pelo menos dois antidepressivos diferentes com dose e duração adequadas para tratar o atual episódio depressivo moderado a grave (depressão resistente ao tratamento) em combinação com antidepressivos orais (tais como ISRS - Inibidores seletivos da recaptação de serotonina e ISRSN - Inibidores da recaptação de serotonina e norepinefrina).
Spravato® é indicado, em conjunto com terapia antidepressiva oral, para a rápida redução dos sintomas depressivos em pacientes adultos com Transtorno Depressivo Maior com comportamento ou ideação suicida aguda. Não foi demonstrada efetividade de Spravato® na prevenção do suicídio ou na redução da ideação ou comportamento suicida. Mesmo que o paciente apresente melhoras com as doses iniciais de Spravato®, o uso de Spravato® não dispensa a necessidade de hospitalização, caso clinicamente justificada.
2. RESULTADOS DE EFICÁCIA
A eficácia e segurança de Spravato® em spray nasal foram avaliadas em cinco estudos clínicos de Fase 3 em pacientes adultos (18 a 86 anos) com depressão resistente ao tratamento (TRD) que atenderam aos critérios do DSM-5 [Manual Diagnóstico e Estatístico de Transtornos Mentais - 5ª edição] para transtorno depressivo maior e que foram não respondedores a pelo menos dois tratamentos com antidepressivos (ADs) orais, com dosagem e duração adequadas, no atual episódio depressivo maior. Um mil e oitocentos e trinta e três (1833) pacientes adultos foram incluídos, dos quais 1601 pacientes foram expostos a Spravato®.
Depressão resistente ao tratamento - Estudos a curto prazo
O Spravato® foi avaliado em dois estudos de Fase 3, a curto prazo (4 semanas), randomizados, duplo-cegos, multicêntricos, controlados ativamente em pacientes com TRD. Os Estudos TRANSFORM-1 (TRD3001) e TRANSFORM-2 (TRD3002) foram conduzidos em adultos (18 a < 65 anos). Os pacientes no TRD3001 e no TRD3002 iniciaram o tratamento com Spravato® com 56 mg mais um AD oral diário recém-iniciado ou um AD oral diário recém-iniciado mais placebo em spray nasal no Dia 1 e as dosagens de Spravato® foram, então, mantidas em 56 mg ou tituladas até 84 mg, com administração duas vezes por semana durante uma fase duplo-cega de indução de 4 semanas. As doses de Spravato® de 56 mg ou 84 mg foram fixas no Estudo TRD3001 e flexíveis no Estudo TRD3002. Um novo AD oral aberto [(IRSN): duloxetina, venlafaxina de liberação prolongada; (ISRS): escitalopram, sertralina)] foi iniciado no Dia 1 em todos os estudos. A seleção do AD oral recém-iniciado foi determinada pelo pesquisador com base no histórico de tratamento anterior do paciente. As características demográficas e da doença do período basal dos pacientes nos estudos TRD3001 e TRD3002 foram semelhantes entre os grupos de Spravato® mais AD oral e AD oral mais placebo em spray nasal. A idade mediana dos pacientes era de 47 anos (faixa de 18 a 64 anos), 67% eram do sexo feminino; 83% eram caucasianos e 5% de descendência africana, e a duração média do tratamento com AD anterior era de aproximadamente 425 dias. No momento da triagem, a duração média do episódio atual de depressão era de 168 semanas. No momento da triagem, 90% dos pacientes não tiveram resposta a ≥ 2 ADs orais e o restante dos pacientes precisavam de confirmação da não-resposta ao segundo AD durante a fase prospectiva de triagem de 4 semanas. Nos estudos TRD3001 e TRD3002, o novo AD oral aberto iniciado durante a fase duplo-cega de indução de 4 semanas foi um ISRS em 38% dos pacientes e um IRSN em 62% dos pacientes. O desfecho primário de eficácia foi a alteração em relação ao valor basal na pontuação total da Escala de Classificação de Depressão de Montgomery-Åsberg (MADRS) no final da fase duplo-cega de indução de 4 semanas. A MADRS é uma escala classificada clinicamente de dez itens usada para avaliar a severidade de sintomas depressivos. As pontuações na MADRS variam de 0 a 60, com pontuações mais altas indicando depressão mais severa. No estudo de doses flexíveis TRD3002, para o desfecho primário de eficácia de melhora de sintomas depressivos (alteração nas pontuações totais da MADRS em relação ao valor basal no final da fase de indução de 4 semanas), Spravato® mais um AD oral recém-iniciado demonstrou superioridade estatística e clinicamente significativa em comparação ao tratamento padrão (AD oral recém-iniciado) mais placebo em spray nasal. No estudo TRD3001, foi observado um efeito do tratamento clinicamente significativo na alteração das pontuações totais da MADRS em relação ao valor basal no final da fase de indução de 4 semanas a favor de Spravato® mais AD oral recém-iniciado em comparação ao padrão de tratamento (AD oral recém-iniciado) mais placebo em spray nasal (Tabela 1). No TRD3002, as melhoras na pontuação total da Escala de Incapacidade de Sheehan (SDS), que avalia o comprometimento funcional global, e na pontuação total do Questionário de Saúde do Paciente-9 (PHQ-9), que avalia sintomas de depressão, favoreceram numericamente Spravato® mais um AD oral recém-iniciado em comparação ao tratamento padrão (AD oral recém-iniciado) mais placebo em spray nasal.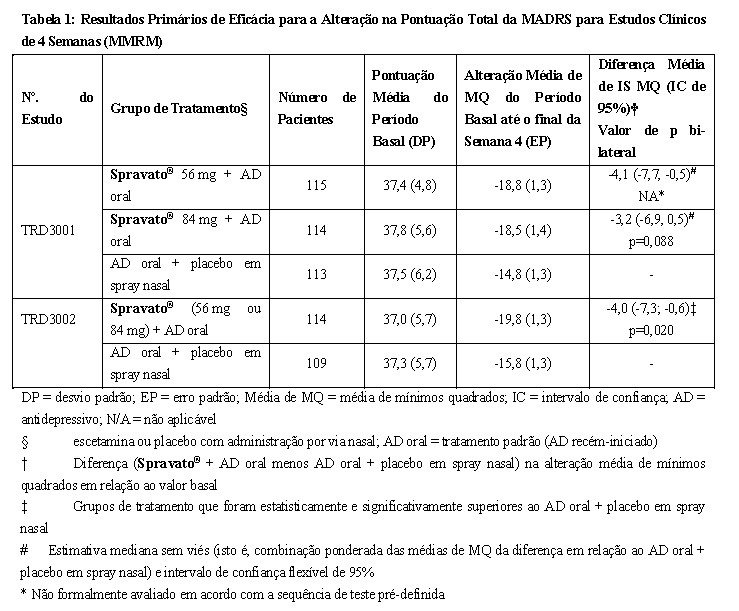
-Evolução no Tempo da Resposta ao Tratamento No Estudo TRD3002, foi observado um efeito de Spravato® sobre a redução de sintomas logo em 24 horas pósdose e aumentou nas semanas subsequentes, com o efeito antidepressivo completo de Spravato® observado até o Dia 28. Ao longo de toda a fase duplo-cega de indução de 4 semanas do Estudo TRD3002, a alteração média na pontuação total da MADRS para Spravato® com dose flexível (56 mg ou 84 mg) mais AD oral foi maior do que para AD oral mais placebo com administração por via nasal. No Dia 28, 67% dos pacientes randomizados para Spravato® estavam recebendo a administração de 84 mg. A Figura 1 (Análise de MMRM) descreve a evolução no tempo da resposta no desfecho primário de eficácia (MADRS) no Estudo TRD3002. Um efeito do tratamento consistente foi observado no Estudo TRD3001.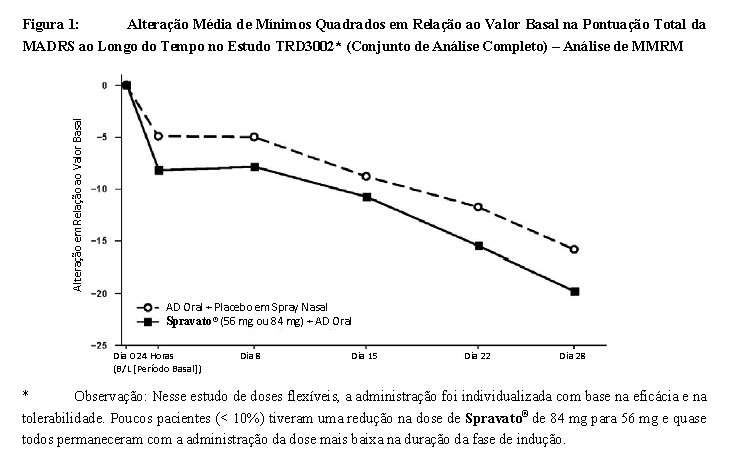
Além disso, no estudo TRD3002, a associação de Spravato® mais AD oral foi superior à associação de AD oral mais placebo em spray nasal em termos da alteração média em relação ao valor basal na pontuação total da MADRS no final da Semana 4. Uma diferença consistente entre os tratamentos foi observada no Estudo TRD3001 [Figura 2 (MMRM)].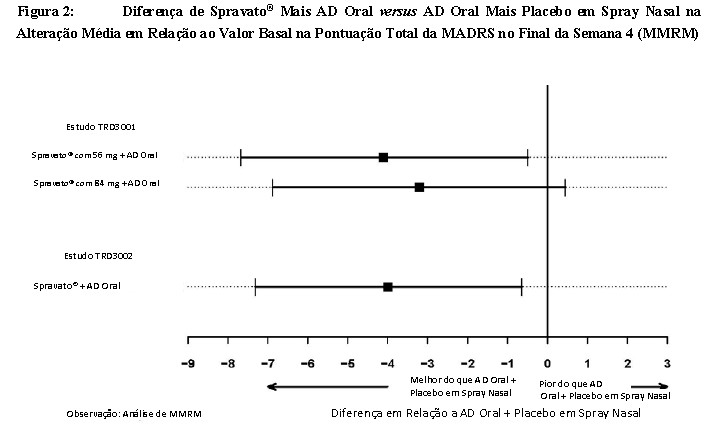
Taxas de resposta e remissão
A resposta foi definida como uma redução de ≥ 50% na pontuação total da MADRS em relação ao valor basal da fase de indução. Com base na redução na pontuação total da MADRS em relação ao valor basal, a proporção de pacientes nos Estudos TRD3001 e TRD3002 que demonstraram resposta ao tratamento com Spravato® mais AD oral foi maior do que aquela para AD oral mais placebo em spray nasal ao longo de toda a fase duplo-cega de indução de 4 semanas (Tabela 2). A remissão foi definida como uma pontuação total da MADRS de ≤ 12. Em todos os três estudos, uma maior proporção de pacientes tratados com Spravato® mais AD oral estava em remissão no final da fase duplo-cega de indução de 4 semanas do que para AD oral mais placebo em spray nasal (Tabela 2).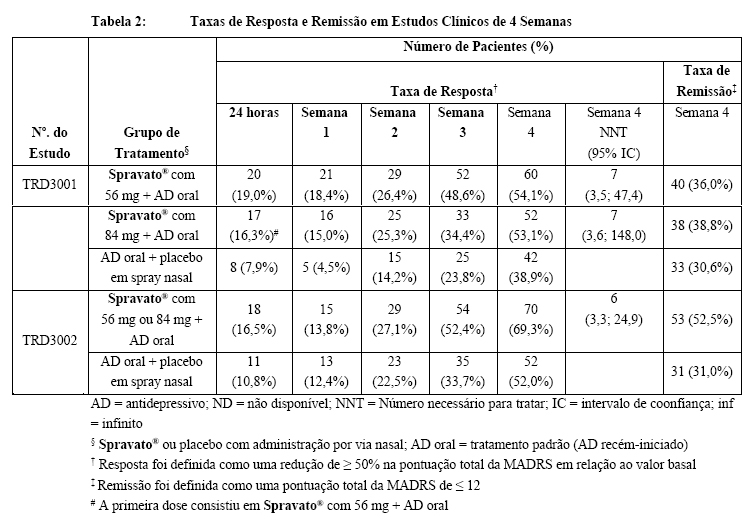
Depressão resistente ao tratamento - Estudos a longo prazo
Estudo de prevenção de recidiva
O Estudo SUSTAIN-1 (TRD3003) foi um estudo de prevenção de recidiva a longo prazo, randomizado, duplo-cego, de grupos paralelos, ativamente controlados, multicêntrico. Ao total, 705 pacientes foram incluídos; 437 incluídos diretamente; 150 transferidos do TRD3001 e 118 transferidos do TRD3002. Os pacientes incluídos diretamente receberam a administração de Spravato® (56 mg ou 84 mg duas vezes por semana) mais AD oral em uma fase aberta de indução de 4 semanas. Os pacientes que foram respondedores (redução da pontuação total da MADRS de ≥ 50% em relação ao valor basal) continuaram a receber o tratamento com Spravato® mais AD oral em uma fase de otimização de 12 semanas. No final da fase aberta de indução, 52% dos pacientes estavam em remissão (pontuação total da MADRS de ≤ 12) e 66% dos pacientes foram respondedores (melhora de ≥ 50% na pontuação total da MADRS). Quatrocentos e cinquenta e cinco (455) pacientes tratados com escetamina entraram na fase de otimização, pacientes em remissão estável ou resposta estável foram randomizados para continuar com Spravato® ou parar Spravato® e mudar para o placebo de spray nasal. Após um tratamento inicial de 16 semanas com Spravato® mais AD oral, 176 (39%) dos pacientes estavam em remissão estável e 121 (27%) estavam em resposta estável (mas não em remissão estável). A remissão estável foi definida como uma pontuação total da MADRS de ≤ 12 em pelo menos 3 das últimas 4 semanas da fase de otimização e a resposta estável foi definida como uma redução de ≥ 50% na pontuação total da MADRS em relação ao valor basal nas últimas 2 semanas da fase de otimização, mas não em remissão estável. As características demográficas e da doença do período basal dos pacientes randomizados para a fase duplo-cega de manutenção foram semelhantes entre os grupos de Spravato® mais AD oral e AD oral mais placebo, a idade mediana dos pacientes era de 48 anos (faixa de 19 a 64 anos), 66% eram do sexo feminino; 90% eram caucasianos e 4% de descendência africana.
-Remissão Estável
Os pacientes em remissão estável que continuaram o tratamento com Spravato® mais AD oral apresentaram um tempo até a recidiva de sintomas depressivos estatística e significativamente mais longo do que os pacientes com a administração de tratamento padrão (AD oral) mais placebo em spray nasal (Figura 3). A recidiva foi definida como uma pontuação total da MADRS de ≥ 22 por 2 semanas consecutivas ou hospitalização em razão de agravamento da depressão ou qualquer outro evento clinicamente relevante indicativo de recidiva. O tempo mediano até a recidiva para o grupo de tratamento padrão (AD oral) mais placebo em spray nasal foi de 273 dias, enquanto que a mediana não foi estimável para Spravato® mais AD oral, uma vez que esse grupo nunca alcançou uma taxa de recidiva de 50%.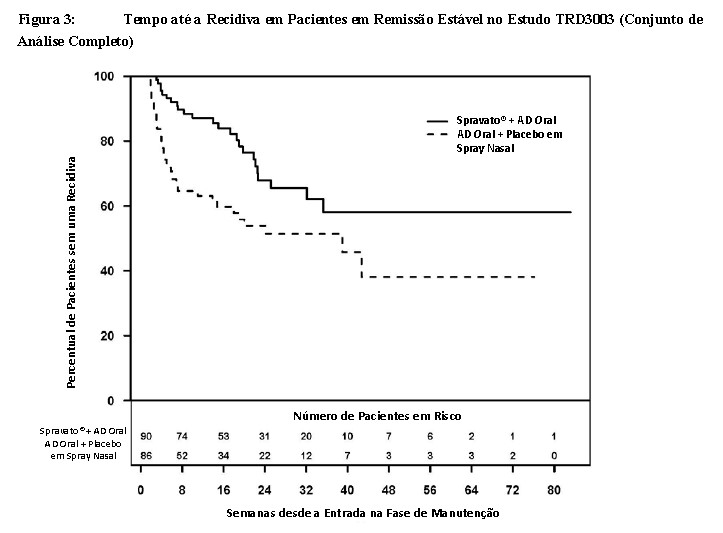
Para pacientes em remissão estável, a razão de risco estimada (IC de 95%) de Spravato® mais AD oral em relação ao tratamento padrão (AD oral) mais placebo em spray nasal, com base em estimativas ponderadas, foi de 0,49 (IC de 95%: 0,29; 0,84), indicando que pacientes que estavam em remissão estável e continuaram o tratamento com Spravato® mais AD oral nesse grupo tiveram, em média, 51% menos probabilidade de apresentar recidiva do que pacientes que trocaram para o tratamento padrão (AD oral) mais placebo em spray nasal.
-Resposta Estável
Os resultados de eficácia também foram consistentes para pacientes em resposta estável que continuaram o tratamento com Spravato® mais AD oral; os pacientes apresentaram um tempo até a recidiva de sintomas depressivos estatística e significativamente mais longo do que os pacientes com a administração de tratamento padrão (AD oral) mais placebo em spray nasal (Figura 4). O tempo mediano até a recidiva para o grupo de tratamento padrão (AD oral) mais placebo em spray nasal (88 dias) foi mais curto em comparação àquele para o grupo de Spravato® mais AD oral (635 dias).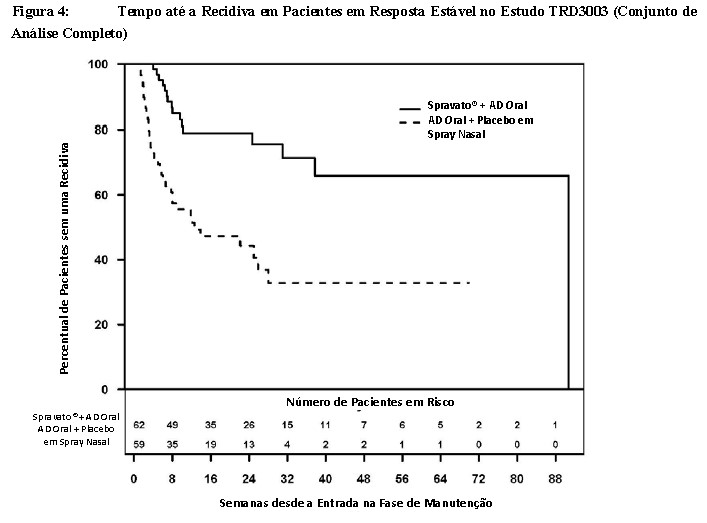
Para pacientes em resposta estável, a razão de risco estimada (IC de 95%) de Spravato® mais AD oral em relação ao tratamento padrão (AD oral) mais placebo em spray nasal, com base no modelo de riscos proporcionais de Cox, foi de 0,30 (IC de 95%: 0,16; 0,55), indicando que pacientes que eram respondedores estáveis e continuaram o tratamento com Spravato® mais AD oral nesse grupo tiveram, em média, 70% menos probabilidade de apresentar uma recidiva do que pacientes que trocaram para o tratamento padrão (AD oral) mais placebo em spray nasal. A inclusão no TRD3003 foi escalonada ao longo de aproximadamente 2 anos. A fase de manutenção foi de duração variável e continuou até que o paciente individual apresentasse uma recidiva de sintomas depressivos, ou descontinuasse por qualquer outro motivo, ou o estudo terminasse em razão da ocorrência do número necessário de eventos de recidiva. Os números de exposição foram influenciados pela interrupção do estudo com um número predeterminado de recidivas, com base na análise interina. Após um tratamento inicial de 16 semanas com Spravato® mais AD oral, a duração mediana da exposição a Spravato® na fase de manutenção foi de 4,2 meses (faixa: 1 dia a 21,2 meses) em pacientes tratados com Spravato® (remissão estável e resposta estável). Nesse estudo, 31,6% dos pacientes receberam Spravato® por mais de 6 meses e 7,9% dos pacientes receberam Spravato® por mais de 1 ano na fase de manutenção.
Frequência de Administração
A partir da Semana 8, um algoritmo (com base na MADRS) foi usado para determinar administração da dose. Os pacientes em remissão (isto é, a pontuação total da MADRS era de ≤ 12) recebiam a administração da dose em semanas alternadas; mas, se a pontuação total da MADRS aumentasse para > 12, então a frequência era aumentada para administração semanal pelas próximas 4 semanas; com o objetivo de manter o paciente na menor frequência de administração da dose a fim de conservar a resposta/remissão. A frequência de doses utilizada na maior parte do tempo durante a fase de manutenção é mostrada na Tabela 3. Dos pacientes randomizados para Spravato®, 60% receberam 84 mg e 40% receberam 56 mg.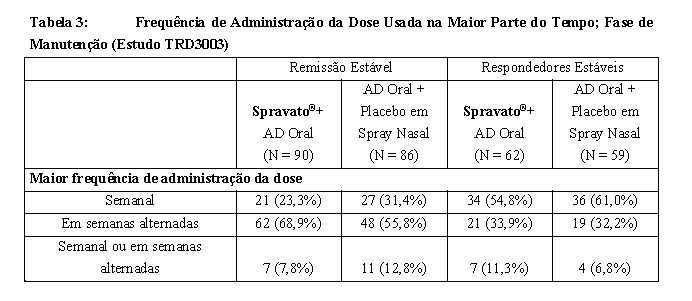
Estudo de Segurança e Eficácia a Longo Prazo Aberto O Estudo SUSTAIN-2 (TRD3004) foi um estudo aberto, a longo prazo de Spravato® mais AD oral em pacientes com TRD. O desfecho primário foi avaliar a segurança e a eficácia a longo prazo (até 52 semanas) de Spravato®.O Spravato® não foi associado a efeitos sobre a função cognitiva ou sintomas emergentes do tratamento de cistite intersticial. No subgrupo de idosos (≥ 65 anos de idade), foi observado retardamento do tempo de reação, com início na Semana 20 e até o final do estudo, no entanto, o desempenho em outros exames cognitivos permaneceu estável. Além disso, não houve nenhuma evidência de sintomas de abstinência e/ou rebote após a cessação do tratamento com Spravato®. Não foi relatado nenhum caso de depressão respiratória e não houve nenhuma evidência de alterações relacionadas ao tratamento em parâmetros laboratoriais. O peso corporal médio permaneceu estável durante o tratamento com Spravato® mais AD oral tanto na fase de indução quanto na fase de manutenção (alteração média em relação ao valor basal ± desvio padrão de -0,29 ± 2,15 kg no Dia 28 e de 0,44 ± 5,83 kg na Semana 48). O TRD3004 também avaliou a eficácia a longo prazo, incluindo os efeitos sobre os sintomas depressivos. No final da fase de indução de 4 semanas, a taxa de resposta (melhora de ≥ 50% em relação ao valor basal na pontuação total da MADRS) foi de 78,4% (593/756) e a taxa de remissão (pontuação total da MADRS de ≤ 12) foi de 47,2% (357/756); dos respondedores que prosseguiram para a fase de manutenção, 76,5% (461/603) estavam em resposta e 58,2% (351/603) estavam em remissão no desfecho.
Rápida Redução dos Sintomas Depressivos em Adultos com Transtorno Depressivo Maior Com Comportamento ou Ideação Suicida Aguda Spravato® foi avaliado em dois estudos de Fase 3 idênticos, de curta duração (4 semanas), randomizados, duplocego, multicêntricos, controlados por placebo, ASPIRE I (SUI3001; NCT03039192) e ASPIRE II (SUI3002; NCT03097133) em pacientes adultos com transtorno depressivo maior (TDM) moderado a grave (pontuação total MADRS > 28) que tiveram ideação suicida ativa com intenção. Nesses estudos, os pacientes receberam tratamento com Spravato® 84 mg ou spray nasal de placebo duas vezes por semana, durante 4 semanas. Todos os pacientes receberam tratamento de cuidado padrão abrangente (SOC), incluindo uma internação inicial e uma terapia antidepressiva oral (AD) recém-iniciada ou otimizada (monoterapia com AD ou AD com regime terapêutico adicional, tais como ISRSs, IRSNs, outros antidepressivos, antipsicóticos atípicos e estabilizadores de humor), conforme determinado pelo investigador. Após a primeira dose, foi permitida uma única redução de dose para Spravato® 56 mg em pacientes que não toleraram a dose de 84 mg.
As características demográficas e basais da doença dos pacientes nos estudos SUI3001 e SUI3002 foram semelhantes entre os grupos Spravato® mais SOC ou spray nasal placebo mais SOC. A idade média dos pacientes foi de 40 anos (faixa de 18 a 64 anos), 61% eram do sexo feminino; 73% caucasianos e 6% negros; e 63% dos pacientes tiveram pelo menos uma tentativa de suicídio anterior. A população de pacientes estudados nos estudos SUI3001 e SUI3002 tiveram uma pontuação média total na escala de avaliação de depressão de Montgomery-Åsberg (MADRS) de 40, 82% dos pacientes tiveram uma pontuação total de MADRS > 34 (considerada grave) e 90% dos pacientes preencheram os critérios de TDM grave do DSM-5. Antes de entrar no estudo, 92% dos pacientes estavam recebendo terapia antidepressiva. Durante o estudo, como parte do tratamento padrão, 40% dos pacientes receberam AD em monoterapia, 54% dos pacientes receberam AD com regime terapêutico adicional e 6% dos pacientes trocaram entre AD em monoterapia ou AD com regime terapêutico adicional em algum momento durante a fase duplo-cega.
O objetivo primário foi avaliar a eficácia do SPRAVATO® 84 mg em comparação com o spray nasal placebo, além do SOC abrangente na redução dos sintomas da TDM, em pacientes que tiveram ideação suicida ativa com a intenção medida pela alteração da pontuação total do MADRS (medida de eficácia primária) basal 24 horas após a primeira dose (Dia 2).
No SUI3001 e SUI3002, o Spravato® mais SOC demonstrou superioridade estatística para o desfecho de eficácia primário em comparação com o spray nasal placebo mais SOC (vide Tabela 4).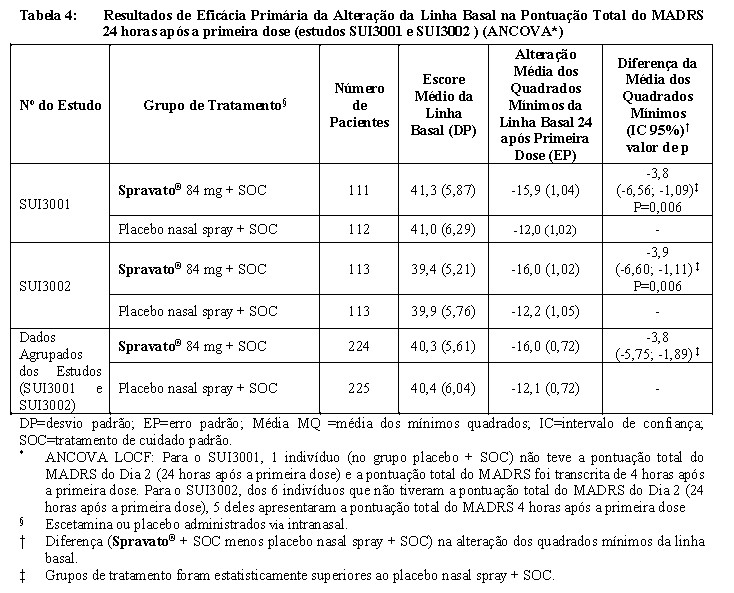
As diferenças do tratamento (IC 95%) na mudança da linha basal na pontuação total do MADRS no Dia 2 (24 horas após a primeira dose) entre Spravato® + SOC e placebo + SOC foram de -4,81 (-7,26; -2,36) para a subpopulação
que relatou uma tentativa anterior de suicídio (N = 282) e -2,32 (-5,54; 0,91) para a subpopulação que não relatou uma tentativa de suicídio anterior (N = 166).
Resposta ao Tratamento ao Longo do Tempo
Tanto no SUI3001 quanto no SUI3002, a diferença do tratamento de Spravato® em comparação ao placebo foi observada a partir de 4 horas. Entre 4 horas e o Dia 25, os grupos Spravato® e placebo continuaram a melhorar; a diferença entre os grupos geralmente permaneceu, mas não pareceu aumentar ao longo do tempo até o Dia 25. A Figura 5 mostra a evolução do desfecho de eficácia primário da mudança na pontuação total do MADRS com os dados agrupados do SUI3001 e do SUI3002.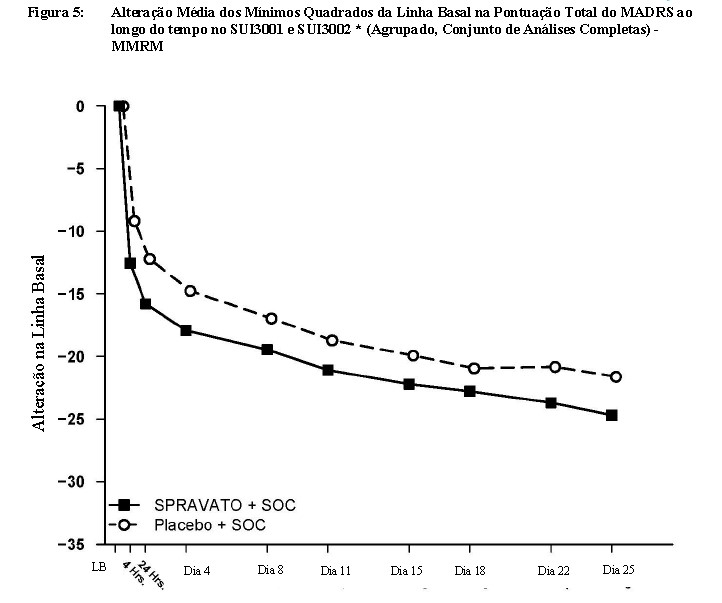
* Nota: Nestes estudos, após a primeira dose, foi permitida uma única redução de dose para Spravato® 56 mg para pacientes que não tolerararam a dose de 84 mg. Aproximadamente 16% dos pacientes tiveram redução na dose de Spravato® de 84 mg para 56 mg duas vezes por semana.
A alteração no escore da escala MADRS no período de 4 semanas, com administração de escetamina duas vezes por semana, foi analisada como desfecho secundário nos estudos clínicos usando um modelo de efeitos mistos para medidas repetidas (MMRM) em dados de casos observados. As diferenças médias de tratamento LS numericamente favoreceram o grupo escetamina + SOC em todos os pontos de tempo durante a fase de tratamento duplo-cego: 4 horas e 24 horas após a primeira dose, bem como nos dias 4, 8, 11, 15, 18, 22 e 25. A diferença média de tratamento LS (IC 95%) no Dia 25 foi -3,1 (-5,41; -0,73).
Taxas de Remissão
Nos estudos de Fase 3, a porcentagem de pacientes que alcançou remissão (pontuação total do MADRS ≤12 em um determinado momento durante o estudo) foi maior no grupo Spravato® + SOC do que no grupo placebo + SOC em todos os momentos durante a fase duplo-cega de tratamento (Tabela 5).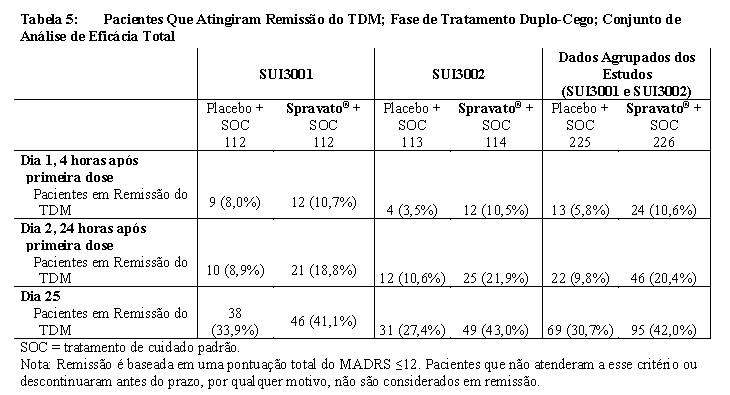
Avaliação da Escala (CGI-SS-r)
A severidade da suicidalidade, conforme medido pela escala Clinical Global Impression -Severity of Suicidality Revised (CGI-SS-r) em 24 horas após a primeira dose foi o desfecho secundário principal. No geral pacientes em ambos os grupos de tratamento tiveram melhoras na CGI-SS-r, embora não houvesse diferença estatisticamente significativa entre os grupos de tratamento. A eficácia a longo prazo do Spravato® na prevenção do suicídio não foi estabelecida.
Referências bibliográficas:
1. FEDGCHIN, M.; TRIVEDI, M.; DALY, E. J. et al. Randomized, Double-Bind Study of Fixed-Dosed Intranasal Esketamine Plus Oral Antidepressant vs. Active Control in Treatment-Resistant Depression. In: 9th Biennial Conference of the International Society for Affective Disorders (ISAD), 2018, Houston.
2. POPOVA, V.; DALY, E. J.; TRIVEDI, M. et al. Randomized, Double-Blind Study of Flexibly-Dosed Intranasal Esketamine Plus Oral Antidepressant vs. Active Control in Treatment-Resistant Depression. In: 2008 Annual Meeting of The American Psychiatric Association (APA), 2018, Nova Iorque.
3. WAJS, E.; ALUISIO, L.; MORRISON, R. et al. Long-Term Safety of Esketamine Nasal Spray Plus Oral Antidepressant in Patients with Treatment-Resistant Depression: Phase 3, Open-Label, Safety and Efficacy Study (SUSTAIN-2). In: 2018 Annual Meeting of The American Society of Clinical Psychopharmacology (ASCP), 2018, Miami.
4. DALY, E.; TRIVEDI, M.; JANIK, A. et al. A Randomized Withdrawal, Double-blind, Multicenter Study of Esketamine Nasal Spray Plus an Oral Antidepressant for Relapse Prevention in Treatment-resistant Depression. In: 2018 Annual Meeting of The American Society Of Clinical Psychopharmacology (ASCP), 2018, Miami.
5. OCHS-ROSS, R.; DALY, E.; LANE, R. et al. Efficacy and safety of intranasal esketamine plus an oral antidepressant in elderly patients with treatment-resistant depression. In: 2018 Annual Meeting of The American Psychiatric Association, 2018, Nova Iorque.
6. FU, D. J.; IONESCU, D.; LI, X. et al. Esketamine Nasal Spray for Rapid Reduction of Major Depressive Disorder Symptoms in Patients Who Have Active Suicidal Ideation With Intent: Double-Blind, Randomized Study (ASPIRE I). In: J Clin Psychiatry 81:3, May/June 2020.
7. IONESCU D.; FU D.J.; QIU X. et al. Esketamine Nasal Spray for Rapid Reduction of Depressive Symptoms in Patients with Major Depressive Disorder Who Have Active Suicide Ideation with Intent: Results of a Phase 3, Double-Blind, Randomized Study (ASPIRE II). The International Journal of Neuropsychopharmacology. https://doi.org/10.1093/ijnp/pyaa068.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
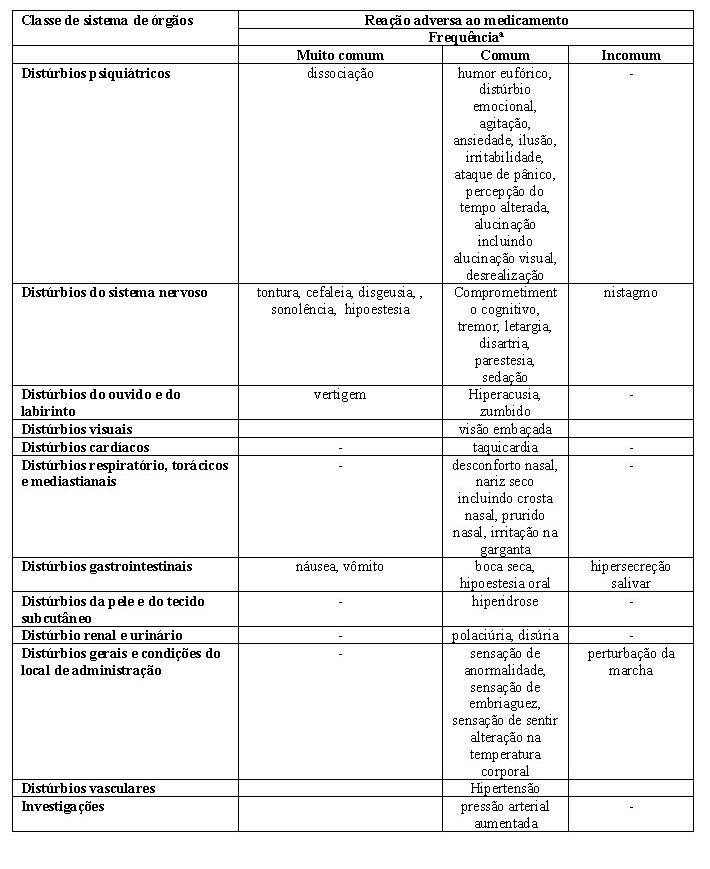
A escetamina, o enantiômero S da cetamina racêmica, é um antidepressivo com um novo mecanismo de ação. Ela é um antagonista não seletivo, não competitivo do receptor N-metil-D-aspartato (NMDA), um receptor ionotrópico do glutamato.Supostos contribuintes etiológicos da depressão, incluindo estresse e outras condições, são conhecidos por causar comprometimento estrutural e funcional de sinapses em regiões cerebrais envolvidas na regulação do humor e do comportamento emocional. Evidências dentro da literatura sugerem que, por meio do antagonismo do receptor NMDA, a escetamina produz um aumento transitório na liberação de glutamato, levando a aumentos na estimulação do receptor ácido a-amino-3-hidroxi-5-metil-4-isoxazol propiônico (AMPAR) e, subsequentemente, a aumentos na sinalização neurotrófica que restauram a função sináptica nessas regiões cerebrais. Ao contrário de outros tratamentos com antidepressivos, a ação antidepressiva primária da escetamina não envolve diretamente receptores de monoaminas, GABA ou opioides.
Efeitos farmacodinâmicos
Efeito sobre a capacidade de dirigir
Os efeitos de Spravato® sobre as habilidades de direção foram avaliados em um estudo em 23 pacientes adultos com transtorno depressivo maior. O estudo, o qual foi simples-cego e controlado por placebo, avaliou os efeitos de uma dose única de 84 mg de escetamina em spray nasal sobre a direção no dia seguinte. O desempenho de direção nas estrada foi avaliado pelo desvio padrão da posição lateral (DPPL), uma medida de comprometimento da direção. Uma bebida contendo etanol foi usada como um controle positivo. O DPPL após a administração da dose única de 84 mg de escetamina em spray nasal foi semelhante àquele do placebo. O limite superior do intervalo de confiança (IC) de 95% bilateral da diferença média entre a dose única de escetamina e o placebo foi de 0,58 cm, que é menor do que a margem de não inferioridade pré-especificada de 2,4 cm. O limite inferior do IC de 95% da diferença média entre o etanol e o placebo foi de 1,03 cm (p < 0,001), confirmando a sensibilidade do ensaio.
Efeito sobre o intervalo QT/QTc e a eletrofisiologia cardíaca
O tratamento com Spravato® não prolongou o intervalo QTc. O efeito de Spravato® (84 mg em spray nasal e 0,8 mg/kg de escetamina com administração por infusão intravenosa ao longo de 40 minutos) sobre o intervalo QTc foi avaliado em um estudo randomizado, duplo-cego, controlado por placebo e por controle positivo (moxifloxacino com 400 mg), de 4 períodos, de cruzamento em 60 indivíduos sadios. As concentrações máximas de escetamina no plasma produzidas pela infusão intravenosa foram aproximadamente 3 vezes mais altas do que as concentrações máximas produzidas pela dose nasal de 84 mg. O limite superior do intervalo de confiança de 90% para o maior intervalo QTc ajustado por placebo e corrigido pelo valor basal permaneceu abaixo de 10 mseg, em todos os tempos avaliados com base no método de correção de Fridericia (QTcF) para ambos os grupos de tratamento.
Propriedades Farmacocinéticas
-Absorção
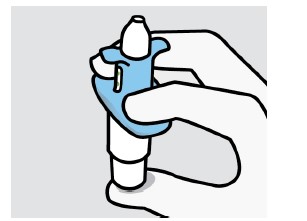
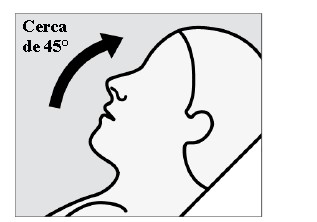
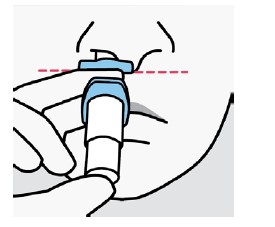
A biodisponibilidade absoluta média de 84 mg de escetamina com administração na forma de spray nasal é de aproximadamente 48%. A escetamina é rapidamente absorvida pela mucosa nasal após a administração nasal e pode ser medida no plasma dentro de 7 minutos após uma dose de 28 mg. O tempo até alcançar a concentração plasmática máxima (tmáx) é tipicamente de 20 a 40 minutos após o último jato nasal de uma sessão de tratamento. (vide "Posologia e Modo de Usar"). Aumentos lineares dependentes da dose na Cmáx e na ASC∞ plasmáticas da escetamina em spray nasal foram produzidos por doses de 28 mg, 56 mg e 84 mg. O perfil farmacocinético da escetamina é semelhante após a administração de dose única e de doses repetidas, sem qualquer acúmulo no plasma quando a escetamina é administrada duas vezes por semana.
-Distribuição
O volume de distribuição em estado de equilíbrio médio da escetamina com administração por via intravenosa é de 709 L. A proporção da concentração total de escetamina que é ligada a proteínas no plasma humano é, em média, de 43 a 45%. O grau em que a escetamina é ligada a proteínas plasmáticas não é dependente da função hepática ou renal. A escetamina não é um substrato dos transportadores de glicoproteína-P (P-gp; proteína de resistência a múltiplos medicamentos 1), proteína de resistência ao câncer de mama (BCRP) ou transportador de ânions orgânicos (OATP) 1B1 ou OATP1B3. A escetamina não inibe esses transportadores ou a proteína de extrusão de múltiplos medicamentos e toxinas 1 (MATE1) e a MATE2-K ou o transportador de cátions orgânicos 2 (OCT2), o OAT1 ou o OAT3.
-Metabolismo
A escetamina é extensivamente metabolizada no fígado. A via metabólica primária da escetamina em microssomos hepáticos humanos é a N-desmetilação para formar a nor-escetamina. As principais enzimas do CYP responsáveis pela N-desmetilação da escetamina são a CYP2B6 e a CYP3A4. Outras enzimas do CYP, incluindo a CYP2C19 e a CYP2C9, contribuem em um grau muito menor. A nor-escetamina é subsequentemente metabolizada por meio de vias dependentes do CYP em outros metabólitos, alguns dos quais passam por glicuronidação.
-Eliminação
O clearance médio da escetamina com administração por via intravenosa foi de aproximadamente 89 L/hora. Depois que a Cmáx foi alcançada após a administração nasal, a diminuição nas concentrações de escetamina no plasma foi rápida nas primeiras poucas horas e, então, mais gradual. A meia-vida terminal mediana após a administração na forma de spray nasal geralmente variou de 7 a 12 horas. Após a administração intravenosa de escetamina radiomarcada, aproximadamente 78% e 2% da radioatividade administrada foi recuperada na urina e nas fezes, respectivamente. Após a administração oral de escetamina radiomarcada, aproximadamente 86% e 2% da radioatividade administrada foi recuperada na urina e nas fezes, respectivamente. A radioatividade recuperada consistiu primariamente em metabólitos da escetamina. Para as vias intravenosa e oral de administração, < 1% da dose foi excretada na urina na forma de medicamento inalterado.
Populações especiais
-Idosos (65 anos de idade e mais velhos)
A farmacocinética da escetamina com administração na forma de spray nasal foi comparada entre indivíduos idosos porém sadios, e adultos sadios mais jovens. Os valores médios de Cmáx e ASC∞ da escetamina produzidos por uma dose de 28 mg foram 21% e 18% mais altos, respectivamente, em indivíduos idosos (faixa de idade de 65 a 81 anos) em comparação a adultos mais jovens (faixa de idade de 22 a 50 anos). Os valores médios de Cmáx e ASC∞ da escetamina produzidos por uma dose de 84 mg foram 67% e 38% mais altos, respectivamente, em indivíduos idosos (faixa de idade de 75 a 85 anos) em comparação a adultos mais jovens (faixa de idade de 24 a 54 anos). A meia-vida terminal da escetamina foi semelhante nos indivíduos idosos e adultos mais jovens.
-Eficácia em Idosos
A eficácia de Spravato® para o tratamento de TRD em pacientes geriátricos foi avaliada no Estudo TRD3005, um estudo duplo-cego, randomizado, de 4 semanas, comparando doses flexíveis de Spravato® intranasal mais um novo antidepressivo oral recém-iniciado em comparação ao placebo intranasal mais um novo antidepressivo oral recéminiciado em pacientes ≥ 65 anos de idade. Spravato® foi iniciado com 28 mg duas vezes por semana e pôde ser titulado para 56 mg ou 84 mg administrado duas vezes por semana. Ao final de quatro semanas, não houve diferença estatisticamente significativa entre os grupos no desfecho primário de eficácia de alteração em relação ao valor basal até a Semana 4 na Escala de Avaliação de Depressão de Montgomery-Åsberg (MADRS).
-Comprometimento renal
Em relação aos indivíduos com função renal normal (clearance de creatinina [CLCR], 88 a 140 mL/min), a Cmáx de escetamina foi, em média, 20 a 26% mais alta em indivíduos com comprometimento renal leve (CLCR, 58 a 77 mL/min), moderado (CLCR, 30 a 47 mL/min) ou severo (CLCR, 5 a 28 mL/min, não em diálise) após a administração de uma dose de 28 mg de escetamina em spray nasal. A ASC∞ foi 13 a 36% mais alta nos indivíduos com comprometimento renal leve a severo. Não há nenhuma experiência clínica com a escetamina com administração na forma de spray nasal em pacientes em diálise.
-Comprometimento hepático
A Cmáx e a ASC∞ da escetamina produzidas por uma dose de 28 mg foram semelhantes entre indivíduos com comprometimento hepático de classe A de Child-Pugh (leve) e indivíduos sadios. A Cmáx e a ASC∞ da escetamina foram 8% mais alta e 103% mais alta, respectivamente, em indivíduos com comprometimento hepático de classe B de Child-Pugh (moderado) em relação a indivíduos sadios. Não há nenhuma experiência clínica com a escetamina com administração na forma de spray nasal em indivíduos com comprometimento hepático de classe C de Child-Pugh (severo).
-Raça
A farmacocinética da escetamina em spray nasal foi comparada entre indivíduos sadios asiáticos e caucasianos. Os valores plasmáticos médios de Cmáx e ASC∞ da escetamina produzidos por uma dose única de 56 mg de escetamina foram aproximadamente 14% e 33% mais altos, respectivamente, em indivíduos chineses em comparação a caucasianos. Ambos os parâmetros foram aproximadamente 40% mais altos em indivíduos japoneses em relação a caucasianos. Em média, a Cmáx de escetamina foi 10% menor e a ASC∞ da escetamina foi 17% maior em indivíduos coreanos em relação a caucasianos. A meia-vida terminal mediana da escetamina no plasma de indivíduos asiáticos variou de 7,1 a 8,9 horas e foi de 6,8 horas em indivíduos caucasianos.
-Sexo
Foi conduzida uma análise farmacocinética da população que incluiu indivíduos sadios (138 homens e 118 mulheres) e pacientes com transtorno depressivo maior (203 homens e 361 mulheres). Os resultados indicaram que a farmacocinética da escetamina com administração na forma de spray nasal não é influenciada pelo sexo.
-Peso Corporal
Foi conduzida uma análise farmacocinética da população que incluiu 256 indivíduos sadios e 564 pacientes com transtorno depressivo maior. O peso corporal total dos indivíduos variava de 39 a 170 kg. Os resultados indicaram que a farmacocinética da escetamina com administração na forma de spray nasal não é influenciada pelo peso corporal.
-Rinite alérgica
A farmacocinética de uma dose única de 56 mg de escetamina com administração na forma de spray nasal foi semelhante em indivíduos com rinite alérgica que foram expostos a pólen de gramíneas em comparação a indivíduos sadios.
Dados de segurança pré-clínicos
Toxicidade Geral
A administração nasal uma vez ao dia de escetamina até 9 mg/dia por 6 meses em ratos, e até 72 mg/dia por 9 meses em cães resultou em sinais clínicos relacionados ao sistema nervoso central não adversos que refletiram as propriedades anestésicas do artigo em teste. Não foi constatada nenhuma lesão notável na cavidade nasal ou em qualquer órgão periférico. Após 3 meses de tratamento diário com 9 mg/dia em ratos, a exposição sistêmica da escetamina (Cmáx e ASC) se assemelhou àquela em humanos com a dose máxima recomendada em humanos (DMRH) de 84 mg, enquanto que as razões de exposição com base na Cmáx e na ASC para escetamina em cães após 3 meses de tratamento diário com 72 mg/dia corresponderam a aproximadamente 4 vezes e 1 vez, respectivamente.
Neurotoxicidade
Em estudos de neurotoxicidade de dose única e doses repetidas de 14 dias com escetamina por administração nasal em ratos, não foi observada nenhuma lesão cerebral histopatológica. Em estudos de neurotoxicidade de dose única, em que ratos receberam administração nasal de escetamina com uma dose de até 72 mg, as margens de segurança com base na Cmáx e na ASC para escetamina corresponderam a aproximadamente 59 vezes e 86 vezes, respectivamente, em comparação à exposição em humanos com a DMRH de 84 mg. Em um estudo de neurotoxicidade de 14 dias, em que ratos receberam escetamina por administração nasal uma vez ao dia até uma dose de 54 mg/dia, as margens de segurança com base na Cmáx e na ASC para escetamina corresponderam a aproximadamente 17 vezes e 11 vezes. Além disso, não foi constatada nenhuma evidência de neurotoxicidade nos estudos de toxicologia de doses repetidas de 6 meses em ratos e de 9 meses em cães com administração nasal uma vez ao dia de escetamina, conforme considerado por avaliações de histopatologia cerebral e funcionais. Semelhantemente, não foi observada nenhuma neurotoxicidade nos estudos de toxicologia em animais a prazo mais curto com a escetamina administrada por via nasal. No geral, espera-se que o risco de neurotoxicidade associado à administração nasal de escetamina aos pacientes seja baixo.
Carcinogenicidade e Mutagenicidade
A administração nasal uma vez ao dia de escetamina não aumentou a incidência de tumores em um estudo de 2 anos de carcinogenicidade em ratos com doses de até 9 mg/dia. Com essa dose, a exposição à escetamina se assemelhou à exposição em humanos com a DMRH de 84 mg. A escetamina também não foi carcinogênica após a administração subcutânea uma vez ao dia em um estudo de 6 meses em camundongos transgênicos (Tg.rasH2) com doses de até 70/40 mg/kg/dia. Com essa dose, as razões de exposição com base na Cmáx e na ASC para a escetamina corresponderam a aproximadamente 20 vezes e 4 vezes, respectivamente, em comparação à exposição com a DMRH de 84 mg. A escetamina não foi mutagênica com ou sem ativação metabólica no teste de Ames. Foram observados efeitos genotóxicos com a escetamina em um teste de micronúcleo in vitro de triagem na presença de ativação metabólica. No entanto, a escetamina administrada por via intravenosa foi desprovida de propriedades genotóxicas em um ensaio in vivo de micronúcleo de medula óssea em ratos e um ensaio Comet in vivo em células hepáticas de ratos. Em líquido gástrico simulado, não há nenhuma evidência de que N-nitroso-escetamina seja formada a partir da fração da dose administrada por via nasal de escetamina que é absorvida por via oral.
Toxicologia Reprodutiva
Em um estudo de toxicidade no desenvolvimento embrionário/fetal com a cetamina administrada por via nasal em ratos, a prole não foi afetada adversamente na presença de toxicidade materna com doses de até 150 mg/kg/dia. Em ratos, a margem de segurança com base na Cmáx e na ASC estimada para a escetamina na dose de 150 mg/kg/dia de cetamina correspondeu a 61 vezes e 12 vezes em comparação à dose máxima recomendada em humanos (DMRH) de escetamina de 84 mg. Em coelhas grávidas, a cetamina racêmica foi administrada p