SOGROYA
NOVO NORDISK
somapacitana
Reposição do hormônio do crescimento (GH)..
Apresentações.
Solução injetável de somapacitana em sistema de aplicação preenchido (multidose e descartável).
Sogroya® 5 mg/1,5 mL (3,3 mg/mL):
Cada embalagem contém: 1 sistema de aplicação preenchido contém um carpule com 1,5 mL, vedado permanentemente no sistema de aplicação, de solução injetável de somapacitana e que libera doses de 0,025 mg a 2,0 mg em incrementos de 0,025 mg.
O botão do sistema de aplicação é de cor verde-claro.
Sogroya® 10 mg/1,5 mL (6,7 mg/mL):
Cada embalagem contém: 1 sistema de aplicação preenchido que contém um carpule com 1,5 mL, vedado permanentemente no sistema de aplicação, de solução injetável de somapacitana e que libera doses de 0,05 mg a 4,0 mg em incrementos de 0,05 mg.
O botão do sistema de aplicação é de cor amarelo.
Sogroya® 15 mg/1,5 mL (10 mg/mL):
Cada embalagem contém: 1 sistema de aplicação preenchido que contém um carpule com 1,5 mL, vedado permanentemente no sistema de aplicação, de solução injetável de somapacitana e que libera doses de 0,10 mg a 8,0 mg em incrementos de 0,10 mg.
O botão do sistema de aplicação é de cor vermelho rubi.
USO SUBCUTÂNEO
USO ADULTO E PEDIÁTRICO ACIMA DE 2 ANOS
Composição.
Sogroya® 5 mg/1,5 mL (3,3 mg/mL):
Cada mL de Sogroya® 5 mg/1,5 mL contém 3,3 mg de somapacitana
Excipientes: histidina, manitol, poloxâmer, fenol, ácido clorídrico (ajuste de pH), hidróxido de sódio (ajuste de pH) e água para injetáveis.
Um sistema de aplicação preenchido contém 5 mg de somapacitana em 1,5 mL.
Sogroya® 10 mg/1,5 mL (6,7 mg/mL):
Cada mL de Sogroya® 10 mg/1,5 mL contém 6,7 mg de somapacitana
Excipientes: histidina, manitol, poloxâmer, fenol, ácido clorídrico (ajuste de pH), hidróxido de sódio (ajuste de pH) e água para injetáveis.
Um sistema de aplicação preenchido contém 10 mg de somapacitana em 1,5 mL.
Sogroya® 15 mg/1,5 mL (10 mg/mL):
Cada mL de Sogroya® 15 mg/1,5 mL contém 10 mg de somapacitana
Excipientes: histidina, manitol, poloxâmer, fenol, ácido clorídrico (ajuste de pH), hidróxido de sódio (ajuste de pH) e água para injetáveis.
Um sistema de aplicação preenchido contém 15 mg de somapacitana em 1,5 mL.
Vide item "5. Advertências e Precauções" para informações sobre teor de sódio.
A somapacitana é produzida com tecnologia de DNA recombinante em Escherichia coli, seguida pela adição de uma porção de ligação à albumina.
INFORMAÇÕES TÉCNICAS AOS PROFISSIONAIS DE SAÚDE
Informações técnicas.
1. INDICAÇÕES
Sogroya® é indicado para a reposição do hormônio do crescimento endógeno em crianças e adolescentes a partir de 2 anos de idade com insuficiência de crescimento devido à deficiência de hormônio do crescimento e em adultos com deficiência do hormônio do crescimento.
2. RESULTADOS DE EFICÁCIA
Crianças e adolescentes
- REAL 4 (Fase 3)
A eficácia e segurança de Sogroya® uma vez por semana (5 mg/1,5 mL (3,3 mg/mL), 10 mg/1,5 mL (6,7 mg/mL) e 15 mg/1,5 mL (10 mg/mL)) foram avaliadas em um estudo de 52 semanas, randomizado, multicêntrico, aberto, com controle ativo, grupo-paralelo de fase 3 (REAL 4) em 200 pacientes pediátricos com deficiência de hormônio do crescimento que nunca receberam tratamento com hormônio do crescimento. Os pacientes foram randomizados para 0,16 mg/kg/semana de Sogroya® uma vez por semana (N=132) ou 0,034 mg/kg/dia de somatropina diária (N=68). 15 pacientes atingiram a puberdade durante o estudo.
No início do estudo, os 200 pacientes tinham uma idade média de 6,4 anos (variação: 2,5 a 11). 25,5% dos pacientes eram do sexo feminino e 74,5% do sexo masculino. 37% dos pacientes eram asiáticos, 0,5% eram negros ou afro-americanos, 57% eram caucasianos e 5,5% foram categorizados como "outros" ou não relatados.
O tratamento com Sogroya® uma vez por semana por 52 semanas resultou em uma velocidade de crescimento anualizada de 11,2 cm/ano. Os pacientes tratados com somatropina diária atingiram uma velocidade de crescimento anualizada de 11,7 cm/ano após 52 semanas de tratamento (Tabela 1).
A altura SDS (alteração do basal) foi de 1,25 no braço Sogroya® uma vez por semana e 1,30 no braço somatropina diária na semana 52 (Tabela 5). A alteração do IGF-I SDS desde o basal na semana 52 foi semelhante nos dois braços com valores de 2,36 para Sogroya® uma vez por semana e 2,33 para somatropina diária. A média de IGF-I SDS também foi semelhante entre Sogroya® e somatropina diária na semana 52.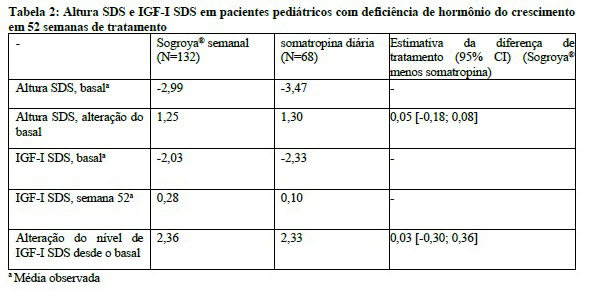
A grande maioria dos pacientes pediátricos (96,9%) no estudo alcançou um nível médio de IGF-I SDS dentro da faixa normal (-2 a +2) após 52 semanas de tratamento com Sogroya® uma vez por semana (Tabela 3). Baixo número de pacientes teve média de IGF-I SDS acima de +2 (2,3%) e nenhum paciente teve média de IGF-I SDS acima de +3.2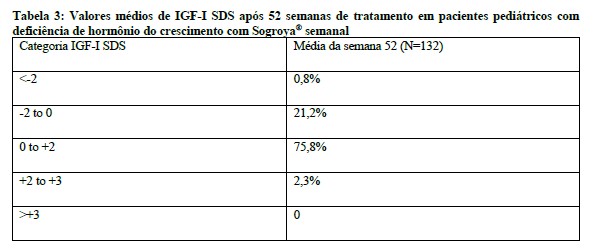
- REAL 3 (Fase 2)
Um total de 59 pacientes pediátricos com deficiência de hormônio do crescimento sem tratamento prévio com hormônio do crescimento completaram um período principal de 26 semanas e uma extensão de 26 semanas em um estudo de grupo paralelo de 4 braços com Sogroya® semanal em doses de 0,04, 0,08 e 0,16 mg/kg /semana e braço de controle ativo de 0,034 mg/kg/dia de somatropina diária. Os pacientes continuaram em um período de extensão para avaliação de segurança, de 104 semanas, aberto, em braços paralelos, com Sogroya 0,16 mg/kg/semana e somatropina diária 0,034 mg/kg/dia. Todos os pacientes foram posteriormente transferidos para Sogroya® 0,16 mg/kg/semana em uma extensão de segurança de longo prazo de 208 semanas. 17 pacientes atingiram a puberdade durante o estudo.
O tratamento com Sogroya® semanal levou a benefícios contínuos do tratamento até pelo menos a semana 208. A SDS de altura foi de -1,06 (alteração do basal: 2,85) em 38 pacientes.
O resultado da altura obtido na semana 208 em pacientes que mudaram de 0,034 mg/kg/dia de somatropina diária para 0,16 mg/kg/semana com Sogroya® na semana 156 indicou que os benefícios do tratamento com somatropina diária são mantidos após a mudança para Sogroya® semanal.
A média de IGF-I SDS permaneceu dentro da faixa normal para todos os grupos.
Amostragem de IGF-I SDS após a aplicação
As amostras de sangue podem ser colhidas em qualquer dia da semana após as aplicações de somapacitana. A amostragem 2 dias após a aplicação aproxima-se muito do valor máximo de IGF-I esperado, enquanto a concentração média de IGF-I ao longo do intervalo de dosagem semanal é mais aproximada com uma amostra colhida 4 dias após a aplicação.
Com base em dados de ensaios clínicos em pacientes pediátricos com deficiência de hormônio do crescimento, uma orientação para calcular o IGF-I SDS médio com base na amostragem de sangue após a aplicação é fornecida na Tabela 4.
Segurança clínica
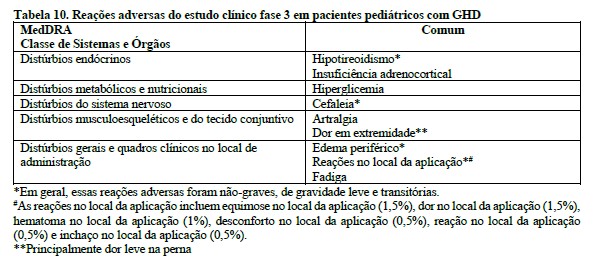
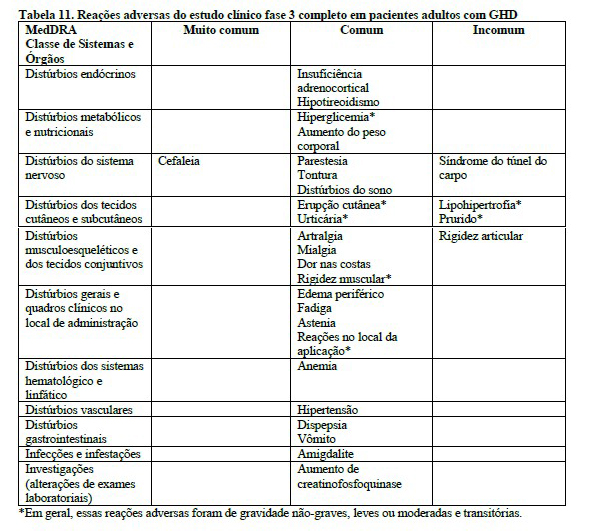
O perfil de segurança da somapacitana foi semelhante ao perfil de segurança bem conhecido da somatropina. Não foram identificados novos problemas de segurança. Não foram identificados problemas de tolerabilidade local.
Imunogenicidade
Um baixo número de pacientes pediátricos testou positivo para anticorpos de ligação à somapacitana em qualquer momento durante o tratamento (16 dentre 132 pacientes). Nenhum desses anticorpos foi neutralizante e não foi observado impacto em farmacocinética, eficácia ou segurança. Nenhum anticorpo foi detectado em pacientes adultos.
O uso de Sogroya® foi avaliado apenas em crianças com deficiência do hormônio de crescimento. Seu uso em crianças nascidas pequenas para a idade gestacional (PIG) não foi avaliado.
Resultados relatados pelo paciente
REAL 4:
Pacientes pediátricos tratados com Sogroya® uma vez por semana relataram uma carga de tratamento menor conforme medido usando o GHD-CTBa na semana 52 em comparação com pacientes tratados com somatropina diária.
Os cuidadores de pacientes pediátricos tratados com Sogroya® uma vez por semana relataram menor carga de tratamento conforme medido usando o GHD-PTBb na semana 52 em comparação com pacientes tratados com somatropina diária.2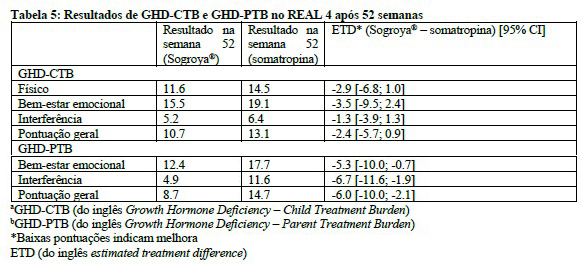
REAL 3:
82% dos cuidadores de pacientes pediátricos que mudaram de somatropina diária preferiram Sogroya® uma vez por semana usando o PPQ (do inglês Patient Preference Questionnaire).
89% dos que preferiram Sogroya® uma vez por semana, indicaram que seriam mais aderentes à terapia do que a somatropina diária.
Adultos
Em um estudo controlado por placebo (duplo-cego) e com controle ativo (aberto) de 34 semanas, 301 pacientes adultos com deficiência do hormônio do crescimento sem tratamento prévio foram randomizados (2:1:2) e 300 foram expostos à somapacitana ou placebo uma vez por semana ou somatropina diariamente por um período de tratamento de 34 semanas (fase principal do estudo clínico). A população de pacientes tinha uma idade média de 45,1 anos (faixa de 23 a 77 anos; 41 pacientes tinham 65 anos ou mais), 51,7% eram do sexo feminino e 69,7% tinham deficiência do hormônio do crescimento de início na fase adulta.
Um total de 272 pacientes adultos com deficiência do hormônio do crescimento que concluíram a fase principal de 34 semanas continuaram em um período de extensão aberto de 53 semanas. Os participantes em uso de placebo foram transferidos para somapacitana e os pacientes em uso de somatropina foram randomizados novamente (1:1) para somapacitana ou somatropina.
Os efeitos clínicos observados para os principais desfechos primários na fase de tratamento principal (Tabela 6) e na fase de tratamento de extensão (Tabela 7) estão apresentados abaixo.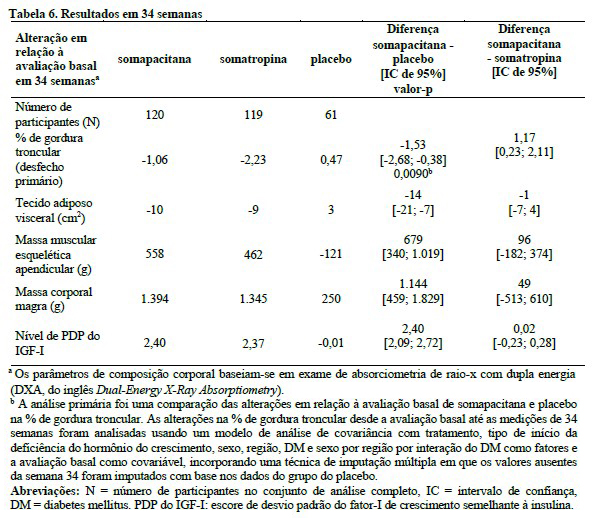
A análise de subgrupos post-hoc das alterações em relação à avaliação basal na porcentagem de gordura troncular (%) em comparação ao placebo na semana 34 revelou uma diferença de tratamento estimada (somapacitana-placebo) de -2,49% [-4,19; -0,79] em homens, -0,80% [-2,99; 1,39] em mulheres que não receberam estrogênio oral, -1,44% [-3,97; 1,09] em mulheres recebendo estrogênio oral.1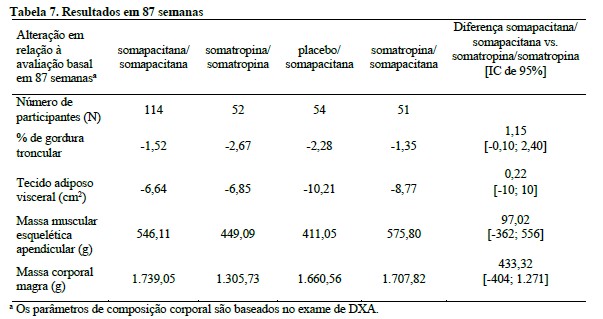
Níveis de PDP do IGF-I observados e simulados no estudo clínico
Na fase principal do estudo clínico, os valores de PDP do IGF-I de 0 e maiores foram, em geral, alcançados em 53% dos participantes do estudo com deficiência do hormônio do crescimento em adultos tratados com somapacitana após um período de titulação da dose de 8 semanas. Essa proporção, no entanto, foi menor em subgrupos específicos, como mulheres em uso de estrogênio oral (32%) e pacientes com início de deficiência do hormônio do crescimento na infância (39%) (Tabela 8). As análises de simulação post-hoc indicaram que as proporções de pacientes adultos com deficiência do hormônio do crescimento que alcançaram níveis de PDP do IGF-I acima de 0 devem ser maiores caso a titulação da dose de somapacitana além de 8 semanas seja permitida. Nesta análise de simulação, presumiu-se que a titulação da dose de somapacitana foi bem tolerada em todos os pacientes até que o intervalo alvo de PDP do IGF-I ou uma dose de somapacitana de 8 mg por semana fosse alcançada.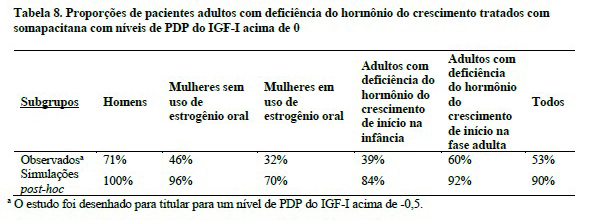
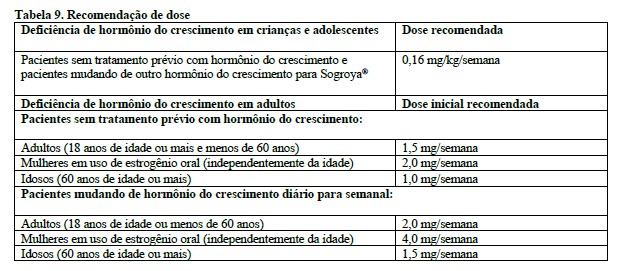
Dose de manutenção
A dose de manutenção varia de pessoa para pessoa e entre pacientes do sexo masculino e feminino. A dose de manutenção média de somapacitana observada nos estudos clínicos de fase 3 foi de 2,4 mg/semana.
Referências:
1. Johannsson G, Gordon MB, Højby Rasmussen M, Håkonsson IH, Karges W, Sværke C, Tahara S, Takano K, Biller BMK. Once-weekly Somapacitan is Effective and Well Tolerated in Adults with GH Deficiency: A Randomized Phase 3 Trial. J Clin Endocrinol Metab. 2020 Apr 1;105(4): e1358-76. doi:10.1210/clinem/dgaa049. PMID: 32022863; PMCID: PMC7076631.
2. A Trial Comparing the Effect and Safety of Once Weekly Dosing of Somapacitan With Daily Norditropin® in Children With Growth Hormone Deficiency (REAL 4).
3. A Trial Investigating Efficacy and Safety of Once-weekly NNC0195-0092 (Somapacitan) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® FlexPro®) in Growth Hormone Treatment naïve Pre-pubertal Children With Growth Hormone Deficiency naïve Pre-pubertal Children With Growth Hormone Deficiency (REAL 3).
3. CARACTERÍSTICAS FARMACOLÓGICAS
• Propriedades Farmacodinâmicas
Mecanismo de ação
A somapacitana é um derivado do hormônio do crescimento humano recombinante de ação prolongada. Ela consiste em 191 aminoácidos semelhantes ao hormônio do crescimento humano endógeno, com uma única substituição na estrutura principal do aminoácido (L101C) à qual um componente de ligação à albumina foi acoplado. O componente de ligação à albumina (cadeia lateral) consiste em um componente de ácido graxo e um espaçador hidrofílico ligado à posição 101 da proteína.
O mecanismo de ação da somapacitana ocorre diretamente via receptor do GH (do inglês growth hormone) e/ou indiretamente via IGF-I produzido nos tecidos em todo o organismo, mas predominantemente pelo fígado.
Quando a deficiência do hormônio do crescimento é tratada com somapacitana, é alcançada uma normalização da composição corporal (ou seja, diminuição da massa de gordura corporal, aumento da massa corporal magra) e da ação metabólica.
A somapacitana distribui-se para a zona hipertrófica e esponjosa primária na epífise da tíbia proximal de ratos hipofisectomizados com deficiência de hormônio do crescimento. A distribuição da somapacitana nos tecidos periféricos é comparável à do hormônio do crescimento endógeno.
A somapacitana estimula o crescimento esquelético em pacientes pediátricos com deficiência de hormônio do crescimento através dos efeitos nas placas de crescimento (epífises) dos ossos.
Efeitos farmacodinâmicos
IGF-I
O IGF-I é um biomarcador geralmente aceito para avaliação de eficácia na deficiência do hormônio do crescimento.
Uma resposta do IGF-I dependente da dose é induzida após a administração de somapacitana. Um padrão de estado de equilíbrio é alcançado nas respostas do IGF-I após 1 a 2 doses semanais.
Os níveis de IGF-I flutuam durante a semana. A resposta do IGF-I é máxima após 2 a 4 dias. Em comparação com o tratamento diário com GH, o perfil do IGF-I da somapacitana difere, consulte a Figura 1.
Em pacientes pediátricos com deficiência de hormônio do crescimento, a somapacitana produz uma resposta de IGF-I linear à dose, com uma alteração de 0,02 mg/kg em média, resultando em uma alteração na pontuação de desvio padrão (SDS) do IGF-I de 0,32.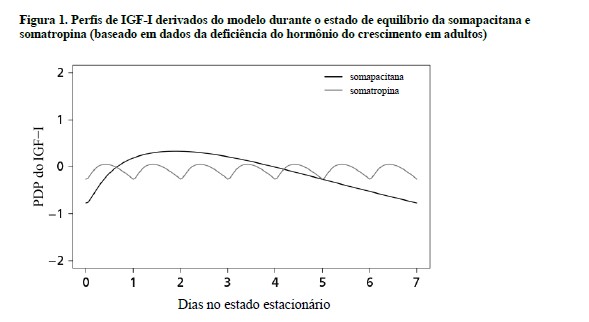
• Propriedades Farmacocinéticas
A somapacitana possui propriedades farmacocinéticas compatíveis com a administração uma vez por semana. A ligação reversível à albumina endógena retarda a eliminação da somapacitana e, portanto, prolonga a meia-vida in vivo e a duração da ação.
A farmacocinética da somapacitana após a administração subcutânea foi investigada em níveis de dose de 0,02 a 0,16 mg/kg/semana na população pediátrica, de 0,01 a 0,32 mg/kg em adultos saudáveis, e em doses de até 0,12 mg/kg em adultos com deficiência do hormônio do crescimento.
Em geral, a somapacitana exibe farmacocinética não linear, mas na faixa de dose clinicamente relevante de somapacitana em adultos com deficiência do hormônio do crescimento, a farmacocinética da somapacitana é aproximadamente linear.
Em pacientes pediátricos, uma dose de somapacitana de 0,16 mg/kg/semana corresponde a uma concentração média de 80,2 ng/mL e, em adultos, as doses de somapacitana na faixa clinicamente relevante correspondem a concentrações médias de 0,1-36,2 ng/mL.
Absorção
Em pacientes adultos e pediátricos com deficiência do hormônio do crescimento, o tmáx mediano variou de 4 a 25,5 horas em doses de 0,02 mg/kg/semana a 0,16 mg/kg/semana.
A exposição em estado de equilíbrio foi alcançada após 1 a 2 administração(ões) semanal(is).
A biodisponibilidade absoluta da somapacitana em humanos não foi investigada.
Distribuição
A somapacitana se liga majoritariamente ( > 99%) às proteínas plasmáticas e espera-se que seja distribuída como a albumina. Com base nas análises da farmacocinética populacional, o volume de distribuição estimado (V/F) foi de 1,7 L em pacientes pediátricos com deficiência do hormônio do crescimento e 14,6 L em adultos.
Eliminação
Após uma dose única de 0,16 mg/kg/semana, a meia-vida terminal foi de 34 horas em pacientes pediátricos com deficiência de hormônio do crescimento.
A meia-vida terminal foi estimada com médias geométricas variando de aproximadamente 2 a 3 dias em estado de equilíbrio em pacientes com deficiência do hormônio do crescimento (doses: 0,02 a 0,12 mg/kg).
A somapacitana estará presente na circulação por aproximadamente 2 semanas após a última dose. Foi observado pouco ou nenhum acúmulo (razão de acúmulo médio: 1 a 2) de somapacitana após doses múltiplas em pacientes com deficiência do hormônio do crescimento.
Biotransformação
A somapacitana é extensivamente metabolizada pela degradação proteolítica e clivagem da sequência ligante entre o peptídeo e o ligante de albumina.
A somapacitana foi extensivamente metabolizada antes da excreção e nenhuma somapacitana intacta foi encontrada na urina, que foi a principal via de excreção (81%), nem nas fezes, onde 13% do material relacionado à somapacitana foi encontrado, indicando biotransformação completa antes da excreção.
Populações especiais
População pediátrica
Com base na análise farmacocinética populacional, sexo, raça e peso corporal não apresentam efeito clinicamente significativo na farmacocinética após a dosagem baseada no peso.
Adultos
- Idade
Indivíduos com mais de 60 anos de idade têm maior exposição (29%) do que indivíduos mais jovens recebendo a mesma dose de somapacitana. Uma dose inicial menor para indivíduos com mais de 60 anos de idade está descrita no item "8. Posologia e Modo de Usar".
- Gênero
Indivíduos do sexo feminino e, em específico, indivíduos do sexo feminino recebendo estrogênio oral, apresentaram exposição menor (53% para mulheres recebendo estrogênio oral e 30% para mulheres não recebendo estrogênio oral) do que indivíduos do sexo masculino na mesma dose de somapacitana. O item "8. Posologia e Modo de Usar" descreve uma dose inicial mais alta para mulheres em uso de estrogênio oral.
- Raça
Não houve diferença na exposição da somapacitana e resposta do IGF-I entre indivíduos japoneses e brancos. Apesar de uma exposição maior em asiáticos não japoneses em comparação com brancos na mesma dose de somapacitana, brancos, japoneses e asiáticos não japoneses precisaram das mesmas doses para atingir níveis semelhantes de IGF-I. Portanto, não há recomendação de ajuste de dose com base na raça.
- Etnia
A etnia (hispânica ou latina 4,5% (15 indivíduos receberam somapacitana)) não foi investigada devido ao pequeno tamanho da amostra no programa de desenvolvimento.
- Peso corporal
Apesar de uma maior exposição em indivíduos adultos com baixo peso corporal em comparação com os de alto peso corporal na mesma dose de somapacitana, os indivíduos precisaram das mesmas doses para atingir níveis semelhantes de IGF-I em toda a faixa de peso corporal de 35 kg a 150 kg. Portanto, não há recomendação de ajuste de dose em adultos com base no peso corporal.
- Insuficiência renal
Uma dose de somapacitana de 0,08 mg/kg em estado de equilíbrio resultou em exposições mais elevadas em indivíduos com insuficiência renal, mais acentuada em indivíduos com insuficiência renal grave e naqueles que necessitavam de hemodiálise, nos quais as razões da AUC0-168h para função renal normal foram de 1,75 e 1,63, respectivamente. Em geral, a exposição da somapacitana tendeu a aumentar com a diminuição da taxa de filtração glomerular (TFG). Foram observados níveis mais elevados de IGF-IAUC0-168h em indivíduos com insuficiência renal moderada e grave e naqueles que necessitavam de hemodiálise, com razões para função renal normal de 1,35, 1,40 e 1,24, respectivamente. Devido ao modesto aumento observado no IGF-I combinado com as baixas doses iniciais recomendadas e a titulação da dose individual de somapacitana, não há recomendação de ajuste da dose em pacientes com insuficiência renal.
- Insuficiência hepática
Uma dose de somapacitana de 0,08 mg/kg em estado de equilíbrio resultou em maior exposição em indivíduos com insuficiência hepática moderada, com razões para função hepática normal de 4,69 para AUC0-168h e 3,52 para Cmáx. Foram observados níveis mais baixos de IGF-I estimulados por somapacitana em indivíduos com insuficiência hepática leve e moderada em comparação com indivíduos com função hepática normal (razão para função normal foi de 0,85 para leve e 0,75 para moderada). Devido à modesta diminuição observada no IGF-I combinado com a titulação da dose individual de somapacitana, não há recomendação de ajuste da dose em pacientes com insuficiência hepática.
• Dados de segurança pré-clínica
Os dados pré-clínicos não revelaram nenhum risco especial para humanos com base em estudos convencionais de farmacologia de segurança, toxicidade de doses repetidas, genotoxicidade ou desenvolvimento pré e pós-natal.
Não foram realizados estudos de carcinogenicidade com a somapacitana.
Não foram observados efeitos adversos na fertilidade masculina e feminina em ratos com uma dose que resultou em exposição pelo menos 13 e 15 vezes maior do que a exposição clínica máxima esperada a 8 mg/semana para machos e fêmeas, respectivamente. No entanto, um ciclo estral irregular em fêmeas foi observado em todas as doses tratadas.
Nenhuma evidência de dano fetal foi identificada quando ratas e coelhas grávidas receberam somapacitana por via subcutânea durante a organogênese em doses que levaram a exposições bem acima da exposição esperada na dose clínica máxima de 8 mg/semana (pelo menos 18 vezes). Em doses altas que levaram a exposições pelo menos 130 vezes acima da exposição clínica máxima esperada de 8 mg/semana, foram observados ossos longos curtos/tortos/espessos em filhotes de ratas fêmeas recebendo somapacitana. Sabe-se que tais achados em ratos se resolveram após o nascimento e devem ser considerados como malformações menores, e não anomalias permanentes.
O crescimento fetal foi reduzido quando coelhas grávidas receberam doses de somapacitana por via subcutânea em exposições pelo menos 9 vezes acima da exposição esperada na dose clínica máxima de 8 mg/semana.
Em ratas lactantes, o material relacionado à somapacitana foi secretado no leite, mas em um nível menor do que o observado no plasma (até 50% do nível no plasma).
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer um dos excipientes listados em "Composição".
Sogroya® não deve ser usado quando houver qualquer evidência de atividade tumoral. Quaisquer tumores pré-existentes devem estar inativos e a terapia antitumoral deve ser concluída antes do início da terapia com Sogroya®. O tratamento deve ser descontinuado se houver evidências de crescimento tumoral (vide item "5. Advertências e Precauções").
Sogroya® não deve ser usado para promoção do crescimento longitudinal em crianças com epífises fechadas.
Adultos:
Pacientes com doença aguda crítica apresentando complicações após a cirurgia cardíaca de peito aberto, cirurgia abdominal, trauma múltiplo acidental, insuficiência respiratória aguda ou quadros clínicos semelhantes não devem ser tratados com Sogroya® (em relação a pacientes submetidos à outras terapias de reposição além do hormônio do crescimento, vide item "5. Advertências e Precauções").
Sogroya® é contraindicado em pacientes com retinopatia diabética proliferativa ativa ou não proliferativa grave.
5. ADVERTÊNCIAS E PRECAUÇÕES
• Hipersensibilidade Grave
Reações de hipersensibilidade sistêmica graves, incluindo reações anafiláticas e angioedema, foram relatadas após a comercialização com o uso de somatropina. Informe os pacientes e/ou cuidadores de que tais reações são possíveis e que atendimento médico imediato deve ser procurado se ocorrer uma reação alérgica. Sogroya® é contraindicado em pacientes com hipersensibilidade conhecida à somatropina ou a qualquer excipiente de Sogroya® (vide item "4. Contraindicações").
• Insuficiência adrenocortical
A introdução do tratamento com hormônio do crescimento pode resultar na inibição de 11bHSD-1 e redução das concentrações séricas de cortisol. Em pacientes tratados com hormônio do crescimento, o hipoadrenalismo central (secundário) não diagnosticado previamente pode ser desmascarado e a reposição de glicocorticoides pode ser necessária. Além disso, os pacientes tratados com reposição de glicocorticoides em decorrência de hipoadrenalismo previamente diagnosticado podem necessitar de um aumento nas doses de manutenção ou reforçadas após o início do tratamento com hormônio do crescimento. É necessário monitorar os pacientes com hipoadrenalismo conhecido quanto à redução dos níveis séricos de cortisol e/ou necessidade de doses aumentadas de glicocorticoide (vide item "6. Interações Medicamentosas").
• Comprometimento do metabolismo da glicose
O tratamento com hormônio do crescimento pode diminuir a sensibilidade à insulina, especialmente em doses mais elevadas em pacientes suscetíveis e, consequentemente, pode ocorrer hiperglicemia em indivíduos com capacidade inadequada de secreção de insulina. Como resultado, o comprometimento da tolerância à glicose não diagnosticada previamente e o diabetes mellitus evidente podem ser desmascarados durante o tratamento com hormônio do crescimento. Portanto, os níveis de glicose devem ser monitorados periodicamente em todos os pacientes tratados com hormônio do crescimento, principalmente em pacientes com fatores de risco de diabetes mellitus, como obesidade ou histórico familiar de diabetes mellitus. Pacientes com diabetes mellitus tipo 1 ou tipo 2 pré-existente ou tolerância à glicose diminuída devem ser monitorados atentamente durante a terapia com hormônio do crescimento. As doses dos medicamentos anti-hiperglicêmicos podem necessitar de ajuste quando a terapia com hormônio do crescimento for instituída nesses pacientes.
• Neoplasias
Existe um risco aumentado de progressão de neoplasias com relação ao uso de somatropina em pacientes com tumores ativos. Quaisquer tumores pré-existentes devem estar inativos e a terapia antitumoral deve ser concluída antes do início da terapia com Sogroya®. O tratamento deve ser descontinuado se houver evidências de atividade tumoral recorrente. (vide item "4. Contraindicações").
Risco de segunda neoplasia em pacientes pediátricos
Em sobreviventes de cânceres pediátricos que receberam radiação no cérebro/cabeça para sua primeira neoplasia e que desenvolveram deficiência do hormônio de crescimento e foram tratados com somatropina, um aumento nas neoplasias secundárias foi reportado. Os tumores intracranianos, em particular os meningiomas, foram os mais comuns dessas segundas neoplasias. Monitore todos os pacientes com história de deficiência do hormônio de crescimento secundária a uma neoplasia intracraniana durante o tratamento com somatropina quanto à progressão ou recorrência do tumor.
Nova malignidade durante o tratamento
Como crianças com certas causas genéticas raras de baixa estatura têm um risco aumentado de desenvolver malignidades, considere cuidadosamente os riscos e benefícios de iniciar Sogroya® nesses pacientes. Se o tratamento com Sogroya® for iniciado, monitore cuidadosamente esses pacientes quanto ao desenvolvimento de neoplasias.
Existe um risco potencial de alterações malignas de nevos pré-existentes em pacientes em tratamento com somatropina. Todos os pacientes recebendo Sogroya® devem ser cuidadosamente monitorados quanto ao aumento do crescimento ou possíveis alterações malignas de nevos pré-existentes. Aconselhe os pacientes a relatarem alterações marcantes no comportamento, aparecimento de dores de cabeça, distúrbios da visão e/ou alterações na aparência de nevos pré-existentes.
• Hipertensão intracraniana benigna
No caso de cefaleias graves ou recorrentes, sintomas visuais, náuseas e/ou vômitos, recomenda-se a realização de fundoscopia para detectar papiledema. Se o papiledema for confirmado, o diagnóstico de hipertensão intracraniana benigna deve ser considerado e, se apropriado, o tratamento com hormônio do crescimento deve ser descontinuado. Atualmente, não há evidências suficientes para orientar a tomada de decisão clínica em pacientes com hipertensão intracraniana resolvida. Se o tratamento com hormônio do crescimento for reiniciado, é necessário um monitoramento cuidadoso dos sintomas de hipertensão intracraniana.
• Função tireoidiana
O hormônio do crescimento aumenta a conversão extratireoidiana de T4 em T3 e pode, como tal, desmascarar o hipotireoidismo incipiente. Como o hipotireoidismo interfere na resposta à terapia com hormônio do crescimento, os pacientes devem ter sua função tireoidiana avaliada regularmente e devem receber terapia de reposição com hormônios da tireoide quando indicado (vide itens "6. Interações Medicamentosas" e "9. Reações Adversas").
• Uso com estrogênio oral
O estrogênio oral influencia a resposta do IGF-I ao hormônio do crescimento, incluindo Sogroya®.
Mulheres que estiverem administrando qualquer forma de estrogênio oral (terapia hormonal ou contracepção) devem considerar mudança da via de administração de estrogênio (por exemplo, medicamentos hormonais transdérmicos ou vaginais) ou usar outra forma de contracepção. Se uma mulher em uso de estrogênio oral iniciar terapia com Sogroya®, podem ser necessárias doses iniciais maiores e um período de titulação mais longo (vide item "8. Posologia e Modo de Usar").
Se uma mulher que estiver administrando Sogroya® começar a terapia com estrogênio oral, a dose de Sogroya® pode precisar ser aumentada para manter os níveis séricos de IGF-I dentro do intervalo normal apropriado para a idade. Por outro lado, se uma mulher em uso de Sogroya® descontinuar a terapia oral com estrogênio, a dose de Sogroya® pode precisar ser reduzida para evitar o excesso de somapacitana e/ou efeitos indesejáveis (vide itens "6. Interações Medicamentosas" e "8. Posologia e Modo de Usar").
• Distúrbios da pele e do tecido subcutâneo
Caso Sogroya® seja administrado no mesmo local por um longo período de tempo, podem ocorrer alterações locais no tecido subcutâneo, como lipohipertrofia, lipoatrofia e lipodistrofia adquirida. O local de aplicação deve ser alternado para reduzir o risco (vide itens "8. Posologia e Modo de Usar" e "9. Reações Adversas").
• Anticorpos
Anticorpos contra somapacitana não foram observados em pacientes adultos com GHD. Poucos pacientes pediátricos com GHD testaram positivo para anticorpos de ligação ao somapacitana. Nenhum desses anticorpos era neutralizante e nenhum impacto nos efeitos clínicos foi observado. Deve-se realizar testes para verificar a presença de anticorpos contra a somapacitana em pacientes que não responderem à terapia.
• Doença aguda crítica
O efeito do hormônio do crescimento na recuperação foi estudado em dois estudos controlados por placebo envolvendo 522 pacientes adultos em estado crítico sofrendo de complicações após cirurgia cardíaca de peito aberto, cirurgia abdominal, trauma múltiplo acidental ou insuficiência respiratória aguda. A mortalidade foi maior em pacientes tratados com 5,3 ou 8,0 mg de hormônio do crescimento diariamente em comparação com pacientes que receberam placebo (42% vs. 19%). Com base nessas informações, esses pacientes não devem ser tratados com Sogroya®. Como não há informações disponíveis sobre a segurança da terapia de reposição do hormônio do crescimento em pacientes com doença aguda crítica, os benefícios da continuidade do tratamento nessa situação devem ser ponderados em relação aos possíveis riscos envolvidos.
A deficiência de hormônio do crescimento em adultos é uma doença crônica e precisa ser tratada de forma adequada. No entanto, a experiência em pacientes com mais de 60 anos de idade e em pacientes com mais de cinco anos de tratamento na deficiência de hormônio do crescimento em adultos ainda é limitada.
• Retenção de fluidos
Pode ocorrer retenção de fluidos durante a terapia de reposição com Sogroya®. As manifestações clínicas da retenção de líquidos (por exemplo, edema e síndromes de compressão nervosa incluindo síndrome do túnel do carpo/parestesia) geralmente são transitórias e dose-dependentes.
• Pancreatite
Casos de pancreatite foram relatados em pacientes recebendo produtos de hormônio de crescimento. A pancreatite deve ser considerada em pacientes que desenvolverem dor abdominal intensa persistente.
• Epifisiólise proximal do fêmur:
Em crianças com rápido crescimento e em pacientes com distúrbios endócrinos, incluindo GHD, a epifisiólise proximal do fêmur pode ocorrer com mais frequência do que na população em geral. Crianças com dor persistente no quadril/joelho e/ou mancando durante o tratamento com somapacitana devem ser examinadas clinicamente.
• População pediátrica
Os riscos associados ao uso de hormônio do crescimento em pacientes pediátricos incluem morte súbita em pacientes com Síndrome de Prader-Willi, aumento do risco de segunda neoplasia em sobreviventes de câncer pediátrico tratados com radiação no cérebro e/ou cabeça, deslizamento epifisário proximal do fêmur, progressão de escoliose pré-existente e pancreatite.
• Teor de sódio
Este medicamento contém menos de 1 mmol de sódio (23 mg) por dose, portanto, é essencialmente "livre de sódio".
Fertilidade, gravidez e amamentação
• Gravidez
Não há dados do uso de Sogroya® em mulheres grávidas. Estudos em animais mostraram toxicidade reprodutiva (vide "Dados de segurança pré-clínica" no item "3. Características Farmacológicas").
Sogroya® não é recomendado durante a gravidez e em mulheres com potencial para engravidar que não utilizam métodos contraceptivos.
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
• Amamentação
Uso criterioso no aleitamento ou na doação de leite humano: O uso deste medicamento no período da lactação depende da avaliação e acompanhamento médico ou do cirurgião-dentista.
Não se sabe se somapacitana e metabólitos são excretados no leite humano.
Os dados farmacodinâmicos/toxicológicos disponíveis em animais mostraram excreção de somapacitana no leite (vide "Dados de segurança pré-clínica" no item "3. Características Farmacológicas").
O risco para recém-nascidos/lactentes não pode ser excluído. A decisão quanto à descontinuação da amamentação ou descontinuação/abstenção da terapia com Sogroya® deve ser ponderada, levando em consideração o benefício da amamentação para a criança e o benefício da terapia para a mulher.
• Fertilidade
Não há experiência clínica com o uso de Sogroya® e seu possível efeito na fertilidade.
Não foram observados efeitos adversos na fertilidade masculina e feminina em ratos (vide "Dados de segurança pré-clínica" no item "3. Características Farmacológicas").
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Sogroya® exerce pouca ou nenhuma influência sobre a capacidade de dirigir veículos e operar máquinas.
Este medicamento pode causar doping.
Crianças tratadas com somapacitana devem ser avaliadas regularmente por um especialista em crescimento infantil. O tratamento com somapacitana sempre deve ser iniciado por um médico especialista na deficiência do
hormônio de crescimento e seu tratamento.
O uso de somapacitana no tratamento da insuficiência de crescimento em crianças só foi avaliado em pacientes
pediátricos com deficiência do hormônio de crescimento, não sendo indicado seu uso para insuficiência de crescimento devido a outras condições.
Sogroya® não é indicado para o tratamento de adultos sem deficiência do hormônio de crescimento.
6. INTERAÇÕES MEDICAMENTOSAS
• Medicamentos metabolizados pelo citocromo P450
Dados de um estudo de interação realizado em adultos com deficiência de hormônio do crescimento sugerem que a administração de hormônio do crescimento pode aumentar a depuração de compostos conhecidos por serem metabolizados pelas isoenzimas do citocromo P450. A depuração dos compostos metabolizados pelo citocromo P450 (por exemplo esteroides sexuais, corticosteroides, anticonvulsivantes e ciclosporina) pode estar especialmente aumentada, resultando em níveis plasmáticos mais baixos desses compostos. A significância clínica desse achado é desconhecida.
• Glicocorticoides
O hormônio do crescimento diminui a conversão de cortisona em cortisol e pode desmascarar o hipoadrenalismo central não descoberto previamente ou tornar ineficazes as doses baixas de reposição de glicocorticoides (vide item "5. Advertências e Precauções").
• Estrogênios orais
Em mulheres recebendo terapia com estrogênio oral, pode ser necessária uma dose maior de Sogroya® para alcançar o objetivo do tratamento (vide itens "5. Advertências e Precauções" e "8. Posologia e Modo de Usar").
• Medicamentos anti-hiperglicêmicos
O tratamento com anti-hiperglicêmicos, incluindo insulina, pode necessitar de ajuste da dose em casos de coadministração com Sogroya®, visto que Sogroya® pode diminuir a sensibilidade à insulina (vide itens "5. Advertências e Precauções" e "9. Reações Adversas").
• Outros
Os efeitos metabólicos de Sogroya® também podem ser influenciados pela terapia concomitante com outros hormônios, por exemplo, testosterona e hormônios da tireoide (vide item "5. Advertências e Precauções").
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Sogroya® deve ser armazenado em geladeira (de 2 °C a 8 °C). Manter distante do compartimento do congelador ou de qualquer sistema de congelamento. Não congelar.
Sogroya® tem validade de 24 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento c