SOTYKTU
B-MS
deucravacitinibe
Antipsoriásico.
Apresentações.
Cada embalagem de SOTYKTU® (deucravacitinibe) contém 28 comprimidos revestidos de 6 mg.
USO ORAL
USO ADULTO
Composição.
SOTYKTU® (deucravacitinibe) 6 mg:
Cada comprimido revestido contém 6 mg de deucravacitinibe e os seguintes excipientes: acetato succinato de hipromelose, lactose, celulose microcristalina, croscarmelose sódica, dióxido de silício, estearato de magnésio e Opadry® II Rosa (álcool polivinílico, dióxido de titânio, macrogol, talco, óxido de ferro vermelho e óxido de ferro amarelo).
Informações técnicas.
1. INDICAÇÕES
SOTYKTU® (deucravacitinibe) é indicado para o tratamento de pacientes adultos com psoríase em placas1 moderada a grave que são elegíveis à terapia sistêmica ou fototerapia.
1CID - L40.0 Psoríase vulgar
2. RESULTADOS DE EFICÁCIA
Psoríase em placas
A eficácia e a segurança de SOTYKTU® (deucravacitinibe) 6 mg, uma vez ao dia, foram avaliadas em dois estudos clínicos multicêntricos, randomizados, duplo-cegos, ativo e placebo controlados, POETYK PSO-1 (NCT03624127) e POETYK PSO-2 (NCT03611751) que incluíram pacientes com 18 anos de idade ou mais com psoríase em placas moderada a grave que eram elegíveis para terapia sistêmica ou fototerapia. Os pacientes tiveram um envolvimento da área de superfície corporal (BSA) de ≥ 10%, uma pontuação do Índice da Gravidade da Psoríase por Área (PASI) ≥ 12 e uma Avaliação Global Estática Realizada pelo Médico (sPGA) ≥ 3 (moderada ou grave).
Em POETYK PSO-1 e POETYK PSO-2, 1.686 pacientes foram randomizados para SOTYKTU® (deucravacitinibe) (6 mg por dia), placebo ou apremilaste (30 mg duas vezes por dia).
Ambos os estudos avaliaram as respostas na semana 16 em comparação com o placebo para os dois desfechos co-primários:
• a proporção de pacientes que alcançaram uma pontuação sPGA de 0 (sem lesão) ou 1 (quase sem lesão);
• a proporção de pacientes que alcançaram uma melhora de pelo menos 75% nos escores PASI
(PASI 75) desde o início.
Outras comparações entre SOTYKTU® (deucravacitinibe) e placebo que foram desfechos secundários na Semana 16:
• a proporção de pacientes que alcançaram PASI 90, PASI 100, sPGA 0, pontuação PGA de gravidade do couro cabeludo (ss-PGA) de 0 (sem lesão) ou 1 (quase sem lesão) e Diário de Sinais e Sintomas em Psoríase (PSSD) Pontuação de Sintoma de 0 (sem sintomas).
As comparações entre SOTYKTU® (deucravacitinibe) e apremilaste foram feitas para os seguintes desfechos secundários, nos seguintes momentos:
• nas semanas 16 e 24 (POETYK PSO-1 e POETYK PSO-2), a proporção de pacientes que alcançaram PASI 75, PASI 90 e sPGA 0/1;
• na semana 16 (POETYK PSO-1 e POETYK PSO-2), a proporção de pacientes que alcançaram sPGA 0 e ssPGA 0/1 (couro cabeludo).
No início do estudo, os pacientes tinham uma BSA afetada mediana de 20% e uma pontuação PASI mediana de 18,7. A proporção de pacientes com escores sPGA de 3 (moderado) e 4 (grave) no início do estudo foi de 79,8% e 20,2%, respectivamente. O escore médio do Índice de Qualidade de Vida em Dermatologia (DLQI) foi 11. Um total de 18,4% dos pacientes tinha história de artrite psoriática.
Em ambos os estudos, 40% dos pacientes haviam recebido fototerapia anterior, 42,4% nunca tinha utilizado nenhuma terapia sistêmica (incluindo tratamento biológico e/ou não biológico), 41% receberam tratamento sistêmico não biológico anterior e 34,8% haviam recebido tratamento biológico anterior (16% inibidores de TNF, 5% inibidores da IL-12/23, 17% inibidores da IL-17 e 4% inibidores de IL-23).
A Tabela 1 apresenta os resultados de eficácia que demonstram a superioridade de SOTYKTU® (deucravacitinibe) em comparação com apremilaste e placebo.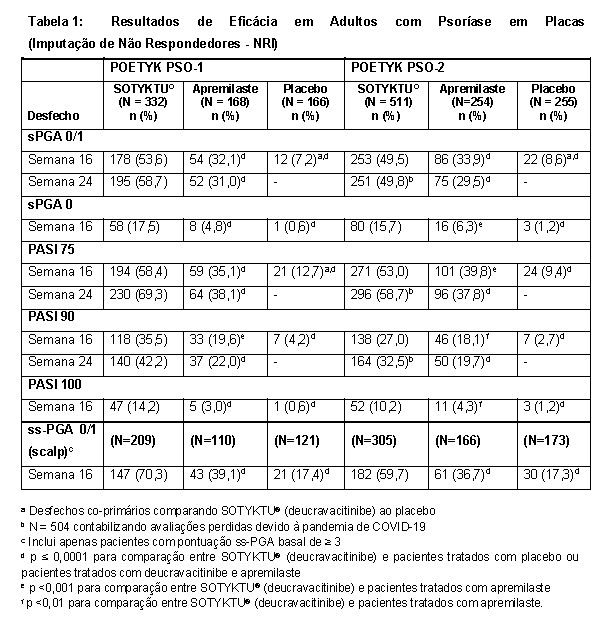
A avaliação de idade, sexo, raça, peso corporal, duração da doença, gravidade da doença de base e tratamento anterior com agentes biológicos ou não biológicos não identificou diferenças na resposta a SOTYKTU® (deucravacitinibe) entre esses subgrupos.
Manutenção e durabilidade de resposta
No POETYK PSO-1, entre os pacientes que receberam SOTYKTU® (deucravacitinibe) e alcançaram a resposta PASI 75 na semana 24, 81,3% dos pacientes que continuaram com SOTYKTU® (deucravacitinibe) mantiveram a resposta PASI 75 na semana 52. Entre os que responderam ao PASI 90 na semana 24, 74,3% dos pacientes mantiveram a resposta PASI 90 na semana 52. Entre os respondentes sPGA 0/1 na semana 24, 77,4% dos pacientes mantiveram a resposta sPGA 0/1 na semana 52.
No POETYK PSO-2, para avaliar a manutenção e durabilidade da resposta, os pacientes que foram originalmente randomizados para SOTYKTU® (deucravacitinibe) e responderam ao PASI 75 na Semana 24, foram randomizados novamente para continuar o tratamento com SOTYKTU® (deucravacitinibe) ou foram retirados da terapia (ou seja, receber placebo).
Na semana 52, 80,4% dos pacientes que continuaram com SOTYKTU® (deucravacitinibe) mantiveram o PASI 75 em comparação com 31,3% dos pacientes que interromperam SOTYKTU® (deucravacitinibe). Para respondentes na Semana 24 que foram retirados do tratamento com SOTYKTU® (deucravacitinibe), o tempo médio para a perda de PASI 75 foi de aproximadamente 12 semanas.
Resultados relatados pelo paciente
Uma proporção maior de pacientes tratados com SOTYKTU® (deucravacitinibe) em comparação com o placebo alcançou a pontuação de 0 (zero) do Diário de Sinais e Sintomas em Psoríase (PSSD) (ausência de coceira, dor, queimação, ardência e rigidez da pele) na Semana 16. Melhorias mais significativas nos sintomas de psoríase em comparação com apremilaste foram observadas em ambos os estudos avaliados pelo PSSD. Uma proporção maior de pacientes tratados com SOTYKTU® (deucravacitinibe) em comparação com placebo e apremilaste alcançou um DLQI 0/1 (sem impacto na vida do paciente).
Dados de segurança pré-clínica
Carcinogenicidade, Mutagenicidade e Comprometimento da Fertilidade
O deucravacitinibe não foi mutagênico em um ensaio de mutagenicidade bacteriana (teste de Ames) ou clastogênico em um ensaio de aberração cromossômica in vitro (células de ovário de hamster chinês em cultura) ou in vivo em um ensaio de micronúcleo do sangue periférico de ratos.
O potencial carcinogênico de deucravacitinibe foi avaliado em estudos de 6 meses em camundongos transgênicos rasH2 e de 2 anos em ratos. Nenhuma evidência de tumorigenicidade foi observada em ratos machos ou fêmeas que receberam deucravacitinibe em doses orais de até 15 mg/kg/dia e exposição de aproximadamente 51 vezes a DHR. Nenhuma evidência de tumorigenicidade foi observada em camundongos Tg.rasH2 machos ou fêmeas que receberam deucravacitinibe em doses orais de até 60 mg/kg/dia e exposição de aproximadamente 185 vezes a DHR.
Em ratos machos, o deucravacitinibe não teve efeitos nos parâmetros reprodutivos (acasalamento, fertilidade e morfologia do esperma), nem sobre o desenvolvimento embrionário inicial de sua prole com doses orais de até 50 mg/kg/dia e exposição aproximadamente 247 vezes a DHR.
Em ratos fêmea, o deucravacitinibe não teve efeitos sobre o acasalamento, fertilidade ou parâmetros embrionários iniciais com doses orais de até 50 mg/kg/dia e exposição de aproximadamente 171 vezes a DHR.
Toxicologia Animal
Nenhuma informação adicional.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O deucravacitinibe é uma molécula sintética que inibe seletivamente a enzima tirosina quinase 2 (TYK2). O deucravacitinibe se liga ao domínio regulador da TYK2, estabilizando uma interação inibitória entre os domínios reguladores e catalíticos da enzima. Isso resulta na inibição alostérica da ativação de TYK2 mediada pelo receptor e suas funções descendentes nas células. A TYK2 realiza a mediação da sinalização de citocina interleucina-23 (IL-23), citocina interleucina-12 (IL-12) e interferons tipo I (IFN), que são citocinas de ocorrência natural envolvidas em respostas inflamatórias e imunes. O deucravacitinibe inibe a liberação de citocinas e quimiocinas pró-inflamatórias.
Propriedades Farmacodinâmicas
Em voluntários saudáveis, a administração de deucravacitinibe resultou em uma inibição dependente da dose e da concentração de duas vias dependentes da TYK2, indicando um envolvimento robusto do alvo. Estes incluem fosforilação de STAT5 induzida por IFN-alfa (mediada por TYK2/JAK1) e produção de IFN-gama induzida por IL-12 (mediada por TYK2 /JAK2) em ensaios ex vivo de sangue total. A inibição máxima foi observada uma hora após a administração, que retornou próximo ao valor basal ao final do intervalo de administração (12 ou 24 horas). Além disso, a expressão do gene regulada por IFN foi inibida in vivo de uma maneira dependente da dose em indivíduos que receberam IFN-alfa, confirmando que o deucravacitinibe inibe TYK2 in vivo.
Em um subestudo de Fase 2 em pacientes com psoríase, o deucravacitinibe reduziu a expressão gênica associada à psoríase na pele com psoríase de uma forma dependente da dose, notavelmente incluindo reduções nos genes regulados pela via IL-23 e IFN tipo I. Nos estudos de Fase 2 e Fase 3, o deucravacitinibe reduziu os níveis de biomarcadores séricos associados à atividade da doença de psoríase. Na fase 3, IL-17A IL-19 e beta-defensina foram reduzidos com o tratamento com deucravacitinibe em 48-50%, 72% e 81-84%, respectivamente.
O tratamento com deucravacitinibe não foi associado a alterações na hemoglobina média, contagens de plaquetas, contagens de linfócitos, contagens de neutrófilos, contagens de células NK, contagens de células T, imunoglobulinas, transaminases hepáticas, creatinina ou lipídios no sangue.
Eletrofisiologia Cardíaca
Com 7 vezes a exposição máxima alcançada com a dose de 6 mg uma vez ao dia em pacientes com psoríase, não houve efeito clinicamente relevante no intervalo QTc.
Propriedades Farmacocinéticas
O deucravacitinibe apresentou absorção oral consistente, aumento na exposição relacionado à dose e nenhuma farmacocinética evidente dependente do tempo. A farmacocinética do deucravacitinibe administrado na forma de comprimidos foi linear em uma faixa de doses de 3 mg a 36 mg.
A meia-vida terminal do deucravacitinibe foi de 10 horas e um acúmulo discreto ( < 1,4 vezes) foi observado após a administração de uma dose diária.
Absorção
Após a administração oral de comprimidos, o deucravacitinibe apresentou absorção rápida e quase completa. O ®ax mediano variou de 2 a 3 horas e a biodisponibilidade oral absoluta foi de 99%.
O deucravacitinibe pode ser administrado sem considerar a alimentação ou moduladores de pH gástrico como famotidina e rabeprazol. A administração concomitante de alimentos ou moduladores do pH gástrico (bloqueadores dos receptores H2 e inibidores da bomba de prótons) não afetou a exposição total (AUC [INF]) de deucravacitinibe. Em estudos clínicos, o deucravacitinibe foi administrado independentemente das refeições.
Distribuição
O volume de distribuição no estado estacionário (Vss) de 140 L é superior ao volume corporal total de água (42 L), indicando distribuição extravascular. O deucravacitinibe liga-se à 81,6% às proteínas plasmáticas humanas. O deucravacitinibe é distribuído de maneira semelhante entre o plasma e os componentes dos glóbulos vermelhos, com uma razão de concentração de sangue/plasma de 1,26.
Metabolismo
Em humanos, o deucravacitinibe é metabolizado por meio de quatro vias de biotransformação primárias, que incluem N-desmetilação na porção triazol pelo citocromo (CYP) P-450 1A2 para formar o metabólito principal BMT-153261, hidrólise da ciclopropil carboxamida pela carboxilesterase 2 (CES2) para formar o principal metabólito BMT-158170, N-glucuronidação pela uridina glucuronil transferase (UGT) para formar BMT-334616 e mono-oxidação por CYP2B6/2D6 no grupo metila deuterado para formar M11.
No estado estacionário, o deucravacitinibe é a principal espécie circulante, constituindo 49% dos componentes medidos relacionados ao medicamento. Dois principais metabólitos circulantes, BMT-153261 e BMT-158170, foram identificados, ambos com meias-vidas semelhantes ao composto original deucravacitinibe. BMT-153261 apresenta potência semelhante ao fármaco original e BMT-158170 não é farmacologicamente ativo. A exposição circulante do BMT-153261 é muito mais baixa do que a do composto original e, portanto, a atividade farmacológica predominante é atribuída ao medicamento original deucravacitinibe. Além disso, nenhum metabólito humano exclusivo e nenhum metabólito circulante de longa vida foram identificados.
Eliminação
O deucravacitinibe é eliminado por múltiplas vias, incluindo o metabolismo da Fase I e II, juntamente com a eliminação renal e fecal direta. Além disso, nenhuma enzima ou via única contribuiu com mais de 26% da depuração total. O deucravacitinibe foi extensamente metabolizado, com 59% da dose de [14C] -deucravacitinibe administrada por via oral eliminados como metabólitos na urina (37% da dose) e nas fezes (22% da dose). O deucravacitinibe inalterado na urina e nas fezes representou 13% e 26% da dose, respectivamente, com depuração renal variando de 27 a 54 mL/ minuto.
A meia-vida terminal em voluntários humanos saudáveis é de 10 horas.
Deucravacitinibe é um substrato de transportadores de efluxo, glicoproteína-P e proteína de resistência ao câncer de mama (BCRP) e do transportador de captação OCT1. Devido à alta permeabilidade passiva, alta biodisponibilidade oral e baixa afinidade para esses transportadores, a contribuição desses transportadores para a farmacocinética do deucravacitinibe é mínima. Deucravacitinibe não é um substrato dos transportadores OATP, NTCP, OAT1, OAT3, OCT2, MATE1 ou MATE2K.
Populações específicas
Insuficiência Renal
O comprometimento renal não tem efeito clinicamente significativo nas exposições ao deucravacitinibe e, portanto, nenhum ajuste de dose é necessário para pacientes com comprometimento renal leve, moderado ou grave ou em pacientes com ESRD em diálise.
Em comparação com o grupo de função renal normal, a Cmax de deucravacitinibe foi alterada em até 13,8% e a AUC [INF] em até 39% nos grupos de insuficiência renal (leve, moderado, grave e ESRD).
A diálise não elimina substancialmente o deucravacitinibe da circulação sistêmica (5,4% da dose eliminada por diálise).
Insuficiência hepática
A insuficiência hepática leve (Child-Pugh Classe A) e moderada (Child-Pugh Classe B) não tem efeito clinicamente significativo nas exposições ao deucravacitinibe e, portanto, nenhum ajuste de dose é necessário para esses pacientes. SOTYKTU® (deucravacitinibe) não é recomendado em pacientes com insuficiência hepática grave (Child-Pugh Classe C).
Em comparação com o grupo de função hepática normal, a Cmax total de deucravacitinibe e AUC [INF] no grupo de insuficiência hepática leve e moderada aumentaram até 10% e 40%, respectivamente, enquanto a Cmax de deucravacitinibe não ligado e AUC (INF) aumentaram em até 26% e 60 %, respectivamente.
Em indivíduos com insuficiência hepática grave, a Cmax total de deucravacitinibe foi comparável e a AUC total foi 43% maior em relação a indivíduos saudáveis compatíveis. Nestes indivíduos, Cmax e AUC [INF] não ligados aumentaram 62% e 131%, respetivamente.
Idosos
As exposições ao deucravacitinibe (Cmax e Cavg) não foram significativamente alteradas em pacientes com psoríase > 65 anos em relação a pacientes com 40 a 65 anos.
Pediátrico e adolescente
A segurança e eficácia de SOTYKTU® (deucravacitinibe) em pacientes pediátricos não foram estabelecidas.
Dados em Animais
Em um estudo com animais jovens, a administração oral de deucravacitinibe (5, 15 ou 50 mg/kg/ dia) em ratos por 10 semanas começando no dia 21 pós-natal não resultou em nenhuma toxicidade ou efeitos no crescimento e desenvolvimento; as alterações limitaram-se aos efeitos imunomoduladores esperados e foram, na sua maioria, reversíveis. Todas as mudanças observadas neste estudo foram observadas anteriormente em ratos adultos em uma magnitude semelhante, indicando que ratos jovens não são mais sensíveis e não demonstram toxicidade exclusiva em relação aos achados observados em ratos maduros. Com 50 mg/kg/dia, as exposições ao deucravacitinibe foram aproximadamente 125 vezes a dose humana recomendada (DHR).
Gênero
As exposições ao deucravacitinibe (Cmax e Cavg) não foram significativamente alteradas nas mulheres em relação aos homens.
Peso corporal
As exposições ao deucravacitinib (Cmax e Cavg) não foram significativamente alteradas em indivíduos com um peso corporal superior ( > 90 kg) ou inferior ( < 90 kg).
Raça e Etnia
A raça não foi identificada como uma covariável significativa para nenhum parâmetro farmacocinético do deucravacitinibe.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao deucravacitinibe ou a qualquer um dos excipientes (vide COMPOSIÇÃO).
5. ADVERTÊNCIAS E PRECAUÇÕES
Imunológico
Infecções
SOTYKTU® (deucravacitinibe) pode aumentar o risco de infecções. Em estudos clínicos, as infecções ocorreram em 29,1% do grupo SOTYKTU® (deucravacitinibe) em comparação com 21,5% do grupo placebo durante 16 semanas de tratamento. As infecções mais comuns no grupo SOTYKTU® (deucravacitinibe) foram infecções do trato respiratório superior. A incidência de infecções graves no grupo SOTYKTU® (deucravacitinibe) e no grupo placebo foi semelhante e ≤ 0,6%. O tratamento com SOTYKTU® (deucravacitinibe) não deve ser iniciado em pacientes com qualquer infecção ativa clinicamente importante até que a infecção seja resolvida ou tratada.
Em pacientes com infecção crônica ou história de infecção recorrente, considere os riscos e benefícios antes de prescrever SOTYKTU® (deucravacitinibe). Os pacientes devem ser instruídos a procurar orientação médica se ocorrerem sinais ou sintomas de infecção clinicamente importante. Se um paciente desenvolver uma infecção desse tipo ou não estiver respondendo à terapia padrão, monitore o paciente de perto e interrompa SOTYKTU® (deucravacitinibe) até que a infecção seja solucionada.
Avaliação pré-tratamento para tuberculose
Avaliar os pacientes quanto à infecção por tuberculose antes de iniciar o tratamento com SOTYKTU® (deucravacitinibe). Não administrar SOTYKTU® (deucravacitinibe) a pacientes com tuberculose ativa. Iniciar o tratamento da tuberculose latente antes de administrar SOTYKTU® (deucravacitinibe).
Considerar a terapia anti-tuberculose antes do início de SOTYKTU® (deucravacitinibe) em pacientes com história anterior de tuberculose latente ou ativa nos quais um curso de tratamento adequado não pode ser confirmado. Monitorar os pacientes que recebem SOTYKTU® (deucravacitinibe) quanto a sinais e sintomas de tuberculose ativa durante o tratamento.
Imunizações
Antes de iniciar a terapia com SOTYKTU® (deucravacitinibe), considerar a conclusão de todas as imunizações apropriadas para a idade de acordo com as diretrizes atuais de imunização. Evitar o uso de vacinas vivas em pacientes tratados com SOTYKTU® (deucravacitinibe). A resposta a vacinas vivas ou não vivas não foi avaliada.
Gravidez e Lactação
Dados em Humanos
Não existem estudos adequados e bem controlados do uso de SOTYKTU® (deucravacitinibe) em mulheres grávidas. Os dados com o uso de SOTYKTU® (deucravacitinibe) em mulheres grávidas são insuficientes para informar sobre o risco associado ao medicamento.
Dados em Animais
O deucravacitinibe foi administrado por via oral durante o período de organogênese em doses de 5, 15 ou 75 mg/kg/dia em ratos e 1, 3 ou 10 mg/kg/dia em coelhos. O deucravacitinibe não foi embrioletal nem teratogênico nas doses mais altas testadas em ambas as espécies. Estas doses resultaram em exposições maternas (AUC) que foram aproximadamente 266 vezes (ratos) ou 91 vezes (coelhos) a exposição à dose humana recomendada (DHR).
Em um estudo de desenvolvimento pré e pós-natal em ratos, o deucravacitinibe foi administrado desde o dia 6 de gestação até ao dia 20 de lactação, em doses de 5, 15 ou 50 mg/kg/dia. Com 50 mg/kg/dia, o peso corporal dos filhotes foi reduzido, em relação aos valores de controle, durante o período pré-desmame; durante o pós-desmame, seus pesos aumentaram e eram comparáveis aos da prole de controle nos dias pós-natal 73 ou 35 em machos e fêmeas, respectivamente. Não houve achados adversos adicionais na prole F1, nem na sobrevida intrauterina F2. As exposições maternas a 50 mg/kg/dia foram aproximadamente 110 vezes a DHR.
Não se sabe se SOTYKTU® (deucravacitinibe) é excretado no leite humano. Deve-se ter cautela, pois muitos medicamentos podem ser excretados no leite humano. Uma dose oral única de 5 mg/kg de deucravacitinibe radiomarcado foi administrada em ratas lactantes (dias pós-parto 8 a 12). O deucravacitinibe e/ou seus metabólitos estavam presentes no leite destas ratas, com razões de concentração de leite/plasma de 2,7 a 30,9.
Categoria de Risco na Gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico ou cirurgião-dentista.
Uso pediátrico
A segurança e eficácia de SOTYKTU® (deucravacitinibe) em pacientes pediátricos com menos de 18 anos de idade não foram estabelecidas.
Uso em idosos
Dos 1.519 pacientes com psoríase em placas tratados com SOTYKTU® (deucravacitinibe), 152 pacientes tinham 65 anos ou mais e 21 pacientes tinham 75 anos ou mais.
Nenhuma diferença geral na exposição, segurança ou eficácia do deucravacitinibe foi observada entre pacientes mais velhos e mais jovens que receberam SOTYKTU® (deucravacitinibe).
Endócrino e Metabolismo
Lactose
Atenção: SOTYKTU® (deucravacitinibe) contém 44 mg de lactose. Pacientes com problemas hereditários raros de intolerância à galactose, deficiência total de lactase ou má absorção de glicose-galactose não devem tomar este medicamento.
Hepático/Biliar/Pancreático
SOTYKTU® não é recomendado em pacientes com insuficiência hepática grave.
Efeitos sobre a capacidade de dirigir e operar máquinas
Não foram realizados estudos sobre os efeitos na habilidade de dirigir e operar máquinas.
Pacientes devem ter cuidado ao dirigir ou operar um veículo ou máquina potencialmente perigosa.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro amarelo e óxido de ferro vermelho, que podem, eventualmente, causar reações alérgicas.
Este medicamento não deve ser usado por pessoas com síndrome de má-absorção de glicose-galactose.
Em caso de suspeita de dengue, ou quando associado a outros medicamentos que aumentem o efeito hemorrágico, a prescrição deste medicamento ou a manutenção do tratamento com ele deve ser reavaliada, devido a seu potencial hemorrágico.
Medicamentos imunossupressores podem ativar focos primários de tuberculose. Esteja alerta quanto à possibilidade de surgimento de doença ativa, tomando os cuidados para o diagnóstico precoce e tratamento.
Informe a seu paciente que, durante tratamento, o uso de vacinas exige avaliação do profissional de saúde.
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de outros medicamentos no SOTYKTU® (deucravacitinibe)
SOTYKTU® (deucravacitinibe) pode interagir com:
• Imunossupressores potentes: A segurança e eficácia de SOTYKTU® (deucravacitinibe) em combinação com imunossupressores, incluindo biológicos, não foram avaliadas em pacientes com psoríase. Devido ao risco aumentado de infecção durante o tratamento com SOTYKTU® (deucravacitinibe), não é recomendado o uso em combinação com outros imunossupressores potentes.
• Vacinas vivas: Evite o uso de vacinas vivas em pacientes tratados com SOTYKTU® (deucravacitinibe).
Não se espera que SOTYKTU® (deucravacitinibe) cause interações medicamentosas importantes por meio de inibições enzimáticas, indução enzimática ou inibição de transportador.
O deucravacitinibe é eliminado por múltiplas vias, incluindo o metabolismo das fases I e II, eliminação renal e fecal direta, sem uma via única predominantemente responsável pela eliminação. Portanto, não são previstas interações medicamentosas importantes por meio da inibição ou indução de uma via.
Em estudos específicos de interação medicamentosa, não foram observadas alterações clinicamente significativas em deucravacitinibe após a administração concomitante de ciclosporina (inibidor duplo de Pgp/BCRP), fluvoxamina (inibidor de CYP1A2), ritonavir (indutor de CYP1A2), diflunisal (inibidor de UGT 1A9), pirimetamina (OCT1) e agentes moduladores do pH gástrico como famotidina (antagonista do receptor H2) ou rabeprazol (inibidor da bomba de prótons).
Não é necessário ajuste da dose de deucravacitinibe quando administrado concomitantemente com inibidores da Pgp/BCRP, inibidores fortes da CYP1A2, indutores da CYP1A2, inibidores da UGT1A9, inibidores da OCT1 ou moduladores do pH gástrico.
Efeito do SOTYKTU® (deucravacitinibe) em outros medicamentos
Com base em dados in vitro de deucravacitinibe e seus principais metabólitos circulantes e estudos clínicos de interação medicamentosa, não se espera que a administração concomitante de deucravacitinibe, 6 mg por dia, tenha efeito clinicamente relevante nas exposições de agentes que são substratos das CYPs (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4), UGTs (1A1, 1A4, 1A6, 1A9, 2B7), CES2 e transportadores de drogas (Pgp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1, e MATE2K).
Em estudos específicos de interação medicamentosa, o deucravacitinibe não teve um efeito significativo nas exposições aos medicamentos concomitantes rosuvastatina, metotrexato, micofenolato mofetil (MMF) ou anticoncepcionais orais (acetato de noretindrona e etinilestradiol), e nenhum ajuste de dose para deucravacitinibe é necessário quando coadministrado com eles.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
SOTYKTU® (deucravacitinibe) deve ser armazenado em temperatura ambiente (de 15 a 30°C).
Prazo de Validade: 36 meses após a data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Propriedades organolépticas e físicas
SOTYKTU® (deucravacitinibe) é um comprimido revestido rosa, redondo, biconvexo, para administração oral, impresso a laser com "BMS 895 6 mg" numa das faces em duas linhas e nenhuma inscrição na outra face.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Considerações de Posologia
• SOTYKTU® (deucravacitinibe) não deve ser administrado em pacientes com qualquer infecção ativa clinicamente importante até que a infecção seja resolvida ou tratada adequadamente. SOTYKTU® (deucravacitinibe) não é recomendado para uso em combinação com outros imunossupressores potentes.
• Avaliar os pacientes quanto à infecção por tuberculose (TB) antes de iniciar o tratamento com SOTYKTU® (deucravacitinibe). SOTYKTU® (deucravacitinibe) não deve ser administrado em pacientes com tuberculose ativa.
• Antes de iniciar a terapia com SOTYKTU® (deucravacitinibe), considere a conclusão de todas as imunizações adequadas à idade de acordo com as diretrizes de imunização atuais.
Posologia
Psoríase em placas
A dose recomendada de SOTYKTU® (deucravacitinibe) é de 6 mg, por via oral, uma vez ao dia, com ou sem alimentos.
Populações especiais
Insuficiência renal
Não é necessário ajuste de dose em pacientes com insuficiência renal leve, moderada ou grave ou em pacientes com doença renal em estágio terminal (ESRD) em diálise.
Insuficiência hepática
SOTYKTU® (deucravacitinibe) não é recomendado em pacientes com insuficiência hepática grave (Child-Pugh Classe C). Nenhum ajuste de dose é necessário em pacientes com insuficiência hepática leve ou moderada.
Uso Pediátrico
A segurança e eficácia de SOTYKTU® (deucravacitinibe) em pacientes pediátricos não foram estabelecidas.
Idosos
Nenhum ajuste de dose é necessário para pacientes com 65 anos ou mais (vide 3. CARACTERÍSTICAS FARMACOLÓGICAS - Populações específicas).
Os comprimidos de SOTYKTU® (deucravacitinibe) devem ser engolidos inteiros e podem ser administrados com ou sem alimentos.
Este medicamento não deve ser partido, esmagado, cortado ou mastigado.
Se uma dose de SOTYKTU® (deucravacitinibe) for esquecida, o paciente não deve tomar uma dose dupla para compensá-la.
9. REAÇÕES ADVERSAS
Dados de Estudos Clínicos
Os ensaios clínicos são conduzidos sob condições muito específicas. Portanto, as taxas de reações adversas observadas nos ensaios clínicos podem não refletir as taxas observadas na prática e não devem ser comparadas com as taxas nos ensaios clínicos de outro medicamento. Informações sobre reações adversas de ensaios clínicos podem ser úteis para identificar e aproximar as taxas de reações adversas a medicamentos em uso no mundo real.
Em estudos clínicos, um total de 1.519 pacientes com psoríase em placas moderada a grave receberam SOTYKTU® (deucravacitinibe) 6 mg, uma vez por dia. Destes, 1.141 pacientes foram expostos ao SOTYKTU® (deucravacitinibe) por pelo menos um ano.
Os dados de dois estudos controlados com placebo e ativo (POETYK PSO-1 e POETYK PSO-2) foram agrupados para avaliar a segurança de SOTYKTU® (deucravacitinibe) até 16 semanas. No total, 842 pacientes foram avaliados no grupo SOTYKTU® (deucravacitinibe) 6 mg.
A reação adversa (RA) mais comum foi infecção do trato respiratório superior. Reações adversas graves foram relatadas em 1,8% dos pacientes tratados com SOTYKTU® (deucravacitinibe), 2,9% dos pacientes tratados com placebo e 1,2% dos pacientes tratados com apremilaste até a semana 16. Nenhuma reação adversa grave foi relatada em mais de 1 paciente.
No período de 16 semanas controlado por placebo dos ensaios clínicos agrupados (POETYK PSO-1 e POETYK PSO-2), a descontinuação da terapia devido a eventos adversos em pacientes que receberam SOTYKTU® (deucravacitinibe) foi de 2,4%, em comparação com 3,8% para placebo e 5,2% para apremilaste.
A maioria das reações adversas que levaram à descontinuação do tratamento ocorreu em um único paciente.
Resumo tabulado das reações adversas
As reações adversas que ocorreram em pacientes tratados com SOTYKTU® (deucravacitinibe) durante o período controlado de 16 semanas são apresentadas na Tabela 2 abaixo.
Essas reações são apresentadas por classe de sistema de órgãos e por frequência. As frequências são definidas como: muito comum (≥ 1/10); comum (≥ 1/100 a < 1/10); incomum (≥ 1 / 1.000 a < 1/100); rara (≥ 1 / 10.000 a < 1 / 1.000); muito rara ( < 1 / 10.000); desconhecida (não pode ser calculado a partir dos dados disponíveis).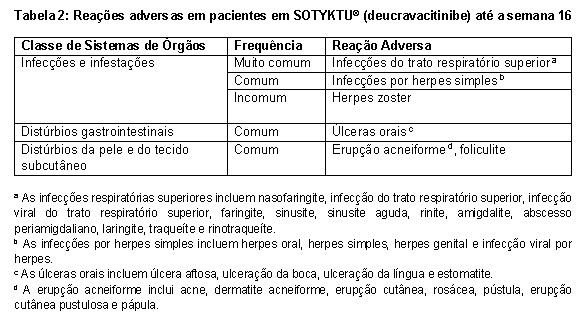
Até a semana 52, nenhuma nova reação adversa foi identificada com SOTYKTU® (deucravacitinibe) e as taxas de incidência de reações adversas comuns não aumentaram em comparação com as observadas durante as primeiras 16 semanas de tratamento.
Infecções
Nas primeiras 16 semanas, as infecções ocorreram em 29,1% do grupo SOTYKTU® (deucravacitinibe) (116 eventos por 100 pessoas/ano) em comparação com 21,5% do grupo de placebo (83,7 eventos por 100 pessoas/ano).
A maioria das infecções foram de gravidade leve a moderada, e não levaram à descontinuação de SOTYKTU® (deucravacitinibe). A incidência de infecções graves no grupo SOTYKTU® (deucravacitinibe) foi de 0,6% (2,0 eventos por 100 pessoas/ano) e no grupo do placebo foi de 0,5% (1,6 eventos por 100 pessoas/ano).
Em POETYK PSO-1 e POETYK PSO-2, até a Semana 52, a taxa de infecções no grupo SOTYKTU® (deucravacitinibe) (95,4 eventos por 100 pessoas/ano) não aumentou em comparação com a taxa observada durante as primeiras 16 semanas de tratamento. A taxa de infecções graves no grupo SOTYKTU® (deucravacitinibe) não aumentou até a Semana 52 (1,7 eventos por 100 pessoas/ano).
Malignidades
Durante o período de tratamento de 0 a 52 semanas dos dois estudos clínicos de psoríase controlados (exposição total de 969 pessoas/ano com SOTYKTU® (deucravacitinibe), doenças malignas (excluindo cânceres de pele não melanoma) foram relatadas em 0,2% dos pacientes tratados com SOTYKTU® (deucravacitinibe) (0,3 eventos por 100 pessoas/ano), incluindo um linfoma. Linfomas também foram relatados com SOTYKTU® (deucravacitinibe) no estudo aberto de extensão em longo prazo e em um estudo regional aberto. O papel potencial do SOTYKTU® (deucravacitinibe) no desenvolvimento de doenças malignas não está claro.
Achados laboratoriais anormais:
Resultados de Ensaios Clínicos
Creatina Fosfoquinase (CPK)
No período controlado por placebo de 16 semanas, foram relatados eventos adversos de CPK aumentada (incluindo Grau 4) em 23 pacientes (9,3 por 100 pacientes-ano) tratados com SOTYKTU® (deucravacitinibe) e 5 pacientes (4,1 por 100 pacientes-ano) tratados com placebo.
Transaminases hepáticas (ALT e AST)
Eventos de aumento das enzimas hepáticas ≥3 vezes o limite superior normal (LSN) foram observados em pacientes tratados com SOTYKTU® (deucravacitinibe). No período de 16 semanas controlado por placebo:
• Elevações de ALT ≥3 vezes, o LSN foram relatadas em 9 pacientes (3,6 por 100 pacientes-ano) tratados com SOTYKTU® (deucravacitinibe) e 2 pacientes (1,6 por 100 pacientes-ano) tratados com placebo.
• Elevações de AST ≥3 vezes, o LSN foram relatadas em 13 pacientes (5,2 pacientes por 100 pacientes-ano) tratados com SOTYKTU® (deucravacitinibe) e 2 pacientes (1,6 por 100 pacientes-ano) tratados com placebo.
Diminuição da taxa de filtração glomerular (TFG)
No período de 16 semanas controlado por placebo em pacientes que apresentavam insuficiência renal moderada (eTFG 30-59 mL/min) na linha basal, a diminuição da TFG foi relatada em 4 pacientes (1,6 por 100 pacientes-ano) tratados com SOTYKTU® (deucravacitinibe) e 1 paciente (0,8 por 100 pacientes-ano) tratados com placebo. Dois dos pacientes tratados com SOTYKTU® (deucravacitinibe) apresentaram piora da proteinúria basal.
Elevações de lipídios
A média de triglicerídeos aumentou 0,12 mmol/L (10,3 mg/dL) durante o período de tratamento de 16 semanas em pacientes tratados com SOTYKTU® (deucravacitinibe) e 0,10 mmol/L (9,1 mg/dL) durante o período de tratamento de 52 semanas.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSAGEM
O deucravacitinibe foi administrado em doses únicas até 40 mg ( > 6 vezes a dose humana recomendada de 6 mg/dia) e em doses múltiplas até 24 mg/dia (12 mg duas vezes ao dia) durante 14 dias, sem toxicidade limitante da dose.
Em caso de sobredosagem, recomenda-se que o paciente seja monitorado para quaisquer sinais ou sintomas de reações adversas e o tratamento sintomático apropriado seja instituído imediatamente.
A diálise não elimina o deucravacitinibe de maneira substancial da circulação sistêmica (5,4% da dose eliminada por tratamento de diálise).
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
11. REFERÊNCIAS
• Armstrong A. et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: Effiacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. Journal of the American Academy of Dermatology 2023;v88;29-39, DOI: 10.1016/jaad.2022.07.002
• Strober S. Et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: Efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program for evaluation of TYK2 inhibitor psoriasis second trial. Journal of the American Academy of Dermatology 2023;v88;40-51, DOI: 10.1016/jaad.2022.08.061
Dizeres legais.
Registro: 1.0180.0415
VENDA SOB PRESCRIÇÃO.
Esta bula foi aprovada pela ANVISA em 20/11/2023.