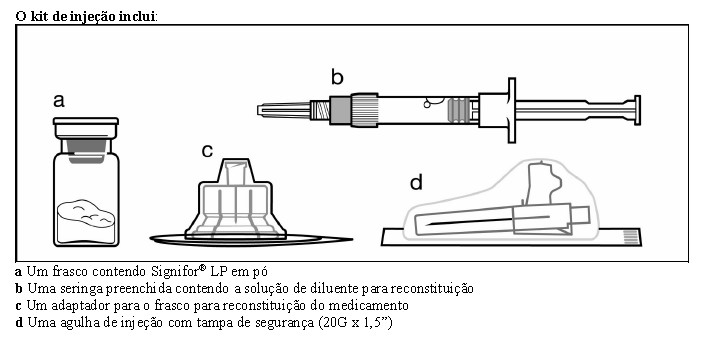
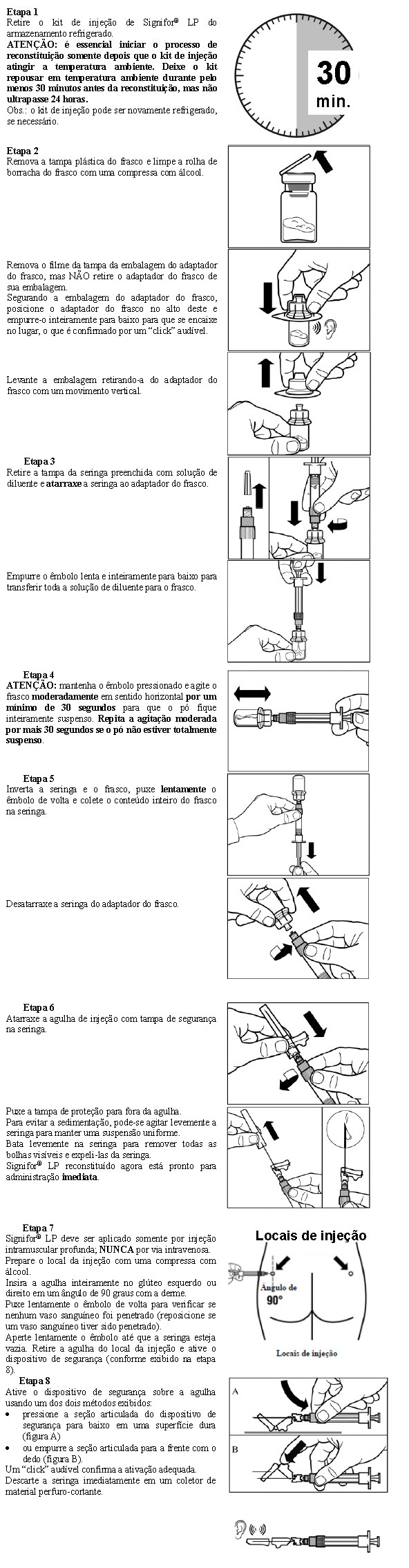
SIGNIFOR LP
RECORDATI
pasireotida
Tratamento da doença de Cushing.
Apresentações.
Signifor® LP 10 mg, 20 mg, 30 mg, 40 mg e 60 mg - embalagens contendo 1 frasco-ampola com pó para suspensão injetável, 1 seringa preenchida com diluente, 1 agulha para injeção com dispositivo de segurança e 1 adaptador para o frasco.
VIA INTRAMUSCULAR
USO ADULTO
Composição.
Cada frasco-ampola de Signifor® LP contém 10 mg de pasireotida (equivalente a 13,71 mg de pamoato de pasireotida). Cada frasco-ampola de Signifor® LP contém 20 mg de pasireotida (equivalente a 27,42 mg de pamoato de pasireotida). Cada frasco-ampola de Signifor® LP contém 30 mg de pasireotida (equivalente a 41,13 mg de pamoato de pasireotida). Cada frasco-ampola de Signifor® LP contém 40 mg de pasireotida (equivalente a 54,84 mg de pamoato de pasireotida). Cada frasco-ampola de Signifor® LP contém 60 mg de pasireotida (equivalente a 82,26 mg de pamoato de pasireotida). Excipientes: Pó para suspensão injetável: copolímero de glicolida e lactida com glicose, copolímero de glicolida e lactida. Diluente: manitol, croscarmelose sódica, poloxâmer, água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Signifor® LP é indicado para o tratamento de pacientes adultos com acromegalia para os quais a cirurgia do tumor hipofisário foi ineficaz ou não é uma opção e que não estão adequadamente controlados com outros análogos da somatostatina. Signifor® LP reduz os sintomas de acromegalia, que inclui dor de cabeça, transpiração excessiva, dormência das mãos e dos pés, cansaço, e dor nas articulações. Signifor® LP é indicado para o tratamento de pacientes adultos com doença de Cushing para os quais a cirurgia não é considerada uma opção ou não foi curativa.
2. RESULTADOS DE EFICÁCIA
Acromegalia Pacientes sem tratamento medicamentoso prévio, estudo C23051
Foi realizado um estudo de fase III multicêntrico, randomizado, cego, para avaliar a segurança e a eficácia do Signifor® LP versus Sandostatin® LAR em pacientes com acromegalia ativa sem tratamento medicamentoso prévio. Um total de 358 pacientes foram randomizados e tratados. Os pacientes foram randomizados na razão de 1:1 em cada um dos dois grupos de tratamento a seguir: 1) pacientes que foram submetidos a uma ou mais cirurgias da hipófise, mas não receberam tratamento medicamentoso prévio ou 2) pacientes novos (de-novo) que apresentaram um adenoma hipofisário visível na ressonância magnética e que recusaram cirurgia da hipófise ou para os quais esta cirurgia é contraindicada.2 Os dois grupos de tratamento estavam bem equilibrados em termos de parâmetros demográficos basais e características da doença. Dos pacientes nos grupos de tratamento com Signifor® LP e Sandostatin® LAR, 59,7% e 56%, respectivamente, eram pacientes que não haviam realizadocirurgia hipófisaria prévia da (de-novo). A média de idade dos pacientes era de aproximadamente 45 anos. As mulheres constituíam 52% dos pacientes em ambos os grupos de tratamento, sendo que 59,7% dos pacientes no grupo do Signifor® LP e 61,0% no grupo do Sandostatin® LAR eram caucasianos. 1,2 A dose inicial foi de 40 mg para Signifor® LP e 20 mg para Sandostatin® LAR. Permitia-se o aumento da dose para obtenção de eficácia, a critério dos investigadores, após três e seis meses de tratamento se os parâmetros bioquímicos indicassem GH médio ≥ 2,5 microgramas/L e/ou IGF-1 > LSN (limite superior da normalidade relacionado a idade e sexo). A dose máxima permitida foi de 60 mg de Signifor® LP e 30 mg de Sandostatin® LAR (Figura 1).2 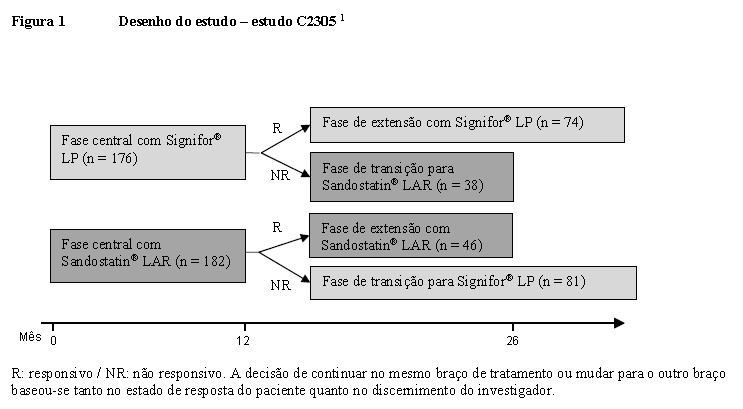
Fase central
O desfecho primário de eficácia foi a proporção de pacientes com redução do nível médio de GH < 2,5 microgramas/L e a normalização do IGF-1 aos limites normais (relacionados a idade e sexo) no mês 12. O desfecho primário de eficácia foi atendido; o percentual de pacientes que alcançou o controle bioquímico foi de 31,3% e 19,2% com Signifor® LP e Sandostatin® LAR, respectivamente, demonstrando um resultado superior estatisticamente significativo a favor do Signifor® LP (valor de p = 0,007) (Tabela 1). 2 
O controle bioquímico foi alcançado cedo no estudo (ou seja, no mês 3) por uma proporção mais elevada de pacientes no braço de Signifor® LP do que no braço de Sandostatin® LAR (30,1% e 21,4%) e foi mantido em todas as avaliações subsequentes durante a fase central. 2 Entre os pacientes com pelo menos um aumento da dose, 12,4% dos pacientes no braço de tratamento com Signifor® LP e 8,9% no braço de tratamento com Sandostatin® LAR, alcançaram o controle bioquímico. 2 No mês 12, a redução no volume tumoral foi comparável entre os grupos de tratamento e em pacientes com e sem cirurgia hipofisária prévia. Noventa e oito por cento dos pacientes tratados com Signifor® LP apresentaram ou redução ou nenhuma alteração no volume do tumor, a partir dos valores basais avaliados por imagem de ressonância magnética no mês 12. A alteração média (variação) no volume do tumor foi uma redução de 39,8% (-97,6% para 16,9 %).A proporção de pacientes com redução do volume tumoral superior em 20% no mês 12 foi de 80,8% com Signifor® LP e de 77,4% com Sandostatin® LAR. 3 A qualidade de vida relacionada à saúde, aferida pelo instrumento AcroQol, foi avaliada na visita inicial e no mês 12. No mês 12, houve melhoras estatisticamente significativas nas pontuações de aparência física e psicológica e nas pontuações globais do AcroQoL, tanto nos grupos de tratamento com Signifor® LP quanto com Sandostatin® LAR. Em média, a melhora observadaem relação ao valor basal foi maior com Signifor® LP do que com Sandostatin® LAR, mas essa diferença não foi estatisticamente significativa. Além disso, o tamanho do anel e cinco sintomas associados a acromegalia (ou seja, cefaleia, fadiga, transpiração, parestesia e osteoartralgia) receberam pontuação 0 (nenhum sintoma) a 4 (muito grave) em cada mês em ambos os pontos de tempo. No mês 12, houve reduções no tamanho do anel e nas pontuações de gravidade de todos os cinco sintomas em ambos os grupos de tratamento comparados ao valor basal, sem diferenças estatisticamente significativas entre os dois grupos de tratamento. 2
Fase de extensão
Ao término da fase central, os pacientes que alcançaram o controle bioquímico ou se beneficiaram do tratamento, conforme avaliado pelo investigador, puderam continuar a ser tratados na fase de extensão com o medicamento do estudo em que foram inicialmente randomizados (Figura 1). 2 Durante a fase de extensão, 74 pacientes continuaram a receber Signifor® LP e 46 pacientes continuaram o tratamento com Sandostatin® LAR.1,2 No mês 25, 48,6% dos pacientes (36/74) no grupo do Signifor® LP e 45,7% (21/46) no grupo do Sandostatin® LAR alcançaram o controle bioquímico. No mesmo ponto de tempo, 70,3% e 80,4% dos pacientes no braço de Signifor® LP e no braço de Sandostatin® LAR, respectivamente, apresentaram valores médios de GH < 2,5 microgramas/L; e a normalização do IGF-1 foi alcançada por 51,4% e 47,8% dos pacientes, respectivamente. O percentual de pacientes que alcançou o controle bioquímico, incluindo pacientes com IGF-1 < LIN, foi de 60,8% (45/74) no grupo do Signifor® LP e 52,2% (24/46) no grupo do Sandostatin® LAR. 2 Durante a fase de extensão, o volume tumoral continuou a diminuir e as melhoras nos sinais e sintomas de acromegalia permaneceram comparáveis entre os dois braços de tratamento. As pontuações do AcroQoL permaneceram numericamente superiores no grupo de Signifor® LP em relação ao grupo de tratamento com Sandostatin® LAR durante toda a fase de extensão. 2
Pacientes com controle inadequado da doença
Estudo C2402 4
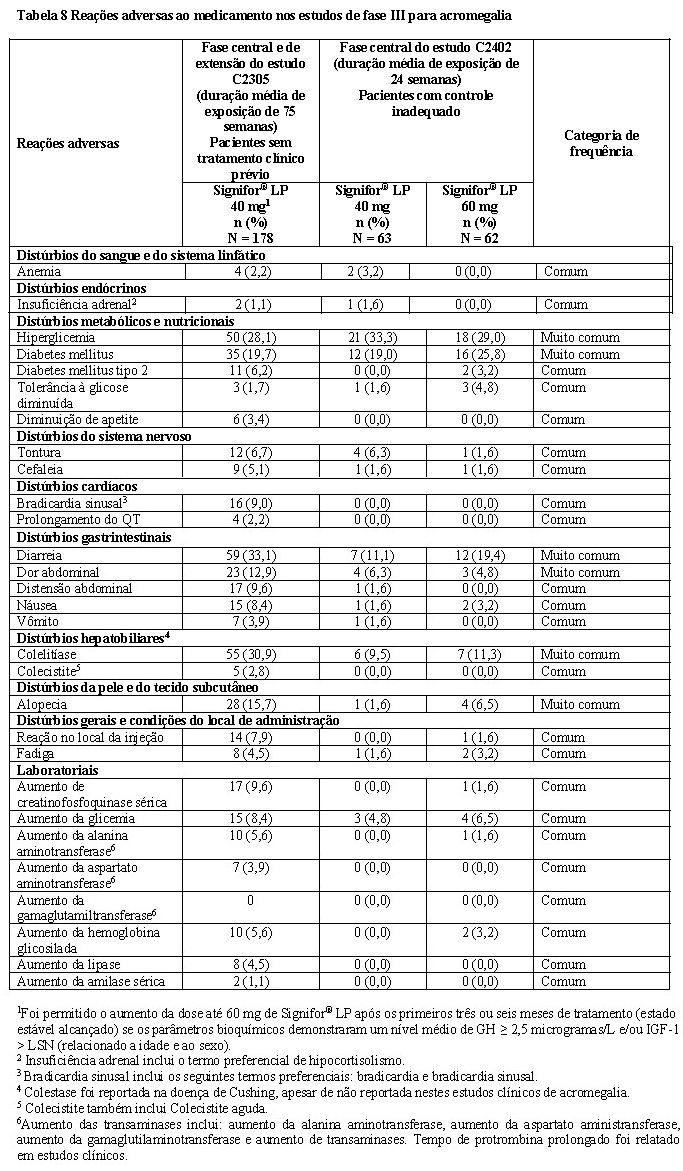
O estudo C2402 foi um estudo de fase III, multicêntrico, randomizado, de grupos paralelos, com três braços, correspondendo a Signifor® LP 40 mg ou Signifor® LP 60 mg em caráter duplo-cego versus Sandostatin® LAR 30 mg ou lanreotida ATG 120 mg em regime aberto em pacientes com acromegalia com controle inadequado da doença. Um total de 198 pacientes foi randomizado para receber Signifor® LP 40 mg (n = 65), Signifor® LP 60 mg (n = 65) ou controle ativo (n = 68). Foram tratados 192 pacientes. Um total de 181 pacientes concluiu a fase central (24 semanas) do estudo. 2,4 Pacientes com controle inadequado no estudo C2402 foram definidos como pacientes com concentração média de GH de um perfil de cinco pontos durante um período de duas horas > 2,5 microgramas/L e IGF-1 ajustado por sexo e idade > 1,3 × limite superior da normalidade (LSN). Os pacientes tinham que ser tratados com as doses máximas indicadas de Sandostatin® LAR (30 mg) ou lanreotida ATG (120 mg) durante pelo menos seis meses antes da randomização. As características basais demográficas e da doença estavam equilibradas entre os braços de tratamento, com média de idade em torno de 45 anos, proporção aproximadamente igual de homens e mulheres e tempo mediano desde o diagnóstico de aproximadamente quatro anos. Três quartos dos pacientes haviam sido anteriormente tratados com Sandostatin® LAR e um quarto com lanreotida ATG. Quase metade dos pacientes havia passado por outro tratamento clínico anterior adicional para acromegalia, além dos análogos da somatostatina. Dois terços de todos os pacientes havia sido submetido a cirurgia anterior. O valor basal médio de GH foi de 17,6 microgramas/L, 12,1 microgramas/L e 9,5 microgramas/L, nos grupos de 40 mg, 60 mg e de controle ativo, respectivamente. Os valores médios de IGF-1 na visita inicial foram de 2,6, 2,8 e 2,9 X LSN respectivamente. 2,4 O desfecho primário de eficácia foi comparar a proporção de pacientes que alcançou o controle bioquímico (definido como níveis médios de GH < 2,5 microgramas/L e normalização de IGF-1 ajustado por sexo e idade) na semana 24 com Signifor® LP 40 mg ou 60 mg versus tratamento contínuo com controle ativo (Sandostatin® LAR 30 mg ou lanreotida ATG 120 mg), separadamente. O estudo atingiu seu desfecho primário de eficácia em ambas as doses de Signifor® LP. A proporção de pacientes que alcançou o controle bioquímico foi de 15,4% (valor de p = 0,0006) e 20,0% (valor de p < 0,0001) com Signifor® LP 40 mg e 60 mg, respectivamente, em 24 semanas comparado a zero no braço de controle ativo (Tabela 2). 2,4 
Em pacientes tratados com Signifor® LP nos quais se observaram reduções dos níveis de GH e IGF-1, essas alterações ocorreram rapidamente e foram mantidas até a semana 24, o que é compatível com o que foi observado em pacientes sem tratamento medicamentoso prévio no estudo C2305. 2,4 A proporção de pacientes com redução ou sem alteração no volume do tumor hipofisário na semana 24 foi de 81,0% e 70,3% com Signifor® LP 40 e 60 mg e de 50,0% com controle ativo. A alteração média (variação) no volume do tumor foi uma redução de -10,4% (-74,5% para 19,4%) e -6,3% (-66,7% para 14,5%), a partir dos valores basais para Signifor® LP 40 mg e 60 mg, respectivamente. Além disso, uma proporção mais elevada de pacientes tratados com Signifor® LP (18,5% e 10,8% com 40 mg e 60 mg, respectivamente) do que o comparador ativo (1,5%) atingiu uma redução no volume tumoral de pelo menos 25%. 2,4 A qualidade de vida relacionada à saúde, aferida pelo instrumento AcroQol, foi avaliada na visita inicial e na semana 24. Na semana 24, houve uma melhora nas pontuações de aparência física e psicológica e nas pontuações globais do AcroQoL nos grupos de tratamento com Signifor® LP, tanto com 40 mg quanto com 60 mg. No grupo de Signifor® LP 40 mg, essas alterações foram estatisticamente significativas na parte da avaliação física do AcroQoL. No grupo de Signifor® LP 60 mg, essas alterações foram estatisticamente significativas nas pontuações de aparência física, psicológica e nas pontuações globais. Não houve diferenças estatisticamente significativas no grupo de Sandostatin® LAR ou lanreotida ATG. A melhora média em relação ao valor basal foi maior no grupo de Signifor® LP 60 mg em todas as pontuações. Entretanto, a diferença nas alterações do valor basal até a semana 24 entre os grupos de tratamento não foi estatisticamente significativa. 2
Fase de transição do estudo C2305 1
Ao término da fase central, os pacientes que não responderam adequadamente à terapia inicial foram autorizados a mudar para o outro tratamento (Figura 1). Houve a transição de 81 pacientes de Sandostatin® LAR para Signifor® LP e de 38 pacientes de Signifor® LP para Sandostatin® LAR. Doze meses após a transição, o percentual de pacientes que alcançou o controle bioquímico foi de 17,3% (14/81) com Signifor® LP e 0% (0/38) com Sandostatin® LAR. 2 O percentual de pacientes que alcançou o controle bioquímico, incluindo pacientes com IGF-1 < LIN, foi de 25,9% no grupo de Signifor® LP e 0% no grupo de Sandostatin® LAR. 2 Doze meses após a transição, as taxas de resposta para redução de GH (GH < 2,5 microgramas/L) foram de 44,4% e 23,7% em pacientes tratados com Signifor® LP e Sandostatin® LAR, respectivamente; as taxas de resposta em relação ao IGF-1 foram de 27,2% e 5,3%, respectivamente. Os níveis médios de GH diminuíram acentuadamente em pacientes que mudaram para Signifor® LP, enquanto o nível médio de GH aumentou ao longo do tempo em pacientes que mudaram para Sandostatin® LAR. Os níveis médios de IGF-1 diminuíram ao longo do tempo em pacientes que mudaram para Signifor® LP, ao passo que o nível médio de IGF-1 em pacientes que mudaram para Sandostatin® LAR permaneceu elevado. 2 Foi observada uma diminuição adicional no volume tumoral 12 meses após a transposição em ambos os grupos de tratamento e ela foi maior em pacientes que mudaram para o Signifor® LP (-24,7%) do que em pacientes que mudaram para o Sandostatin® LAR (-17,9%). 2 Foram observadas melhoras, em relação ao valor basal na transposição, das pontuações de gravidade de sintomas de acromegalia em ambos os tratamentos de transposição. 1
Doença de Cushing
Estudo G2304
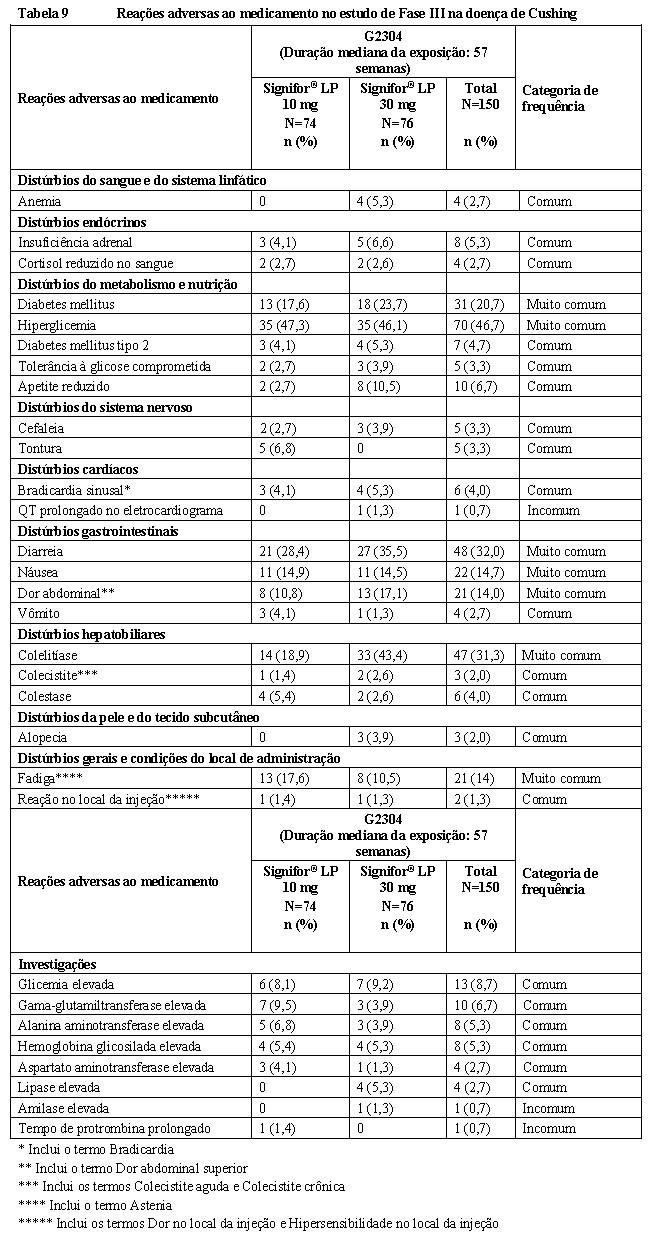
Um estudo de fase III, randomizado, duplo-cego, multicêntrico foi conduzido para avaliar a segurança e eficácia de dois regimes posológicos de Signifor® LP ao longo de um período de tratamento de doze meses em pacientes com doença de Cushing com doença persistente ou recorrente, ou pacientes de novo que não eram considerados candidatos para cirurgia hipofisária.
O estudo incluiu 150 pacientes com um nível de cortisol livre urinário médio (mUFC) na triagem ≥1,5 e ≤5 x LSN, que foram randomizados em uma proporção 1:1 para receber uma dose inicial de Signifor® LP de 10 mg IM uma vez a cada 28 dias ou 30 mg IM uma vez a cada 28 dias. A randomização foi estratificada por valores de mUFC na triagem (1,5 a < 2x LSN versus 2 a 5x LSN, respectivamente).
Após quatro meses de tratamento, os pacientes que apresentavam mUFC ≤1,5 x LSN continuaram na dose cega à qual eles foram randomizados. Os pacientes com mUFC > 1,5 x LSN aos quatro meses tiveram suas doses aumentadas de maneira cega de 10 mg para 30 mg, ou de 30 mg para 40 mg, desde que não houvesse preocupações quanto à tolerabilidade. Aumentos adicionais da dose foram permitidos no mês 7 e mês 9 (em um nível de dose).
A redução da dose em um nível de dose para tolerabilidade foi permitida de maneira cega durante os primeiros sete meses, com um nível mínimo de dose de 5 mg. Após os primeiros sete meses, a titulação decrescente cega de mais de um nível de dose foi permitida em qualquer mês.
Após doze meses de tratamento, os pacientes tiveram a opção de entrar em uma fase de extensão para continuar recebendo Signifor® LP se eles tivessem se beneficiado do tratamento.
O desfecho primário de eficácia foi a proporção de pacientes em cada braço que eram responsivos mUFC (mUFC ≤LSN) após sete meses de tratamento, independentemente da condição de titulação crescente no mês 4. O principal desfecho secundário foi a proporção de pacientes em cada braço que eram responsivos mUFC após sete meses de tratamento e que não titularam de forma crescente a dose antes do mês 7. O limite pré-especificado do limite inferior do intervalo de confiança de 95% para eficácia para o desfecho primário e principal desfecho secundário foi de 15%. Para os pacientes que descontinuaram entre os meses 4 e 7, o último valor observado de mUFC foi usado para determinar a resposta. Os pacientes que descontinuaram antes do mês 4 foram automaticamente classificados como não responsivos. Outros desfechos secundários incluíram alterações desde o período basal no UFC de 24 horas, ACTH plasmático, níveis séricos de cortisol, sinais e sintomas clínicos da doença de Cushing e qualidade de vida relacionada à saúde (HRQL), conforme medida pelas ferramentas SF-12v2 e CushingQoL. Todas as análises foram conduzidas com base nos grupos de dose randomizados.
As características demográficas basais e o histórico da doença estavam bem equilibrados entre os dois grupos de dose randomizados e eram consistentes com a epidemiologia da doença. A idade média dos pacientes era de aproximadamente 38,5 anos, com uma predominância de pacientes do sexo feminino (78,7%). A maioria dos pacientes tinha doença de Cushing persistente ou recorrente (82,0%). 5
Resultados
O estudo atendeu ao objetivo primário de eficácia para ambos os grupos de dose. No mês 7, a resposta mUFC foi atingida em 41,9% (IC de 95% 30,5 a 53,9) e 40,8% (IC de 95% 29,7 a 52,7) dos pacientes randomizados para pasireotida LAR em uma dose inicial de 10 mg uma vez a cada 28 dias e 30 mg uma vez a cada 28 dias, respectivamente, independentemente da titulação crescente no mês 4. 6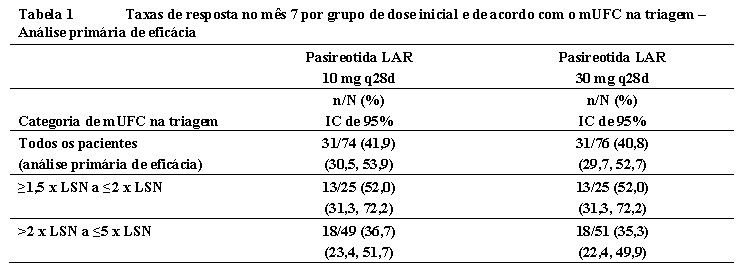
No mês 4, 31/74 (41,9%) e 28/76 (36,8%) pacientes realizaram titulação crescente nos braços de pasireotida LAR 10 mg e 30 mg, respectivamente. Com todos os pacientes que realizaram titulação crescente antes do mês 7 contados como não responsivos, a resposta mUFC no mês 7 foi observada em 28,4% (IC de 95% 18,5 a 40,1) e 31,6% (IC de 95% 21,4 a 43,3) dos pacientes randomizados para pasireotida LAR em uma dose inicial de 10 mg uma vez a cada 28 dias e 30 mg uma vez a cada 28 dias, respectivamente. O estudo atendeu ao principal objetivo secundário de eficácia para ambos os grupos de dose. 5,6
Uma análise secundária de eficácia foi conduzida para a proporção combinada de pacientes que obtiveram mUFC ≤1,0 X LSN (controlado) ou apresentaram uma redução de pelo menos 50% no UFC (parcialmente controlado) na fase principal do estudo. A taxa combinada de responsivos controlados ou parcialmente controlados no mês 7 constituiu 50,0% e 56,6% dos pacientes randomizados para os grupos de dose de 10 mg e 30 mg, respectivamente (Tabela 4). 6
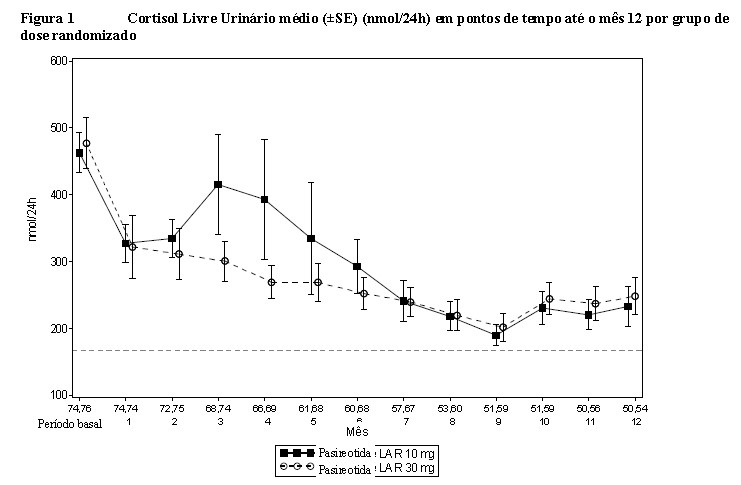
O número de pacientes contribuindo para a média e erro padrão (SE) para cada mês é exibido abaixo ao eixo X (10 mg/30 mg). Esta análise inclui apenas visitas programadas. ---é o LSN para o ensaio de UFC (166,48 nmol/24h)
Diminuições também foram demonstradas pelo percentual total de alteração nos níveis médios e medianos de UFC nos meses 7 e 12 em comparação aos valores basais (Tabela 5). Reduções nos níveis de cortisol sérico e ACTH plasmático também foram observadas nos meses 7 e 12 para cada grupo de dose. 6,7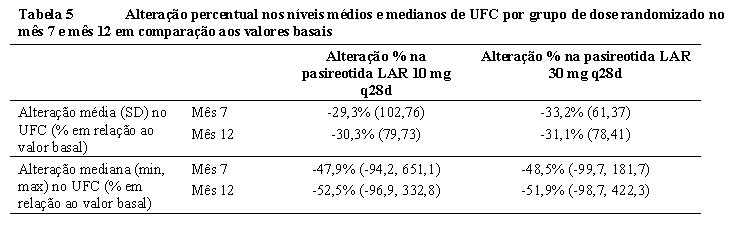
Reduções clinicamente significativas do volume do tumor hipofisário (diminuição ≥20% desde o período basal) foram observadas em ambos os braços de dose. No mês 12, mês 18 e mês 24, a diminuição em % mediana (min, max) em todos os pacientes foi de 17,2% (-92,4, 81,8 [n=73]), 23,3% (-66,7, 62,5 [n=36]) e 25,2% (-77,2, 103,8 [n=26]), respectivamente. 6
Diminuições clinicamente significativas na pressão arterial sistólica e diastólica na posição supina e no peso corporal foram observadas em ambos os grupos de dose no mês 7. Reduções globais nestes parâmetros tenderam a ser maiores em pacientes que eram responsivos mUFC. Tendências semelhantes foram observadas no mês 12. 6
No mês 7, a maioria dos pacientes demonstrou melhora ou sinais estáveis da doença de Cushing em comparação ao período basal. O rubor facial melhorou em 43,5% (47/108) dos pacientes, e mais de um terço dos pacientes demonstrou melhora na gordura supraclavicular (34,3%) e gordura dorsal (34,6%). Resultados semelhantes foram registrados no mês 12. A Tabela 6 apresenta os resultados dos sinais da doença de Cushing por percentual do número de pacientes avaliados no mês 7 para cada sinal. 6 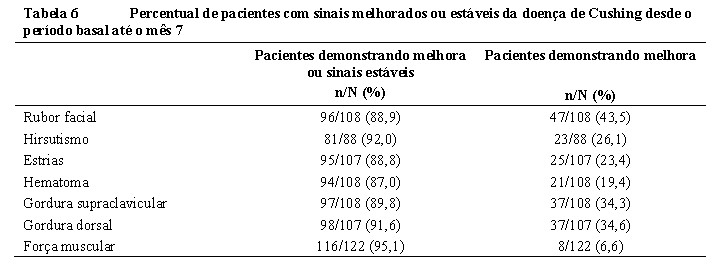
A qualidade de vida foi avaliada usando os questionários SF-12v2 e CushingQoL. Uma associação entre a redução no mUFC e a melhora nas pontuações do Resumo do Componente Mental (MCS) do SF-12v2 e CushingQoL desde o período basal foi observada (p < 0,05, com base em uma análise de medidas repetidas de dados combinados de todas as visitas programadas durante os primeiros 12 meses do estudo), corroborando que as melhoras no mUFC estavam associadas a benefícios na qualidade de vida. 5,6
Referências bibliográficas
1. Study SOM230C2305 CSR - A multicenter, randomized, blinded study to assess safety and efficacy of pasireotida LAR versus. octreotida LAR in patients with active acromegaly. [13]
2. Clinical Overview - Acromegaly. Novartis Pharma AG. Oct 2013. [1]
3. Colao A, Bronstein M, Freda P, et al (2012) Pasireotida LAR is significantly more effective than octreotida LAR at inducing biochemical control in patients with acromegaly: results of a 12-month randomized, double-blind, multicenter, Phase III study; presented at ICE/ECE congress 2012 in Florence/Italy; Endocrine Abstracts 2012, 29 Oct 2012. [21]
4. Study SOM230C2402 CSR - A phase III, multicenter, randomized, parallel-group study to assess the efficacy and safety of double-blind pasireotida LAR 40 mg and pasireotida LAR 60 mg versus open-label octreotida LAR or lanreotida ATG in patients with inadequately controlled acromegaly. [14]
5. Study CSOM230G2304 CSR - A randomized, double-blind, multicenter, phase III study to evaluate the efficacy and safety of pasireotide LAR in patients with Cushing's disease. [38]6. Clinical Overview - Cushing's disease. Novartis Pharma AG. Oct 2016. [36]
7. Summary of Clinical Efficacy - Cushing's disease. Novartis Pharma AG. Oct 2016. [43]
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: ATC: H01CB05, somatostatina e análogos
Mecanismo de ação
A pasireotida é um novo ciclo-hexapeptídeo injetável análogo da somatostatina. Assim como os hormônios peptídicos naturais somatostatina 14 e somatostatina 28 (também conhecidos como Fator de inibição da liberação da somatotropina, SRIF) e outros análogos da somatostatina, a pasireotida exerce a sua atividade farmacológica via ligação a receptores da somatostatina (SSTR). São conhecidos cinco subtipos de receptores da somatostatina humana: SSTR 1, 2, 3, 4 e 5. Esses subtipos de receptores são expressos em diferentes tecidos sob condições fisiológicas normais. Os análogos da somatostatina se ligam a receptores SSTR com potências diferentes (Tabela 7). A pasireotida se liga com alta afinidade a quatro dos cinco SSTRs.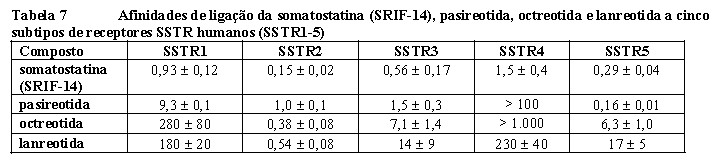
Os resultados são os valores médios + EPM (erro-padrão médio) dos valores da CI50 expressos em nmol/L
(nM).
Farmacodinâmica
Os receptores da somatostatina são expressos em muitos tecidos, sobretudo em tumores neuroendócrinos nos quais hormônios são secretados em excesso, incluindo o hormônio do crescimento na acromegalia e hormônio adrenocorticotrófico (ACTH) na doença de Cushing. Em função de seu amplo perfil de ligação a receptores da somatostatina, a pasireotida tem o potencial de estimular tanto os subtipos receptores do SSTR2 como do SSTR5 relevantes para a inibição da secreção de GH e IGF-1. Na doença de Cushing, estudos in vitro demonstraram que células tumorais corticotróficas de pacientes com doença de Cushing exibem uma elevada expressão de hsst5, enquanto os outros subtipos do receptor não são expressados ou são expressados em níveis menores. A pasireotida se liga aos receptores hsst dos corticotrofos nos adenomas produtores de ACTH, ativando-os, resultando na inibição da secreção de ACTH. A alta afinidade da pasireotida por quatro dos cinco hssts, especialmente ao hsst5 (vide Tabela 7), fornece os fundamentos para que a pasireotida seja um tratamento eficaz para pacientes com doença de Cushing.
Metabolismo da glicose
Em um estudo de mecanismo, randomizado, duplo-cego, conduzido em voluntários saudáveis, o desenvolvimento de hiperglicemia com pasireotida administrada na forma de Upelior® subcutâneo em doses de 600 e 900 microgramas duas vezes ao dia, estava relacionado a diminuições significativas na secreção de insulina, bem como dos hormônios incretinas (ou seja, peptídeo-1 semelhante ao glucagon [GLP-1] e polipeptídeo insulinotrófico glicose dependente [GIP]). A pasireotida não afetou a sensibilidade à insulina. Em outro estudo randomizado conduzido em voluntários saudáveis, foram investigados os efeitos da pasireotida na glicemia por comparação entre a administração de Upelior® subcutâneo 600 microgramas duas vezes ao dia isoladamente com a coadministração de um medicamento antihiperglicêmico (metformina, nateglinida, vildagliptina ou liraglutida, respectivamente. A insulina não foi estudada) durante um período de sete dias. A terapia à base de incretinas (agonistas do GLP-1 e inibidores DDP-IV) foi extremamente eficaz no tratamento da hiperglicemia associada a pasireotida em voluntários saudáveis.
Eletrofisiologia cardíaca
O efeito da pasireotida (administrada como Upelior® subcutâneo) no intervalo QT foi avaliado em dois estudos cruzados completos e controlados do QT. No primeiro estudo, que investigou uma dose de 1.950 microgramas administrada duas vezes ao dia, a alteração média máxima do QTcF em relação ao valor basal, subtraído o placebo (DDQTcF), foi de 17,5 ms (IC de 90%: 15,53; 19,38). No segundo estudo, que investigou doses de 600 microgramas e
1.950 microgramas administradas duas vezes ao dia, as alterações médias máximas do QTcI em relação ao valor basal, subtraído o placebo (DDQTcI), foram de 13,19 ms (IC de 90 %: 11,38; 15,01) e 16,12 ms (IC de 90 %: 14,30; 17,95), respectivamente. Em ambos os estudos, a alteração média em relação ao valor basal, subtraído o placebo, ocorreu em duas horas pós-dose. Ambas as doses de Signifor® LP diminuíram a frequência cardíaca, com uma diferença máxima em relação ao placebo observada em uma hora na dose de 600 microgramas duas vezes ao dia (-10,39 bpm) e em 0,5 hora com 1.950 microgramas duas vezes ao dia (-14,91 bpm). Não se observou nenhum episódio de Torsades de points. Os picos de concentração previstos na dose máxima de Signifor® LP de 60 mg em pacientes com acromegalia com função hepática normal e de 40 mg em pacientes com acromegalia com insuficiência hepática moderada de 25,8 ng/mL e 28,8 ng/mL, respectivamente, são semelhantes ao pico de concentração observado (24,3 mg/mL) do Upelior® subcutâneo 600 microgramas duas vezes ao dia e abaixo do pico de concentração observado (80,6 ng/mL) com a dose de 1.950 microgramas duas vezes ao dia. As concentrações máximas previstas para a dose máxima de Signifor® LP de 40 mg em pacientes com doença de Cushing com função hepática normal e de 20 mg em pacientes com doença de Cushing com comprometimento hepático moderado são de 14 ng/mL e 11,7 ng/mL, respectivamente, sendo que ambas estão abaixo das concentrações máximas observadas de Upelior® 600 microgramas duas vezes ao dia e 1950 microgramas duas vezes ao dia descritas acima. O aumento do intervalo QT com a administração de pasireotida não é mediado por um efeito no canal de potássio do hERG. A restituição cardíaca, a capacidade do coração de recuperar-se de cada batimento precedente, foi medida nos ECGs contínuos de 24 horas para determinar o efeito da pasireotida na vulnerabilidade à arritmia. A pasireotida melhorou significativamente todos os parâmetros de restituição na presença de prolongamento QT indicando que o prolongamento QT mediado pela pasireotida pode não estar associado a um aumento no risco pró-arrítmico. Além disso, a análise morfológica quantitativa das ondas T não mostrou alterações indicativas de diminuição da heterogeneidade espacial da repolarização cardíaca durante o tratamento com pasireotida.
Farmacocinética
A pasireotida para uso intramuscular é formulada como microesferas para liberação prolongada. Após uma única injeção, a concentração plasmática da pasireotida apresenta um pico de liberação inicial uma liberação inicial acelerada no dia da injeção, seguida por uma queda a partir do dia 2 ao 7, e então um aumento lento até a concentração máxima em torno do dia 21, e uma fase de declínio lento ao longo das próximas semanas, concomitante à fase de degradação final da matriz polimérica da forma farmacêutica.
- Absorção
A biodisponibilidade relativa da pasireotida administrada na forma de Signifor® LP em relação a pasireotida administrada na forma de Upelior® subcutâneo é completa. Com base nos dados de biodisponibilidade absoluta de ~100% de pasireotida subcutânea provenientes de estudos pré-clínicos em ratos e macacos, prevê-se que a biodisponibilidade absoluta da pasireotida administrada na forma de Signifor® LP seja completa em humanos. É improvável que haja efeito de alimentos, uma vez que o Signifor® LP é administrada por via parenteral.
- Distribuição
Em voluntários saudáveis, a pasireotida administrada na forma de Signifor® LP é amplamente distribuída, com um grande volume de distribuição aparente (Vz/F > 100 L). A distribuição entre o sangue e o plasma é independente da concentração e mostra que a pasireotida se localiza principalmente no plasma (91%). A ligação às proteínas plasmáticas é moderada (88%) e independente da concentração. A pasireotida tem permeabilidade passiva reduzida e é provavelmente um substrato de Pgp, mas prevê-se que o impacto da Pgp na ADME (absorção, distribuição, metabolismo, excreção) da pasireotida seja reduzida. Em níveis de dose terapêuticos, não se prevê que a pasireotida seja um substrato da BCRP (proteína de resistência ao câncer de mama), do OCT1 (transportador 1 de cátions orgânicos) nem dos OATP (polipeptídeos transportadores de ânions orgânicos) 1B1, 1B3 ou 2B1.
- Biotransformação/metabolismo
A pasireotida demonstrou ser altamente estável metabolicamente em microssomos hepáticos e renais humanos. Em voluntários saudáveis, a pasireotida em sua forma inalterada é a forma predominante encontrada no plasma, na urina e nas fezes.
- Eliminação
A pasireotida é eliminada principalmente por clearance (depuração) hepática (excreção biliar) com uma pequena contribuição da via renal. No estudo de ADME humana com pasireotida administrada na forma de Upelior® subcutâneo em uma dose única de 600 microgramas, 55,9 ± 6,63% da dose de radioatividade foi recuperada durante os primeiros dez dias após a administração, incluindo 48,3 ± 8,16% da radioatividade nas fezes e 7,63 ± 2,03% na urina. O clearance (depuração) aparente (CL/F) da pasireotida administrada na forma de Signifor® LP em voluntários saudáveis é, em média, de 4,5 a 8,5 L/h.
Farmacocinética no estado de equilíbrio
O estado de equilíbrio farmacocinético da pasireotida administrada na forma de Signifor® LP é alcançado após três doses. Após doses intramusculares múltiplas a cada quatro semanas, Signifor® LP demonstra exposições PK aproximadamente proporcionais à dose (concentração mínima no estado de equilíbrio; Cmin, ss) na faixa de dose de 10 mg a 60 mg a cada quatro semanas.
Populações especiais:
- Pacientes geriátrico (65 anos de idade ou mais)
A idade não é uma covariante significativa na análise farmacocinética da população. Os dados referentes aos pacientes com mais de 65 anos de idade são limitados, mas não sugerem nenhuma diferença clinicamente importante de segurança e eficácia em relação aos pacientes mais jovens.
- Pacientes pediátricos
Não foram realizados estudos com pacientes pediátricos.
- Pacientes com insuficiência renal
O clearance (depuração) renal teve uma contribuição pequena na eliminação da pasireotida em humanos. Em um estudo clínico com administração de uma dose única de 900mg de pasireotida, como Signifor® LP, em indivíduos com função renal deficiente, a insuficiência renal leve, moderada ou grave, ou falência renal terminal não tiveram um impacto significativo sobre a farmacocinética de pasireotida. A função renal (clearance (depuração) de creatinina e taxa de filtração glomerular estimada) não é uma covariante na análise farmacocinética da população. Portanto, não se prevê que a função renal venha a afetar de maneira significativa os níveis circulantes de pasireotida.
- Pacientes com insuficiência hepática
Em um estudo clínico com administração de dose única de 600mg de pasireotida administrada na forma de Upelior® subcutâneo em indivíduos com insuficiência da função hepática, os indivíduos com insuficiência hepática moderada e grave (Child-Pugh B e C) apresentaram exposições significativamente mais elevadas do que os indivíduos com função hepática normal. Houve aumento de 60% e 79% na AUCinf, aumento de 67% e 69% na Cmáx e diminuição de 37% e 44% no CL/F, respectivamente, nos grupos com insuficiência hepática moderada e grave em relação ao grupo de controle.
- Dados demográficos
As análises farmacocinéticas da população de pasireotida administrada na forma de Signifor® LP sugerem que raça, sexo e peso corporal não exercem influência clinicamente relevante nos parâmetros farmacocinéticos. Não há necessidade de ajustes de dose em relação aos parâmetros demográficos.
Dados de segurança pré-clínicos
Os estudos de segurança pré-clínicos conduzidos com pasireotida por via subcutânea incluíram farmacologia de segurança, toxicidade de doses repetidas, genotoxicidade e potencial carcinogênico, toxicidade para a reprodução e desenvolvimento. Além disso, foram conduzidos estudos de tolerância e toxicidade de doses repetidas com pasireotida LAR por via intramuscular. A maioria dos achados observados nos estudos de toxicidade de doses repetidas foi reversível e atribuível à farmacologia da pasireotida. Foram observados efeitos em estudos pré-clínicos em exposições consideradas semelhantes ou além da exposição humana máxima. Nos estudos da farmacologia de segurança (com pasireotida por via subcutânea), a pasireotida não exerceu efeitos adversos nas funções respiratória ou cardiovascular. Foram observadas reduções da atividade geral e comportamental em camundongos na dose de 12 mg/kg, equivalente a aproximadamente 32 vezes a dose terapêutica humana máxima recomendada (MHRD) de pasireotida subcutânea, ou 27 vezes a dose diária máxima estimada de pasireotida LAR com base na área de superfície. A pasireotida não foi genotóxica em uma bateria de ensaios in vitro (teste de mutação de Ames em Salmonella e E. coli e teste de mutação em linfócitos periféricos humanos). A pasireotida não foi genotóxica em um teste in vivo com núcleo de medula óssea de ratos com doses de até 50 mg/kg, aproximadamente 250 vezes a dose terapêutica humana máxima recomendada (MHRD) de pasireotida subcutânea, ou 224 vezes a dose diária máxima estimada de pasireotida LAR com base na área de superfície, mg/m2. Estudos da carcinogenicidade conduzidos em ratos e camundongos transgênicos não identificaram nenhum potencial carcinogênico. Em estudos de desenvolvimento embriofetal em ratos e coelhos com pasireotida por via subcutânea, a pasireotida não foi teratogênica em doses maternalmente tóxicas (respectivamente 10 e 5 mg/kg/dia), resultando em exposições (AUC0 a 24 horas) respectivamente 144 e 40 vezes maiores do que a MHRD de pasireotida subcutânea ou 106 e 29,6 vezes maiores do que a MHRD de pasireotida LAR. A uma razão de 10 mg/kg/dia em ratos, a frequência de reabsorções precoces/totais e membros com má rotação foi elevada. A uma razão de 5 mg/kg/dia em coelhos foram observados mais abortos, fetos com pesos menores e variações esqueléticas. Observou-se redução do peso fetal e retardo consequente da ossificação a 1 mg/kg/dia (exposição 4,8 vezes maior do que a MHRD de pasireotida LAR). A pasireotida não exerceu efeito no parto de ratos que receberam até 10 mg/kg/dia (45 vezes mais do que a MHRD de pasireotida LAR com base na área de superfície, mg/m2). Os dados toxicológicos disponíveis em animais mostraram a excreção de pasireotida no leite. Foi observado retardo do crescimento fisiológico, atribuído à inibição do GH, a 2 mg/kg/dia (dez vezes mais do que a MHRD de pasireotida subcutânea ou nove vezes mais do que a dose diária máxima estimada de Signifor® LP, com base na área de superfície, mg/m2) durante um estudo pré e pós-natal em ratos. Após o desmame, os ganhos de peso corporal nos filhotes de ratos expostos à pasireotida foram comparáveis aos dos controles, mostrando reversibilidade. A pasireotida não afetou a fertilidade em ratos machos em doses de até 10 mg/kg/dia (uma dose 52 vezes maior do que a MHRD de pasireotida subcutânea ou 45 vezes maior do que a dose diária máxima estimada de Signifor® LP, com base na área de superfície, mg/m2). Nas ratas, conforme esperado pela farmacologia da pasireotida, houve diminuição da fertilidade em doses diárias de 0,1 mg/kg/dia (0,6 vez a dose terapêutica humana máxima recomendada para pasireotida subcutânea ou 0,5 vez a dose diária máxima estimada de pasireotida LAR, com base na área de superfície, mg/m2), conforme demonstrado pela redução dos números de corpos lúteos e locais de implantação. Foram observados ciclos anormais ou aciclicidade a 1 mg/kg/dia (cinco vezes maior do que a MHRD de pasireotida subcutânea ou 4,5 vezes maior do que a dose diária máxima estimada de pasireotida LAR, com base na área de superfície, mg/m2).
4. CONTRAINDICAÇÕES
Este medicam