SCEMBLIX
NOVARTIS
asciminibe
Tratamento da leucemia mieloide crônica.
Apresentações.
ScemblixTM comprimidos revestidos de 20 mg e 40 mg - embalagens contendo 60 comprimidos.
VIA ORAL
USO ADULTO
Composição.
Cada comprimido revestido de ScemblixTM 20 mg contém 21,62 mg de cloridrato de asciminibe, que é equivalente a 20 mg de asciminibe como base livre. Excipientes: lactose monoidratada, celulose microcristalina, hiprolose, croscarmelose sódica, álcool polivinílico, dióxido de titânio, estearato de magnésio, talco, dióxido de silício, óxido de ferro (amarelo e vermelho), lecitina e goma xantana.
Cada comprimido revestido de ScemblixTM 40 mg contém 43,24 mg de cloridrato de asciminibe, que é equivalente a 40 mg de asciminibe como base livre. Excipientes: lactose monoidratada, celulose microcristalina, hiprolose, croscarmelose sódica, álcool polivinílico, dióxido de titânio, estearato de magnésio, talco, dióxido de silício, óxido de ferro (preto e vermelho), lecitina e goma xantana.
Informações técnicas.
1. INDICAÇÕES
ScemblixTM é indicado para o tratamento de pacientes adultos com leucemia mieloide crônica cromossomo Philadelphia positivo (LMC Ph+) em fase crônica (FC), previamente tratados com dois ou mais inibidores da tirosina quinase (ITQ).
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança clínicas de ScemblixTM no tratamento de pacientes com leucemia mieloide crônica cromossomo Philadelphia positivo (LMC Ph+ FC), previamente tratados com dois ou mais inibidores da tirosina quinase (ITQ) foram demonstradas no estudo multicêntrico, randomizado, com controle ativo e aberto de fase III ASCEMBL.
Neste estudo, um total de 233 pacientes foram randomizados em uma proporção 2:1 e estratificados de acordo com o estado de resposta citogenética maior (RCM) na avaliação basal para receber ScemblixTM 40 mg duas vezes ao dia (N = 157) ou bosutinibe 500 mg uma vez ao dia (N = 76). Os pacientes continuaram o tratamento até toxicidade inaceitável ou insucesso do tratamento.
Os pacientes com LMC Ph+ FC eram 51,5% do sexo feminino e 48,5% do sexo masculino com idade mediana de 52 anos (intervalo: 19 a 83 anos). Dos 233 pacientes, 18,9% tinham 65 anos ou mais, enquanto 2,6% tinham 75 anos ou mais. Os pacientes eram caucasianos (74,7%), asiáticos (14,2%) e negros (4,3%). Dos 233 pacientes, 80,7% e 18% tinham estado de desempenho 0 ou 1 do Eastern Cooperative Oncology Group (ECOG), respectivamente. Os pacientes que haviam recebido anteriormente 2, 3, 4, 5 ou mais linhas prévias de ITQs foram 48,1%, 31,3%, 14,6% e 6%, respectivamente. A duração mediana do tratamento foi de 156 semanas (intervalo: 0,1 a 256,3 semanas) para os pacientes que receberam ScemblixTM e 30,5 semanas (intervalo: 1 a 239,3 semanas) para os pacientes que receberam bosutinibe.
O desfecho primário do estudo foi a taxa de resposta molecular maior (RMM) em 24 semanas e o desfecho secundário principal foi a taxa de RMM em 96 semanas. A RMM é definida como proporção BCR::ABL1 ≤ 0,1% pela Escala Internacional [IS]. Desfechos secundários foram a taxa de resposta citogenética completa (RCC) em 24 e 96 semanas, definida como ausência de metáfases na medula óssea com um mínimo de 20 metáfases examinadas.
Os principais resultados de eficácia do estudo ASCEMBL estão resumidos na Tabela 1.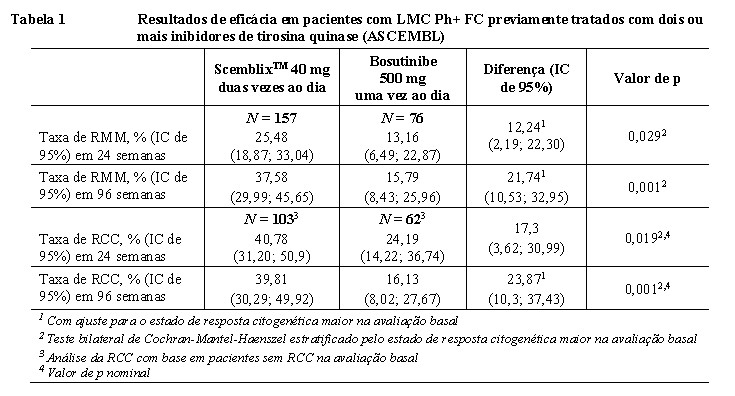
A taxa de RMM prevista em 24 semanas para a dose de 80 mg de ScemblixTM uma vez ao dia é comparável à taxa de RMM em 24 semanas observada no estudo ASCEMBL com a dose de 40 mg de ScemblixTM duas vezes ao dia, com base na análise de resposta à exposição.
No estudo ASCEMBL, 12,7% dos pacientes tratados com ScemblixTM e 13,2% dos pacientes que receberam bosutinibe apresentaram uma ou mais mutações BCR::ABL1 detectadas na avaliação basal. Foi observada uma RMM em 24 semanas em 35,3% e 24,8% dos pacientes que receberam ScemblixTM com ou sem qualquer mutação BCR::ABL1 na avaliação basal, respectivamente. Foi observada uma RMM em 24 semanas em 25% e 11,1% dos pacientes que receberam bosutinibe com ou sem qualquer mutação na avaliação basal, respectivamente. A taxa de RMM em 24 semanas em pacientes nos quais o tratamento randomizado representou a terceira, quarta, quinta ou mais linha de ITQ foi de 29,3%, 25% e 16,1% em pacientes tratados com ScemblixTM e 20%, 13,8% e 0% em pacientes que receberam bosutinibe, respectivamente.
A taxa de RMM em 48 semanas foi de 29,3% (IC de 95%: 22,32, 37,08) em pacientes que receberam ScemblixTM e 13,2% (IC de 95%: 6,49, 22,87) em pacientes que receberam bosutinibe.
A proporção estimada de Kaplan-Meier de pacientes que receberam ScemblixTM e mantiveram RMM por pelo menos 120 semanas foi de 97% (IC de 95%: 88,6, 99,2).
Referências bibliográficas
1. ABL001 - 2.7.3 Summary of Clinical Efficacy in patients with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP) previously treated with two or more tyrosine kinase inhibitors. Novartis. Mar-2021.
2. Phase III study CABL001A2301 (ASCEMBL) - Clinical study report: A phase 3, multi-center, open-label, randomized study of oral ABL001 (asciminib) versus bosutinib in patients with chronic myelogenous leukemia in chronic phase (CMLCP), previously treated with 2 or more tyrosine kinase inhibitors. Novartis. Mar-2021.
3. ABL001 - 2.7.3 Summary of Clinical Efficacy in patients with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP) previously treated with two or more tyrosine kinase inhibitors - Update. Novartis. Jun-2021.
4. ABL001 - 2.5 Clinical Overview in Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP) previously treated with two or more tyrosine kinase inhibitors. Novartis. Jun-2021.
5. ABL001 - 2.7.2 Summary of Clinical Pharmacology Studies of asciminib in Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP) previously treated with two or more tyrosine kinase inhibitors. Novartis. Jun-2021.
6. CABL001A2301 Week 96 CSR - Duration of exposure to study drug (Safety set) - Output table 14.3-1.1. Novartis. Fev-2022.
7. CABL001A2301 Week 96 CSR - MMR rate at scheduled time points (Full analysis set) - Output table 14.2-1.1. Novartis. Fev-2022.
8. CABL001A2301 Week 96 CSR - CCyR rate at scheduled time points (CCyR analysis set) - Output table 14.2-2.3. Novartis. Fev-2022.
9. CABL001A2301 Week 96 CSR - Duration of first MMR among subjects who achieved MMR (MMR responder set) - Output table 14.2-1.4. Novartis. Fev-2022.
10. CABL001A2301 Clinical Study Report, End of Treatment - A Phase III, multi-center, open-label, randomized study of oral ABL001 (asciminib) versus bosutinib in patients with chronic myelogenous leukemia in chronic phase (CML-CP), previously treated with 2 or more tyrosine kinase inhibitors. Novartis. Sep-2023.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Agentes antineoplásicos, inibidores da proteína quinase. Código ATC: L01EA06.
Mecanismo de ação
O asciminibe é um inibidor oral potente de tirosina quinase ABL/BCR::ABL1. O asciminibe inibe a atividade da quinase ABL1 da proteína de fusão BCR::ABL1, ao se direcionar especificamente ao bolso de ligação de miristoil ABL.
Propriedades farmacodinâmicas
In vitro, o asciminibe inibe a atividade da tirosina quinase ABL1 em valores médios de CI50 abaixo de 3 nanomolares. Em células cancerosas derivadas de pacientes, o asciminibe inibe especificamente a proliferação de células que possuem BCR::ABL1 com valores de CI50 entre 1 e 25 nanomolares. Em células que expressam a forma selvagem de BCR::ABL1, o asciminibe inibe o crescimento celular com valores médios de CI50 de 0,61 ± 0,21 nanomolar.
Em modelos de xenoenxerto de LMC em camundongo, o asciminibe inibiu de modo dependente da dose o crescimento de tumores que possuem o tipo selvagem de BCR::ABL1, sendo observada regressão tumoral em doses acima de 7,5 mg/kg duas vezes ao dia.
Eletrofisiologia cardíaca
O tratamento com ScemblixTM está associado a um prolongamento relacionado à exposição do intervalo QT. A correlação entre a concentração de asciminibe e a alteração média máxima estimada a partir do valor basal do intervalo QT com a correção de Fridericia (dQTcF) foi avaliada em 239 pacientes com LMC Ph+ ou leucemia linfoblástica aguda (LLA) que receberam ScemblixTM em intervalos de dose de 10 a 280 mg duas vezes ao dia e 80 a 200 mg uma vez ao dia. A dQTcF média estimada foi de 3,35 ms (limite superior de IC de 90%: 4,43 ms) para a dose de 40 mg duas vezes ao dia de ScemblixTM e 3,64 ms (limite superior de IC de 90%: 4,68 ms) para a dose de 80 mg uma vez ao dia de ScemblixTM.
Propriedades farmacocinéticas
- Absorção
O asciminibe é rapidamente absorvido, com os níveis plasmáticos máximos (Tmáx) medianos atingidos 2 a 3 horas após a administração oral, independentemente da dose. A média geométrica (geoCV%) da Cmáx em estado de equilíbrio é de 1781 ng/ml (23%) e 793 ng/ml (49%) após a administração de ScemblixTM em doses de 80 mg uma vez ao dia e 40 mg duas vezes ao dia, respectivamente. A média geométrica (geoCV%) da ASCtau é de 5262 ng*h/ml (48%) após a administração de ScemblixTM na dose de 40 mg duas vezes ao dia.
Os modelos PBPK (physiologically based pharmacokinetic - farmacocinética baseada em fisiologia) preveem que a absorção do asciminibe é de aproximadamente 100%, enquanto a biodisponibilidade é de aproximadamente 73%.
A biodisponibilidade do asciminibe pode ser reduzida pela coadministração de medicamentos orais contendo hidroxipropil-b-ciclodextrina como excipiente. A coadministração de doses múltiplas de itraconazol solução oral contendo hidroxipropil-b-ciclodextrina em um total de 8 g por dose com uma dose de 40 mg de asciminibe diminuiu a ASCinf do asciminibe em 40,2% em participantes saudáveis.
- Efeito da ingestão alimentar
O consumo de alimentos diminui a biodisponibilidade do asciminibe, com uma refeição hiperlipídica tendo maior impacto na farmacocinética do asciminibe que uma refeição hipolipídica. A ASC do asciminibe diminui em 62,3% com uma refeição hiperlipídica e em 30% com uma refeição hipolipídica em comparação ao jejum, independentemente da dose (consulte as seções 6. Interações medicamentosas e 8. Posologia e Modo de usar).
- Distribuição
O volume de distribuição aparente do asciminibe em estado de equilíbrio é 111 L, com base na análise de farmacocinética populacional. O asciminibe é distribuído principalmente ao plasma, com uma proporção média sangue-plasma de 0,58, independente da dose. O asciminibe se liga 97,3% às proteínas do plasma humano, independentemente da dose.
- Biotransformação/metabolismo
O asciminibe é metabolizado principalmente via oxidação mediada por CYP3A4 (36%), glicuronidação mediada por UGT2B7 e UGT2B17 (13,3% e 7,8%, respectivamente). Os modelos de PBPK predizem que a secreção biliar de asciminibe via BCRP representa 31,1% de sua eliminação sistêmica total. O asciminibe é o principal componente circulante no plasma (92,7% da dose administrada).
- Eliminação
O asciminibe é eliminado principalmente por meio de excreção fecal, com uma contribuição menor da via renal. Oitenta e 11% da dose de asciminibe foram recuperados nas fezes e na urina de participantes saudáveis, respectivamente, após a administração oral de uma dose única de 80 mg de asciminibe marcado com [14C]. A eliminação fecal de asciminibe não metabolizado representa 56,7% da dose administrada.
A depuração oral total (CL/F) de asciminibe é 6,31 L/hora, com base na análise de farmacocinética populacional. A meia-vida cumulativa (T1/2) de asciminibe é de 5,2 horas com dose diária total de 80 mg.
- Linearidade/não linearidade
O asciminibe apresenta um leve aumento acima do proporcional à dose na exposição em estado de equilíbrio (ASC e Cmáx) no intervalo de dose de 10 a 200 mg administrado uma ou duas vezes ao dia.
A proporção média de acúmulo da média geométrica é de aproximadamente 2 vezes, independente da dose. As condições de estado de equilíbrio são alcançadas dentro de 3 dias na dose de 40 mg duas vezes ao dia.
Avaliação in vitro do potencial de interações medicamentosas
- Enzimas CYP450 e UGT
In vitro, o asciminibe inibe de forma reversível CYP3A4/5, CYP2C9 e UGT1A1 em concentrações plasmáticas alcançadas em uma dose diária total de 80 mg.
- Transportadores
O asciminibe é um substrato de BCRP e P-gp. O asciminibe inibe BCRP e P-gp, com valores de Ki de 24,3 e 21,7 micromolares, respectivamente. Com base em modelos de PBPK, o asciminibe aumenta a exposição aos substratos P-gp, OATP1B e BCRP (vide seção 6. Interações Medicamentosas).
- Múltiplas vias
O asciminibe é metabolizado por múltiplas vias, incluindo as enzimas CYP3A4, UGT2B7 e UGT2B17 e secretadas por vias biliares pelo transportador BCRP.
Medicamentos que inibem ou induzem várias vias podem alterar a exposição a ScemblixTM.
O asciminibe (inibe várias vias, incluindo CYP3A4, CYP2C9, OATP1B, P-gp e BCRP. Scemblix pode aumentar a exposição de medicamentos, que são substratos dessas vias (vide seção 6. Interações Medicamentosas).
Populações especiais
- Pacientes geriátricos (65 anos de idade ou mais)
No estudo ASCEMBL, 44 dos 233 (18,9%) pacientes tinham 65 anos de idade ou mais, enquanto 6 (2,6%) tinham 75 anos ou mais.
Não foram observadas diferenças gerais na segurança ou eficácia de ScemblixTM entre pacientes com 65 anos de idade ou acima e pacientes mais jovens. Há um número insuficiente de pacientes com 75 anos de idade ou mais para avaliar se há diferenças na segurança ou eficácia.
- Gênero/raça/peso corporal
A exposição sistêmica ao asciminibe não é afetada pelo gênero, raça ou peso corporal em nenhuma extensão clinicamente relevante.
- Insuficiência renal
Foi realizado um estudo dedicado de insuficiência renal, incluindo 6 participantes com função renal normal (taxa de filtração glomerular absoluta [TFGa] ≥ 90 ml/min) e 8 participantes com insuficiência renal grave sem necessidade de diálise (TFGa de 15 a < 30 ml/min). A ASCinf e a Cmáx de asciminibe aumentam em 56% e 8%, respectivamente, em participantes com insuficiência renal grave em comparação a participantes com função renal normal, após a administração oral de uma dose única de 40 mg de ScemblixTM (consulte a seção 8. Posologia e Modo de usar).
Modelos farmacocinéticos populacionais indicam um aumento na ASC0-24h mediana em estado de equilíbrio de asciminibe em 11,5% em pacientes com insuficiência renal leve a moderada, em comparação a participantes com função renal normal.
- Insuficiência hepática
Foi realizado um estudo dedicado de insuficiência hepática, incluindo 8 participantes cada com função hepática normal, insuficiência hepática leve (pontuação de Child-Pugh A 5 a 6), insuficiência hepática moderada (pontuação Child-Pugh B 7 a 9) ou insuficiência hepática grave (pontuação Child-Pugh C 10 a 15). A ASCinf e a Cmáx de asciminibe aumentam em 22%, 3% e 66% em participantes com insuficiência hepática leve, moderada e grave em comparação a participantes com função hepática normal, após a administração oral de uma dose única de 40 mg de ScemblixTM (consulte a seção 8. Posologia e Modo de usar).
Dados de segurança pré-clínico
O asciminibe foi avaliado em estudos de farmacologia de segurança, toxicidade de doses repetidas, genotoxicidade, toxicidade reprodutiva e fototoxicidade.
- Farmacologia de segurança
Em estudos de farmacologia de segurança, o asciminibe não causou nenhum efeito no sistema nervoso central e no sistema respiratório em ratos em doses de até 600 mg/kg/dia.
Em um estudo in vitro, o asciminibe inibiu os canais do gene humano relacionado ao éter-a-go-go (hERG), com uma CI50 de 11,4 micromolares. Esse valor se traduz em uma margem de segurança clínica de pelo menos 200 vezes ou 100 vezes maior quando comparada à Cmáx livre de asciminibe em pacientes nas doses de 40 mg duas vezes ao dia ou 80 mg uma vez por dia, respectivamente.
Efeitos cardiovasculares moderados (aumento da frequência cardíaca, diminuição da pressão sistólica, diminuição da pressão arterial média e diminuição da pressão do pulso arterial) foram observados em estudos de segurança cardíaca in vivo em cães. Nenhum prolongamento QTc foi evidente em cães até a maior exposição livre de asciminibe de 6,3 micromolares.
- Toxicidade de doses repetidas
Estudos de toxicidade de doses repetidas identificaram o pâncreas, o fígado, sistema hematopoiético, glândula adrenal e trato gastrointestinal como órgãos-alvo do asciminibe.
Os efeitos pancreáticos (aumentos de amilase e lipase séricas, lesões de células acinares) ocorreram em cães com exposições de ASC abaixo daquelas alcançadas em pacientes recebendo 40 mg duas vezes ao dia ou 80 mg uma vez ao dia. Foi observada uma tendência à recuperação.
Foram observadas elevações nas enzimas hepáticas e/ou bilirrubina em ratos, cães e macacos. Foram observadas alterações hepáticas histopatológicas (hipertrofia de hepatócitos centrolobulares, leve hiperplasia do ducto biliar, aumento de necrose de hepatócitos individuais e hipertrofia hepatocelular difusa) em ratos e macacos. Essas alterações ocorreram em exposições de ASC equivalentes a (ratos) ou 8 a 18 vezes (cães e macacos) maiores do que as alcançadas em pacientes recebendo 40 mg duas vezes ao dia ou 80 mg uma vez ao dia. Essas alterações foram completamente reversíveis.
Os efeitos no sistema hematopoiético (redução na massa eritrocitária, aumento do pigmento esplênico ou da medula óssea e aumento de reticulócitos) foram condizentes com uma anemia leve, regenerativa, extravascular e hemolítica em todas as espécies. Essas alterações ocorreram em exposições de ASC equivalentes a (ratos) ou 10 a 14 vezes (cães e macacos) maiores do que as alcançadas em pacientes recebendo 40 mg duas vezes ao dia ou 80 mg uma vez ao dia. Essas alterações foram completamente reversíveis.
Houve hipertrofia/hiperplasia mínima da mucosa (aumento da espessura da mucosa com prolongamento frequente dos vilos) no duodeno de ratos, com exposições de ASC 30 ou 22 vezes mais elevadas que as alcançadas em pacientes recebendo 40 mg duas vezes ao dia ou 80 mg uma vez ao dia, respectivamente. Essa alteração foi completamente reversível.
Houve hipertrofia mínima ou discreta da glândula adrenal e redução leve a moderada da vacuolização na zona fasciculada em exposições de ASC equivalentes a (macacos) ou 19 a 13 vezes (ratos) maiores que as alcançadas em pacientes recebendo 40 mg duas vezes ao dia ou 80 mg uma vez ao dia, respectivamente. Essas alterações foram completamente reversíveis.
- Carcinogenicidade e mutagenicidade
O asciminibe não apresentou potencial mutagênico, clastogênico ou aneugênico in vitro ou in vivo.
Em um estudo de carcinogenicidade em ratos com a duração de 2 anos, foram observadas alterações proliferativas não neoplásicas que consistem em hiperplasia das células de Sertoli ovarianas em fêmeas em doses iguais ou superiores a 30 mg/kg/dia. Tumores benignos de células de Sertoli nos ovários foram observados em ratas com a dose mais alta de 66 mg/kg/dia. As exposições da ASC ao asciminib em ratos fêmeas com 66 mg/kg/dia foram geralmente 8 vezes ou 5 vezes superiores às alcançadas em doentes com a dose de 40 mg duas vezes por dia ou 80 mg uma vez por dia, respetivamente. Não foram observados achados neoplásicos ou hiperplásicos relacionados ao asciminibe em ratos machos em qualquer nível de dose.
A relevância clínica desses achados é atualmente desconhecida.
- Toxicidade reprodutiva
Para informações sobre toxicidade reprodutiva, consulte o item "Gravidez, lactação, mulheres e homens com potencial
Reprodutivo" na seção 5. Advertências e Precauções.
- Fototoxicidade
Em camundongos, o asciminibe apresentou efeitos fototóxicos dependentes da dose a partir de 200 mg/kg/dia. No NOAEL (No Observed Adverse Effect Level - Nível Sem Efeitos Adversos Observáveis) de 60 mg/kg/dia, a exposição com base na Cmáx no plasma foi 15 vezes ou 6 vezes maior que a exposição em pacientes tratados com 40 mg duas vezes ao dia ou 80 mg uma vez ao dia, respectivamente.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a algum dos excipientes do medicamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Mielossupressão
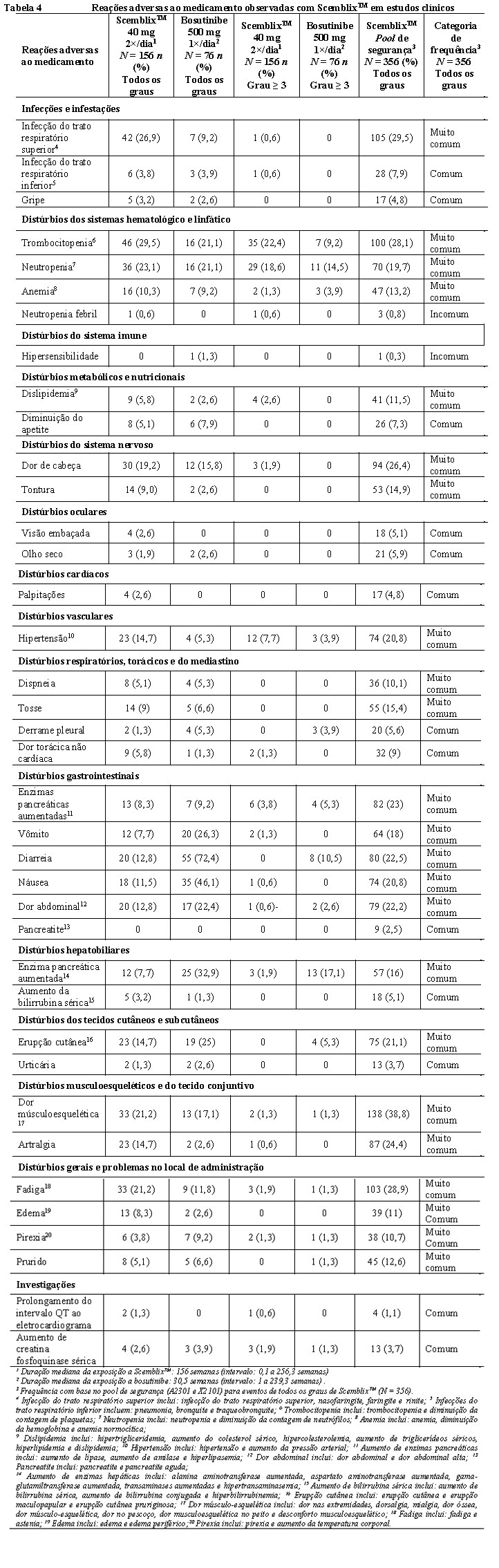
Houve trombocitopenia, neutropenia e anemia em pacientes que receberam ScemblixTM. Trombocitopenia (grau 3 ou 4 CTCAE NCI) e neutropenia grave foram relatados durante o tratamento com ScemblixTM (consulte a seção 9. Reações adversas). A mielossupressão foi geralmente reversível e tratada com suspensão temporária de ScemblixTM. Hemogramas completos devem ser realizados a cada duas semanas nos primeiros 3 meses de tratamento e, a partir de então, mensalmente ou conforme clinicamente indicado. Os pacientes devem ser monitorados quanto a sinais e sintomas de mielossupressão.
Com base na gravidade da trombocitopenia e/ou neutropenia, a dose de ScemblixTM deve ser reduzida, temporariamente interrompida ou permanentemente descontinuada, conforme descrito na Tabela 3 (consulte a seção 8. Posologia e Modo de usar).
Toxicidade pancreática
Houve pancreatite em 9 dos 356 (2,5%) dos pacientes que receberam ScemblixTM, com reações de grau 3 ocorrendo em 4 (1,1 %) pacientes. Todas essas reações ocorreram no estudo de fase I (X2101). Dos 9 pacientes com pancreatite, 2 (0,6%) descontinuaram permanentemente ScemblixTM, enquanto ScemblixTM foi temporariamente interrompido em 5 (1,4%) pacientes devido à reação adversa ao medicamento. Houve elevação assintomática da lipase e amilase séricas em 82 de 356 (23%) pacientes que receberam ScemblixTM, com eventos graus 3 e 4 ocorrendo em 37 (10,4%) e 9 (2,5%) dos pacientes, respectivamente. Dos 82 pacientes com elevação de enzimas pancreáticas, ScemblixTM foi permanentemente descontinuado em 8 (2,2%) pacientes devido a reação adversa ao medicamento.
Os níveis séricos de lipase e amilase devem ser avaliados mensalmente durante o tratamento com ScemblixTM ou conforme clinicamente indicado. Os pacientes devem ser monitorados quanto a sinais e sintomas de toxicidade pancreática. Deve ser realizado um monitoramento mais frequente em pacientes com histórico de pancreatite. Se o aumento da lipase e amilase séricas for acompanhado por sintomas abdominais, o tratamento deve ser temporariamente interrompido e os testes diagnósticos apropriados devem ser considerados para excluir pancreatite (consulte a seção 8. Posologia e Modo de usar.
Com base na gravidade da elevação da lipase e da amilase séricas, a dose de ScemblixTM deve ser reduzida, temporariamente interrompida ou permanentemente descontinuada, conforme descrito na Tabela 3 (consulte a seção 8. Posologia e Modo de usar).
Prolongamento do QT
O prolongamento do QT ao eletrocardiograma ocorreu em 4 de 356 (1,1%) pacientes que receberam ScemblixTM (consulte a seção 9. Reações adversas). No estudo clínico ASCEMBL, um paciente apresentou um QTcF (intervalo QT por Fredericia) prolongado superior a 500 ms, bem como um aumento superior a 60 ms de QTcF em relação ao valor basal e um paciente apresentou um QTcF prolongado com um aumento superior a 60ms de QTcF em relação ao valor basal.
Recomenda-se que um eletrocardiograma seja realizado antes do início do tratamento com ScemblixTM e monitorado durante o tratamento, conforme clinicamente indicado. Hipocalemia e a hipomagnesemia devem ser corrigidas antes da administração de ScemblixTM e monitoradas durante o tratamento, conforme clinicamente indicado.
Deve-se ter cautela ao administrar ScemblixTM na dose diária de 80 mg, concomitantemente com medicamentos com risco conhecido de Torsades de pointes (consulte a seção 6. Interações medicamentosas, e a seção 3,Características farmacológicas).
Hipertensão
Houve hipertensão em 74 dos 356 (20,8%) pacientes que receberam ScemblixTM, com reações de graus 3 e 4 relatados em 39 (11%) e 1 (0,3%) pacientes, respectivamente. Entre os pacientes com hipertensão grau ≥ 3, o tempo mediano até a primeira ocorrência de eventos foi de 29,21 semanas (intervalo: 0,14 a 365 semanas). Dos 74 pacientes com hipertensão, ScemblixTM foi temporariamente interrompido em 3 (0,8%) pacientes devido à reação adversa ao medicamento.
Hipertensão deve ser monitorada e tratada usando terapia anti-hipertensiva padrão durante o tratamento com ScemblixTM conforme clinicamente indicado.
Hipersensibilidade
Os eventos de hipersensibilidade ocorreram em 119 dos 356 (33,4%) pacientes recebendo ScemblixTM, com eventos grau ≥ 3 relatados em 6 (1,7 %) pacientes. Os pacientes devem ser monitorados quanto a sinais e sintomas de hipersensibilidade e o tratamento adequado deve ser iniciado conforme clinicamente indicado.
Reativação da hepatite B
A reativação do vírus da hepatite B (HBV) ocorreu em pacientes portadores crônicos deste vírus após a administração de outros inibidores da tirosina quinase BCR::-ABL1 (ITQs). Os pacientes devem ser testados quanto a infecção por HBV antes do início do tratamento com ScemblixTM. Portadores de HBV que precisam de tratamento com ScemblixTM devem ser atentamente monitorados quanto a sinais e sintomas de infecção ativa por HBV durante toda a terapia e por vários meses após o término da terapia.
Toxicidade embriofetal
Com base nos achados de estudos em animais, ScemblixTM pode causar danos fetais quando administrado a gestantes. Gestantes e mulheres com potencial reprodutivo devem ser aconselhadas sobre o possível risco para o feto se ScemblixTM for usado durante a gravidez ou se a paciente engravidar enquanto estiver tomando ScemblixTM. O estado de gravidez de mulheres com potencial reprodutivo deve ser verificado antes de se iniciar o tratamento com ScemblixTM. Mulheres sexualmente ativas com potencial reprodutivo devem usar contracepção eficaz durante o tratamento com ScemblixTM e por pelo menos 3 dias após a última dose (consulte o item "Gravidez, lactação, mulheres e homens com potencial reprodutivo" na seção 5. Advertências e Precauções).
Gravidez, lactação, mulheres e homens com potencial reprodutivo
- Gravidez
Resumo dos riscos
Com base nos achados de estudos em animais, ScemblixTM pode causar dano fetal quando administrado a gestantes. Não há estudos adequados e bem controlados em gestantes para informar um risco associado ao produto.
Estudos de reprodução animal em ratas e coelhas prenhas demonstraram que a administração oral de asciminibe durante a organogênese induziu embriotoxicidade, fetotoxicidade e teratogenicidade.
Gestantes e mulheres com potencial reprodutivo devem ser aconselhadas sobre o risco potencial para o feto se ScemblixTM for usado durante a gravidez ou se a paciente engravidar enquanto estiver tomando ScemblixTM.
- Dados em animais
Em estudos de desenvolvimento embriofetal, os animais prenhes receberam doses orais de asciminibe de 25, 150 e 600 mg/kg/dia em ratos e 15, 50 e 300 mg/kg/dia em coelhos durante o período de organogênese.
Em ratos, o asciminibe não foi tolerado em animais maternos na dose de 600 mg/kg/dia e resultou na eutanásia precoce do grupo de dose. Não houve evidência de morte embriofetal relacionada ao asciminibe em doses inferiores ou iguais a 150 mg/kg/dia. Foi observado um aumento relacionado à dose nos pesos fetais com 25 e 150 mg/kg/dia. Variações fetais no trato urinário e esqueleto (crânio, coluna vertebral e costelas), indicativas de alterações na taxa de desenvolvimento, foram observadas principalmente na dose de 150 mg/kg/dia. Também foi observado um discreto aumento na taxa de malformação (anasarca e malformações cardíacas) e algumas variantes viscerais indicativas de efeitos adversos no desenvolvimento embriofetal com 150 mg/kg/dia. O nível materno sem efeitos adversos observados (NOAEL) foi de 150 mg/kg/dia e o NOAEL fetal foi de 25 mg/kg/dia. No NOAEL fetal de 25 mg/kg/dia, as exposições de ASC foram equivalentes ou inferiores às atingidas em pacientes nas doses de 40 mg duas vezes ao dia ou de 80 mg uma vez ao dia, respectivamente.
Em coelhos, 300 mg/kg/dia causaram morbidade nos animais maternos e resultaram na eutanásia precoce do grupo de dose. Observou-se aumento da incidência de reabsorções, um indicativo de mortalidade embriofetal, e baixa incidência de malformações cardíacas, um indicativo de teratogenicidade, com doses de 50 mg/kg/dia. Não houve efeito no crescimento fetal. O NOAEL para toxicidade materna foi de 50 mg/kg/dia e o NOAEL fetal foi de 15 mg/kg/dia. No NOAEL fetal de 15 mg/kg/dia, as exposições de ASC foram equivalentes ou inferiores às atingidas em pacientes nas doses de 40 mg duas vezes ao dia ou de 80 mg uma vez ao dia, respectivamente.
ScemblixTM pertence à categoria B de risco na gravidez, portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
- Lactação
Resumo dos riscos
Não se sabe se o asciminibe é transferido para o leite humano após a administração de ScemblixTM. Não há dados sobre os efeitos de asciminibe em lactentes ou na produção de leite.
Devido ao potencial de reações adversas ao medicamento graves em lactentes, a amamentação não é recomendada durante o tratamento com ScemblixTM e por pelo menos 3 dias após a última dose.
- Homens e mulheres com potencial reprodutivo
Testes de gravidez
O estado de gravidez de mulheres com potencial reprodutivo deve ser verificado antes de se iniciar o tratamento com ScemblixTM.
Contracepção
Mulheres sexualmente ativas e com potencial reprodutivo devem usar contracepção eficaz (métodos que resultem em taxas de gravidez inferiores a 1%) durante o tratamento com ScemblixTM e por pelo menos 3 dias após a última dose.
Infertilidade
Não há dados sobre o efeito de ScemblixTM na fertilidade humana.
No estudo de fertilidade em ratos, o asciminibe não afetou a função reprodutiva em ratos machos e fêmeas. Observou-se um efeito discreto na motilidade e contagem de espermatozoides em machos em doses de 200 mg/kg/dia, provavelmente com exposições de ASC 19 vezes ou 13 vezes mais elevadas do que as alcançadas em pacientes que recebem 40 mg duas vezes ao dia ou 80 mg uma vez ao dia, respectivamente.
6. INTERAÇÕES MEDICAMENTOSAS
Agentes que podem reduzir as concentrações plasmáticas de asciminibe
- Indutores potentes de CYP3A4
A coadministração de um indutor potente da CYP3A4 (rifampicina) diminuiu a ASCinf de asciminibe em 14,9%, enquanto aumenta a Cmáx de asciminibe em 9% em participantes saudáveis que receberam uma dose única de 40 mg de ScemblixTM.
Os modelos de PBPK preveem que a coadministração de asciminibe na dose de 80 mg uma vez ao dia com rifampicina diminuiria a ASCtau e a Cmáx do asciminibe em 52% e 23%, respectivamente.
Deve-se ter cautela durante a administração concomitante de ScemblixTM com indutores potentes de CYP3A4, incluindo, entre outros, carbamazepina, fenobarbital, fenitoína ou erva-de-são-joão (Hypericum perforatum). Não é necessário ajustar a dose de ScemblixTM.
Agentes cujas concentrações plasmáticas podem ser alteradas pelo asciminibe
- Substratos de CYP3A4 com índice terapêutico estreito
A coadministração de asciminibe com substrato de CYP3A4 (midazolam) aumentou a ASCinf e a Cmáx de midazolam em 28% e 11%, respectivamente, em participantes saudáveis que receberam ScemblixTM na dose de 40 mg duas vezes ao dia.
Os modelos de PBPK preveem que a coadministração de asciminibe na dose de 80 mg uma vez ao dia aumentaria a ASCinf e a Cmáx de midazolam em 24% e 17%, respectivamente.
Deve-se ter cautela durante a administração concomitante de ScemblixTM com substratos da CYP3A4 conhecidos por terem um índice terapêutico estreito, incluindo, entre outros, substratos da CYP3A4 fentanila, alfentanila, diidroergotamina ou ergotamina (consulte a seção 3 Características farmacológicas). Não é necessário ajustar a dose de ScemblixTM.
- Substratos de CYP2C9
A coadministração de asciminibe com um substrato da CYP2C9 (varfarina) aumentou a ASCinf e a Cmáx de S-varfarina em 41% e 8%, respectivamente, em participantes saudáveis que receberam ScemblixTM na dose de 40 mg duas vezes ao dia.
Os modelos de PBPK preveem que a coadministração de asciminibe na dose de 80 mg uma vez ao dia aumentaria a ASCinf e a Cmáx de S-varfarina em 52% e 4%, respectivamente.
Deve-se ter cautela durante a administração concomitante de ScemblixTM com substratos da CYP2C9 conhecidos por terem um índice terapêutico estreito, incluindo, entre outros, fenitoína ou varfarina (consulte a seção 3 Características farmacológicas). Não é necessário ajustar a dose de ScemblixTM.
Substratos de OATP1B, de BCRP ou de ambos os transportadores
Modelos PBPK predizem que a coadministração de asciminibe 40 mg duas vezes ao dia e 80 mg uma vez ao dia com um substrato de OATP1B (pravastatina) aumentaria a Cmáx da pravastatina em 43% e 63% e a ASCinf em 37% e 51%, respectivamente.
Modelos PBPK predizem que a coadministração de asciminibe 40 mg duas vezes ao dia e 80 mg uma vez ao dia com um substrato de OATP1B, CYP3A4 e Pg-p (atorvastatina) aumentaria a Cmax da atorvastatina em 97% e 143% e a ASCinf em 81% e 122%, respectivamente.
Modelos PBPK predizem que a coadministração de asciminibe 40 mg duas vezes ao dia e 80 mg uma vez ao dia com um substrato BCRP (sulfassalazina) aumentaria a Cmax da sulfasalazina em 334% e 342% e a ASCinf em 333% e 340%, respectivamente.
Modelos PBPK predizem que a coadministração de asciminibe 40 mg duas vezes ao dia e 80 mg uma vez ao dia com um substrato BCRP e OATP1B (rosuvastatina) aumentaria a Cmax da rosuvastatina em 453% e 530% e a ASCinf em 190% e 202%, respectivamente.
Deve-se ter cautela durante a administração concomitante de Scemblix em todas as doses recomendadas com substratos de OATP1B, BCRP ou ambos os transportadores, incluindo, entre outros, sulfassalazina, metotrexato, pravastatina, atorvastatina, pitavastatina, rosuvastatina e sinvastatina. Consulte as reduções de dose dos substratos de OATP1B e BCRP, conforme recomendado em suas informações de prescrição.
A administração concomitante de Scemblix com rosuvastatina deve ser evitada e estatinas alternativas devem ser consideradas. Se a coadministração não puder ser evitada, a dose de rosuvastatina deve ser reduzida, conforme recomendado em suas informações de prescrição.
Substratos P-gp de índice terapêutico estreito
Os modelos PBPK preveem que a coadministração de 40 mg duas vezes ao dia de asciminibe e 80 mg uma vez ao dia com um substrato P-gp (digoxina) aumentaria a Cmáx da digoxina em 30% e 38% e a ASCinf em 20% e 22%, respectivamente.
Deve-se ter cautela durante a administração concomitante de ScemblixTM em todas as doses recomendadas com substratos da gp-P conhecidos por terem um índice terapêutico estreito, incluindo, mas não limitado a digoxina, dabigatrana e colchicina.
Agentes que prolongam o intervalo QT
Deve-se ter cautela durante a administração concomitante de ScemblixTM e os medicamentos com risco conhecido de Torsades de pointes, incluindo, entre outros, bepridil, cloroquina, claritromicina, halofantrina, haloperidol, metadona, moxifloxacino ou pimozida (consulte a seção 3. Características farmacológicas).
Interações medicamento-alimento
A biodisponibilidade de asciminibe diminui com o consumo de alimentos (consulte as seções 3. Características farmacológicas e 8. Posologia e Modo de usar).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de conservação
Conservar este medicamento em temperatura ambiente (entre 15 e 30°C). Armazenar na embalagem original para proteger da umidade.
O prazo de validade é de 24 meses a partir da data de fabricação.
Contém um dessecante. Não comer. Não remover o dessecante do frasco.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspectos físicos/características organolépticas
Comprimidos revestidos de 20 mg: comprimidos revestidos amarelo-claro, redondos, biconvexos, com bordas biseladas, com aproximadamente 6,2 mm de diâmetro, sem sulco, gravados em relevo com o logotipo "Novartis" de um lado e "20" do outro lado.
Comprimidos revestidos de 40 mg: comprimidos revestidos violeta-branco, redondos, biconvexos, com bordas biseladas, com aproximadamente 8,2 mm de diâmetro, sem sulco, gravados em relevo com o logotipo "Novartis" de um lado e "40" do outro lado.
Antes de usar, observe o aspecto do medicamento.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
8. POSOLOGIA E MODO DE USAR
O tratamento com ScemblixTM deve ser iniciado por um médico com experiência no uso de terapias oncológicas.
Regime de dosagem
População-alvo geral
A dose diária total recomendada de ScemblixTM é de 80 mg. ScemblixTM pode ser administrado por via oral como 80 mg uma vez ao dia, aproximadamente no mesmo horário todos os dias, ou como 40 mg duas vezes ao dia em intervalos de aproximadamente 12 horas.
Os pacientes que passam de 40 mg duas vezes ao dia para 80 mg uma vez ao dia devem começar a tomar ScemblixTM uma vez ao dia, aproximadamente 12 horas após a última dose duas vezes ao dia, e, em seguida, continuar com 80 mg uma vez ao dia.
Os pacientes que passam de 80 mg uma vez ao dia para 40 mg duas vezes ao dia devem começar a tomar ScemblixTM, aproximadamente 24 horas após a última dose de 80 mg (uma vez ao dia), e, em seguida, continuar com 40 mg duas vezes ao dia em intervalos de aproximadamente 12 horas.
Qualquer alteração no regime de dosagem fica a critério do responsável pela prescrição, conforme necessário para o tratamento do paciente.
O tratamento com ScemblixTM deverá continuar contanto que seja observado benefício clínico ou até a ocorrência de toxicidade inaceitável.
Dose perdida
Regime de dosagem uma vez ao dia: Se uma dose de ScemblixTM não for tomada por mais de 12 horas, deverá ser pulada e a próxima dose deve ser administrada de acordo com o cronograma.
Regimes de dosagem duas vezes ao dia: Se uma dose de ScemblixTM não for tomada por mais de 6 horas, deverá ser pulada e a próxima dose deve ser administrada de acordo com o cronograma.
Modificações de dose
Para o gerenciamento de reações adversas ao medicamento, a dose de ScemblixTM pode ser reduzida com base na segurança e tolerabilidade individuais, conforme descrito na Tabela 2. Se as reações adversas ao medicamento forem tratadas de maneira eficaz, ScemblixTM pode ser reiniciado conforme descrito na Tabela 2.
ScemblixTM deve ser permanentemente descontinuado em pacientes incapazes de tolerar uma dose diária total de 40 mg.
Populações especiais
- Insuficiência renal
Não é necessário ajustar a dose de pacientes com insuficiência renal leve, moderada ou grave que recebem ScemblixTM (consulte a seção 3. Características farmacológicas).
- Insuficiência hepática
Não é necessário a