SARCLISA
SANOFI MEDLEY
isatuximabe
Anticorpo monoclonal.
Apresentações.
Solução para diluição para infusão 100 mg/5 mL: embalagem contendo 1 frasco-ampola com 5 mL Solução para diluição para infusão 500 mg/25 mL: embalagem contendo 1 frasco-ampola com 25 mL
USO INTRAVENOSO (IV)
USO ADULTO
Composição.
Cada mL da solução para diluição para infusão contém 20 mg de isatuximabe. Excipientes: sacarose, histidina, cloridrato de histidina monoidratado, polissorbato 80 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
SARCLISA é indicado:
-Em combinação com pomalidomida e dexametasona, para o tratamento de pacientes adultos com mieloma múltiplo recidivado e refratário que receberam pelo menos duas terapias anteriores, incluindo lenalidomida e um inibidor de proteassoma, e demonstraram progressão da doença na última terapia.
-Em combinação com carfilzomibe e dexametasona para o tratamento de pacientes adultos com mieloma múltiplo recidivado e/ou refratário que receberam pelo menos uma terapia anterior.
2. RESULTADOS DE EFICÁCIA
ICARIA-MM (EFFC14335)
A eficácia e segurança de SARCLISA em combinação com pomalidomida e dexametasona foram avaliadas no ICARIA-MM (EFC14335), um estudo multicêntrico, multinacional, randomizado, aberto, de dois braços, fase III, em pacientes com recidiva e mieloma múltiplo refratário. Os pacientes haviam recebido pelo menos duas terapias anteriores, incluindo lenalidomida e um inibidor de proteassoma, com progressão da doença em ou dentro de 60 dias após o final da terapia anterior. Pacientes com doença refratária primária foram excluídos. Um total de 307 pacientes foram randomizados na proporção de 1:1 para receber SARCLISA em combinação com pomalidomida e dexametasona (Isa-Pd, 154 pacientes) ou pomalidomida e dexametasona (Pd, 153 pacientes). O tratamento foi administrado em ambos os grupos em ciclos de 28 dias até a progressão da doença ou toxicidade inaceitável. SARCLISA 10 mg/kg foi administrado como uma infusão IV semanal no primeiro ciclo e depois a cada duas semanas. Pomalidomida 4 mg foi tomada por via oral uma vez ao dia, do dia 1 ao dia 21 de cada ciclo de 28 dias. Dexametasona (via oral/IV) 40 mg (20 mg para pacientes com idade ≥ 75 anos) foi administrada nos dias 1, 8, 15 e 22 para cada ciclo de 28 dias. No geral, as características demográficas e da doença no início do estudo foram semelhantes entre os dois grupos de tratamento. A idade mediana dos pacientes foi de 67 anos (variação de 36 a 86), 19,9% dos pacientes tinham idade ≥75 anos, 10,4% dos pacientes entraram no estudo com histórico de DPOC ou asma. A proporção de pacientes com insuficiência renal (depuração da creatinina < 60 mL/min / 1,73 m²) foi de 38,7% no grupo Isa-Pd "versus" 33,8% no grupo Pd. O estágio do International Staging System (ISS) no diagnóstico inicial foi de I em 25,1%, II em 31,6% e III em 28,0% dos pacientes. No geral, 19,5% dos pacientes apresentaram anormalidades cromossômicas de alto risco na entrada do estudo; del (17p), t (4; 14) e t (14; 16) estavam presentes em 12,1%, 8,5% e 1,6% dos pacientes, respectivamente. O número mediano de linhas de terapia anteriores foi de 3 (intervalo 2-11). Todos os pacientes receberam um inibidor prévio do proteassoma, todos os pacientes receberam anteriormente lenalidomida e 56,4% dos pacientes receberam transplante prévio de células-tronco. A maioria dos pacientes eram refratários à lenalidomida, (92,5 %) a um inibidor de proteassoma (75,9%) e a ambos um imunomodulador e um inibidor de proteassoma (72,6%). A duração mediana do tratamento foi de 41,0 semanas para o grupo Isa-Pd em comparação com 24,0 semanas para o grupo Pd. A sobrevida livre de progressão (PFS) foi o desfecho primário de eficácia do ICARIA-MM. PFS foi significativamente prolongado no grupo Isa-Pd em comparação com o grupo Pd. A PFS mediana foi de 11,53 meses (IC 95%: 8,936-13,897) no grupo Isa-Pd "versus" 6,47 meses (IC 95%: 4,468-8,279) no grupo Pd (razão de risco [RR] = 0,596; IC 95%: 0,436-0,814, p = 0,0010), representando uma redução de 40,4% no risco de progressão da doença ou morte em pacientes tratados com resultados de Isa-Pd. Os resultados de PFS foram avaliados por um Comitê de Resposta Independente com base em dados de laboratório central para a proteína M e análise de imagens radiológica central usando os critérios do International Myeloma Working Group (IMWG).
Os resultados de eficácia são apresentados na tabela 1 e as curvas de Kaplan-Meier para PFS e SG são fornecidas nas Figuras 1 e 2: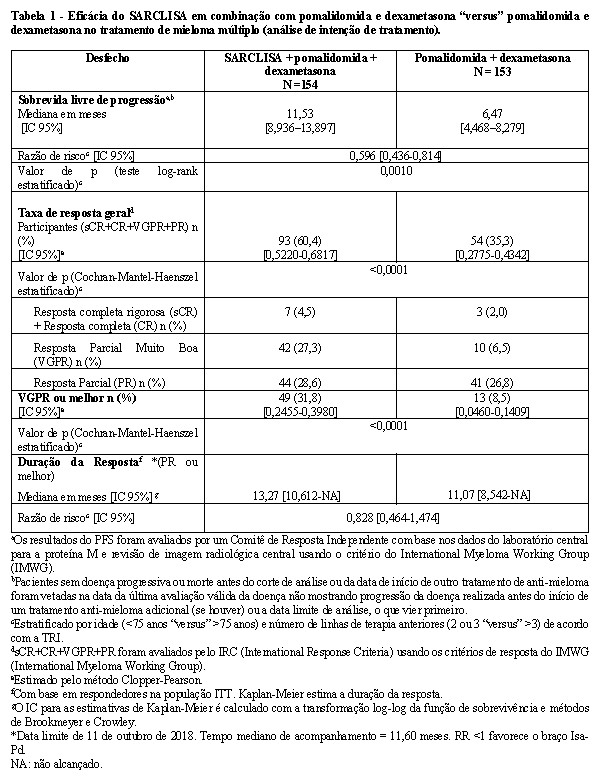
Em pacientes com citogenética de alto risco (avaliação laboratorial central), a PFS mediana foi de 7,49 (IC 95%: 2,628 a NC) no grupo Isa-Pd e 3,745 (IC 95%: 2,793 a 7,885) no grupo Pd (RR = 0,655; IC 95%: 0,334 a 1,283). Melhorias da PFS no grupo Isa-Pd também foram observadas em pacientes > 75 anos (RR = 0,479; IC 95%: 0,242 a 0,946), com ISS estágio III na entrada do estudo (RR = 0,635; IC 95% 0,363 a 1,110), com depuração basal de creatinina < 60 ml/min / 1,73 m² (RR = 0,502; IC 95%: 0,297 a 0,847), com > 3 linhas de terapia anteriores (RR = 0,590; IC 95%: 0,356 a 0,977), em pacientes refratários à terapia anterior com lenalidomida (RR = 0,593; IC 95%: 0,431 a 0,816) ou inibidor de proteassoma (RR = 0,578; IC95 %: 0,405 a 0,824) e naqueles refratários à lenalidomida no final linha anterior à entrada do estudo (RR = 0,601; IC 95%: 0,436 a 0,828). Dados suficientes não estão disponíveis para concluir sobre a eficácia do regime de isatuximabe em pacientes tratados previamente com daratumumabe (1 paciente no braço do isatuximabe e nenhum paciente no braço comparador).
O tempo mediano para a primeira resposta nos respondedores foi de 35 dias no grupo Isa-Pd "versus" 58 dias no grupo Pd. Em um tempo mediano de acompanhamento de 52,44 meses, a sobrevida global mediana final foi de 24,57 meses no grupo Isa-Pd e 17,71 meses no grupo Pd (RR = 0,776; IC de 95%: 0,594 a 1,015). O tempo mediano até o próximo tratamento antimieloma foi de 15,51 meses no grupo Isa-Pd e 8,87 meses no grupo Pd (RR = 0,548; IC de 95%: 0,417 a 0,718).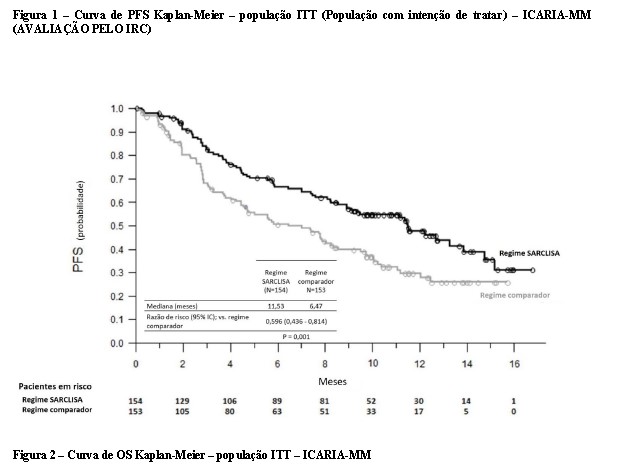
No estudo ICARIA-MM (EFC14335), um volume baseado em peso foi usado para a infusão de isatuximabe. O método de infusão de volume fixo, conforme descrito na seção 8. Posologia e modo de usar, foi avaliado no estudo TCD14079 Parte B e as simulações farmacocinéticas confirmaram diferenças mínimas entre a farmacocinética após a injeção, aplicando um volume baseado no peso do paciente e um volume fixo de 250 mL (ver seção 3. Características Farmacológicas). No estudo TCD14079 parte B, não houve novos sinais de segurança ou diferenças na eficácia em comparação com o estudo ICARIAMM.
IKEMA (EFC15246)
A eficácia e segurança de SARCLISA em combinação com carfilzomibe e dexametasona foram avaliadas no IKEMA (EFC15246), um estudo multicêntrico, multinacional, randomizado, aberto, de dois braços, de fase III, em pacientes com recidiva e/ou mieloma múltiplo refratário. Os pacientes haviam recebido de uma a três terapias anteriores.
Um total de 302 pacientes foram randomizados na proporção de 3:2 para receber SARCLISA em combinação com carfilzomibe e dexametasona (Isa-Kd, 179 pacientes) ou carfilzomibe e dexametasona (Kd, 123 pacientes). O tratamento foi administrado em ambos os grupos em ciclos de 28 dias até a progressão da doença ou toxicidade inaceitável. SARCLISA 10 mg/kg foi administrado como uma infusão IV semanal no primeiro ciclo e depois a cada duas semanas. Carfilzomibe foi administrado como uma infusão IV na dose de 20 mg/m² nos dias 1 e 2; 56 mg/m² nos dias 8, 9, 15 e 16 do primeiro ciclo; e na dose de 56 mg/m² nos dias 1, 2, 8, 9, 15 e 16 para os ciclos subsequentes de 28 dias. Dexametasona (IV nos dias de infusões de isatuximabe e/ou carfilzomibe, e via oral nos outros dias) 20 mg foram administrados nos dias 1, 2, 8, 9, 15, 16, 22 e 23 para cada ciclo de 28 dias.
No geral, as características demográficas e da doença no início do estudo foram semelhantes entre os dois grupos de tratamento. A idade mediana dos pacientes foi de 64 anos (variação de 33 a 90), 8,9% dos pacientes tinham idade ≥75 anos. A proporção de pacientes com insuficiência renal (eTFG < 60 mL/min/1,73m2) foi de 24,0% no grupo Isa-Kd versus 14,6% no grupo Kd. O estágio do International Staging System (ISS) no início do estudo foi I em 53,0%, II em 31,1% e III em 15,2% dos pacientes. No geral, 24,2% dos pacientes tinham cromossomos de alto risco de anormalidades no início do estudo; del (17p), t (4; 14), t (14; 16) estavam presentes em 11,3%, 13,9% e 2,0% dos pacientes, respectivamente. Além disso, o ganho (1q21) estava presente em 42,1% dos pacientes. O número mediano de linhas de terapia anteriores foi dois (intervalo de 1 a 4) com 44,4% dos pacientes que receberam uma linha de terapia anterior. No geral, 89,7% dos pacientes receberam inibidores de proteassoma prévio, 78,1% receberam imunomoduladores anterior (incluindo 43,4% que receberam lenalidomida anterior) e 61,3% receberam transplante prévio de células-tronco. No geral, 33,1% dos pacientes eram refratários aos inibidores de proteassoma anteriores, 45,0% eram refratários a imunomoduladores anteriores (incluindo 32,8% refratários à lenalidomida), e 20,5% eram refratários a um inibidor de proteassoma e a um imunomodulador.
A duração mediana do tratamento foi de 80,0 semanas para o grupo Isa-Kd em comparação com 61,4 semanas para o grupo Kd.
A sobrevida livre de progressão (PFS) foi o desfecho primário de eficácia do IKEMA. Com um tempo médio de acompanhamento de 20,73 meses, a análise primária da PFS mostrou uma melhora estatisticamente significante na PFS representada por uma redução de 46,9% no risco de progressão da doença ou morte em pacientes tratados com Isa-Kd em comparação com pacientes tratados com Kd.
Os resultados de eficácia são apresentados na Tabela 2 e as curvas de Kaplan-Meier para PFS são fornecidos na Figura 3: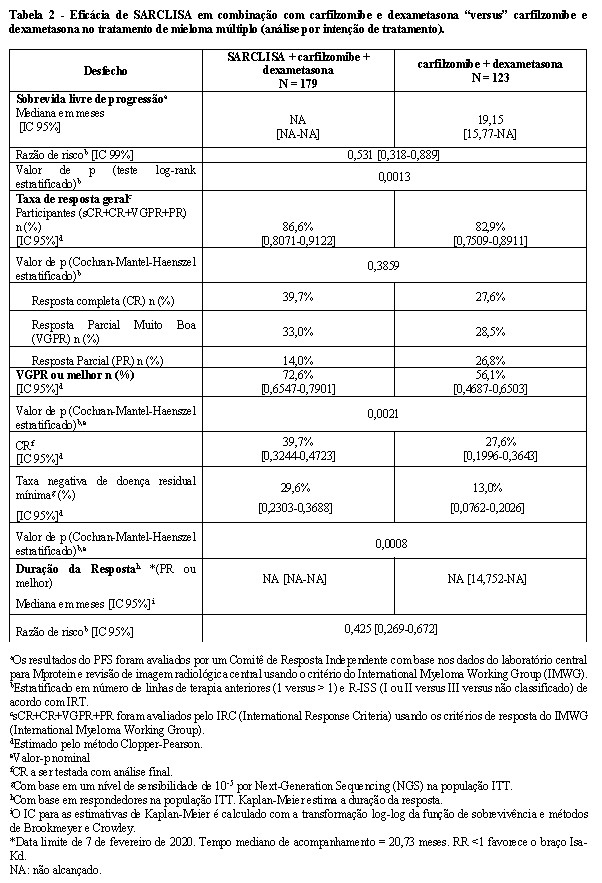
Melhoria no PFS no grupo Isa-Kd foram observadas em pacientes com alto risco citogenético (avaliação do laboratório central, RR = 0,724; IC de 95%: 0,361 a 1,451), com ganho (1q21) de anormalidade cromossômica (RR = 0,569; IC 95%:
0,330 a 0,981), ≥65 anos (RR = 0,429; IC 95%: 0,248 a 0,742), com eTFG basal (MDRD) < 60 mL min/1,73 m² (RR = 0,273;
IC 95%: 0,113 a 0,660), com > 1 linha anterior de terapia (RR = 0,479; IC 95%: 0,294 a 0,778), com ISS estágio III no início do estudo (RR = 0,650; IC 95%: 0,295 a 1,434), e em pacientes refratários à terapia anterior com lenalidomida (RR = 0,598; IC de 95%: 0,339 a 1,055).
O tempo mediano para a primeira resposta foi 1,08 meses no grupo Isa-Kd e 1,12 meses no grupo Kd. O tempo médio para progressão não foi alcançado no grupo Isa-Kd e foi de 20,27 meses (95% CI: 16,986-NA) no grupo Kd (RR = 0,495; IC 95%: 0,324-0,757). Em um tempo mediano de acompanhamento de 56,61 meses, a sobrevida global mediana não foi alcançada no grupo Isa-Kd (IC de 95%: 52,172-NR) e foi de 50,60 meses no grupo Kd (IC de 95%: 38,932-NR) (HR = 0,855; IC de 95%: 0,608-1,202). O tempo mediano até o próximo tratamento antimieloma foi de 43,99 meses no grupo Isa-Kd e 25,00 meses no grupo Kd (HR = 0,583; IC de 95%: 0,429-0,792). O benefício de Isa-Kd em relação a Kd foi mantido após a primeira terapia antimieloma subsequente com uma PFS2 mediana avaliada pelo investigador (tempo desde a randomização até o evento de SLP após a primeira terapia antimieloma subsequente) de 47,18 meses no grupo Isa-Kd (IC de 95%: 38,96557.922) e 32,36 meses no grupo Kd (IC de 95%: 23,129-40.016) (HR = 0,663; IC de 95%: 0,491 - 0,895).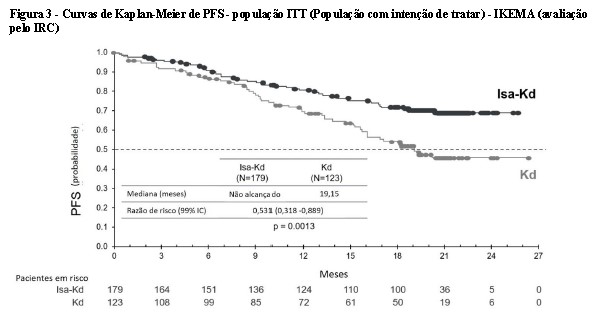
Data de corte = 07 de Fevereiro de 2020.
Entre os pacientes com eTFG (MDRD) < 50 mL/ min/1,73 m2 no início do estudo, resposta renal completa (≥60mL/min/1,73 m2 em ≥1 avaliação pós-basal) foi observada para 52,0% dos pacientes no grupo Isa-Kd e 30,8% no grupo Kd. Resposta renal completa sustentada (≥60 dias) ocorreu em 32,0% dos pacientes no grupo Isa-Kd e em 7,7% no grupo Kd. Nos 4 pacientes no grupo Isa-Kd e os 3 pacientes no grupo Kd com insuficiência renal grave no início do estudo (eTFG (MDRD) > 15 a < 30 mL/min/1,73 m2), a resposta renal mínima (≥30 a < 60 mL/min/1,73 m2 em ≥1 avaliação pós-basal) foi observado para 100% dos pacientes no grupo Isa-Kd e 33,3% dos pacientes no grupo Kd.
No geral, a qualidade de vida avaliada pelo Global Health Status Score (QLQ-C30) foi mantida durante o período de tratamento.
Em um tempo médio de acompanhamento de 43,96 meses, a análise final de PFS mostrou uma PFS mediana de 35,65 meses para o grupo Isa-Kd em comparação com 19,15 meses para o grupo Kd, com uma razão de risco de 0,576 (IC 95,4%: 0,418 a 0,792). A resposta final completa, determinada usando um ensaio IFE isatuximabe-específico validado (Sebia Hydrashift) (vide item 5. Advertências e Precauções), foi de 44,1% no grupo Isa-Kd em comparação com 28,5% no grupo Kd, com probabilidade de ocorrência de 2,094 (IC 95%: 1,259 a 3,482, descritivo p=0,0021). Em 26,3% dos pacientes do grupo Isa-Kd, tanto a DRM negativa (doença residual mínima) quanto a RC foram atingidas em comparação com 12,2% no grupo Kd, com probabilidade de ocorrência de 2,571 (IC 95%: 1,354 a 4,882, p descritivo = 0,0015).
REFERÊNCIAS BIBLIOGRÁFICAS
Attal, Michel et al. "Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study." Lancet (London, England) vol. 394,10214 (2019): 2096-2107. doi:10.1016/S0140-6736(19)32556-5
Moreau, Philippe; et al. "Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial." The Lancet, [S.L.], v. 397, n. 10292, p. 2361-2371, jun. 2021. Elsevier BV. http://dx.doi.org/10.1016/s0140-6736(21)00592-4.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O isatuximabe é um anticorpo monoclonal derivado de IgG1 que se liga a um epítopo extracelular específico do receptor CD38 e desencadeia vários mecanismos que levam à morte de células tumorais que expressam CD38. O CD38 é uma glicoproteína transmembranar com atividade ectoenzimática, expressa em neoplasias hematológicas, e é expressa altamente e uniformemente em várias células do mieloma. O isatuximabe atua através de mecanismos dependentes de IgG Fc, incluindo: citotoxicidade mediada por células dependentes de anticorpos (CMCDA), fagocitose celular dependente de anticorpos (ADCP) e citotoxicidade dependente de complemento (CDC). O isatuximabe também pode desencadear a morte de células tumorais por indução de apoptose por meio de um mecanismo independente de Fc. Nas células mononucleares do sangue periférico humano (CMSPh), as células exterminadoras naturais (NK) expressam os níveis mais altos de CD38. "In vitro", o isatuximabe pode ativar células NK na ausência de células tumorais alvo CD38 positivas através de um mecanismo dependente da porção Fc isatuximabe. Além disso, o isatuximabe inibe Tregs (Células T reguladoras) que expressam níveis mais altos de CD38 em pacientes com mieloma múltiplo em comparação com indivíduos saudáveis. O isatuximabe bloqueia a atividade enzimática do CD38, que catalisa a síntese e a hidrólise da ADP-ribose cíclica, um agente mobilizador de cálcio, e isso pode contribuir para as funções imunorregulatórias. O isatuximabe inibe a produção de cADPR do NAD extracelular nas células do mieloma múltiplo. A combinação de isatuximabe e pomalidomida "in vitro" melhora a lise celular de CD38 que expressa células de mieloma múltiplo por células efetoras (ADCC) e pela morte direta de células tumorais em comparação com o isatuximabe sozinho. Experiências "in vivo" usando um modelo de xenoenxerto de mieloma múltiplo humano demonstraram que a combinação de isatuximabe e pomalidomida resulta em atividade antitumoral aumentada em comparação com a atividade de isatuximabe ou pomalidomida isolada.
Propriedades farmacodinâmicas
A atividade farmacodinâmica do isatuximabe foi caracterizada em monoterapia. Observou-se uma diminuição nas contagens
absolutas de células NK totais (incluindo CD16 inflamatório CD16+baixo CD56+brilhante e citotóxico CD16+brilhante + CD56+dim de células NK), células B CD19+, células T CD4+ e TREG (CD3+, CD4+, CD25+, CD127-) em sangue periférico. A diminuição do TREG foi maior nos pacientes respondedores em comparação aos pacientes não respondedores. O sequenciamento de DNA do receptor de células T (RCT) foi usado para quantificar a expansão de clones de células T individuais, cada um deles com uma RCT única que confere especificidade ao antígeno. Em pacientes com mieloma múltiplo, a monoterapia SARCLISA induziu a expansão clonal do repertório de receptores de células T. Dois pacientes com mieloma múltiplo que tiveram uma resposta clínica sob o tratamento de SARCLISA desenvolveram respostas de células T contra CD38 e antígenos associados ao tumor. No mesmo estudo em monoterapia, dois pacientes que não responderam ao SARCLISA não desenvolveram essa resposta de células T.
Em pacientes com mieloma múltiplo tratados com SARCLISA combinados com pomalidomida e dexametasona, foi observada no sangue periférico uma diminuição nas contagens absolutas de células NK totais (incluindo CD16 inflamatório CD16+baixo CD56+brilhante e citotóxico CD16+brilhante CD56+dim de células NK) e células CD19+ de células B. Um aumento de células T CD4+ e TREG (CD3+, CD4+, CD25+, CD127-) foi observado em toda a população tratada e em pacientes não respondedores.
Os efeitos farmacodinâmicos do SARCLISA em pacientes com mieloma múltiplo suportam seu mecanismo de ação imunomodulador. Além de suas funções efetoras, a resposta de células T induzida por SARCLISA indica uma resposta imune adaptativa.
Propriedades farmacocinéticas
A farmacocinética de SARCLISA foi avaliada em 476 pacientes com mieloma múltiplo tratados com infusão intravenosa de SARCLISA como um agente único ou em combinação com pomalidomida / dexametasona, em doses variando de 1 a 20 mg/kg, administradas uma vez por semana; a cada 2 semanas; ou a cada 2 semanas por 8 semanas, seguido por a cada 4 semanas; ou a cada semana, durante 4 semanas, seguido por cada 2 semanas. SARCLISA exibe farmacocinética não linear com disposição de medicamentos mediada por alvo devido à sua ligação ao receptor CD38. A exposição à SARCLISA (área sob a curva de concentração plasmática-tempo ao longo do intervalo de dosagem da AUC) aumenta de maneira proporcional à dose de 1 a 20 mg/kg, seguindo o esquema a cada 2 semanas, enquanto não é observado nenhum desvio na proporcionalidade da dose entre 5 e 20 mg/kg após cada semana, durante 4 semanas, seguido de um programa a cada 2 semanas. Após a administração de SARCLISA 10 mg/kg, semanalmente, durante 4 semanas, seguidas a cada 2 semanas, o tempo mediano para atingir o estado estacionário foi de 18 semanas com uma acumulação de 3,1 vezes. No ICARIA-MM, a concentração plasmática máxima prevista média (CV%) Cmax e AUC no estado estacionário foi de 351 mg/mL (36,0%) e 72.600 mg.h/mL (51,7%), respectivamente. No IKEMA, a concentração plasmática máxima prevista média (CV%) Cmax e AUC no estado estacionário foram 637 mg/mL (30,9%) e 152.000 mg.h/mL (37,8%), respectivamente. A diferença na exposição é devido a uma mudança no método de ensaio e em um efeito de tratamento superior com Isa-Kd em comparação com Isa-Pd.
Absorção
Não há absorção, pois SARCLISA é administrado por via intravenosa.
Distribuição
O volume total estimado de distribuição de SARCLISA é de 8,75 L.
Metabolismo
Como uma proteína grande, espera-se que o isatuximabe seja metabolizado por processos de catabolismo proteolítico não saturável.
Eliminação
SARCLISA é eliminado por duas vias paralelas, uma via mediada por alvo não linear predominando em baixas concentrações e uma via linear inespecífica predominando em concentrações mais altas. Na faixa de concentrações terapêuticas no plasma, a via linear é predominante e diminui ao longo do tempo em 50% para um valor de estado estacionário de 0,00955 L/h (0,229 L/dia). Isso está associado a uma meia-vida terminal de 28 dias.
Interações medicamentosas
A farmacocinética de SARCLISA e pomalidomida, ou de SARCLISA e carfilzomibe, não foi influenciada por sua coadministração.
Populações especiais
Idade
As análises farmacocinéticas populacionais de 476 pacientes com idade entre 36 e 85 anos mostraram exposição comparável à SARCLISA em pacientes com idade igual ou menor a 75 anos "versus" com idade igual ou maior a 75 anos (n = 70).
Gênero
O sexo não teve efeito clinicamente significativo na farmacocinética de SARCLISA.
Raça
A raça (caucasiana, negra, asiática e outras raças) não teve efeito clínico significativo na farmacocinética de SARCLISA.
Peso
A exposição de SARCLISA (AUC) no estado estacionário aumentou com o aumento do peso corporal, suportando a dose baseada em peso.
Pediatria
SARCLISA não foi avaliado em pacientes com menos de 18 anos de idade.
Insuficiência hepática
Não foram realizados estudos formais de isatuximabe em pacientes com insuficiência hepática. Dos 476 pacientes da análise farmacocinética da população, 65 pacientes apresentaram comprometimento hepático leve [bilirrubina total 1 a 1,5 vezes o limite superior do normal (LSN) ou aspartato amino transferase (AST) > LSN] e 1 paciente apresentou comprometimento hepático moderado (bilirrubina total ≥ 1,5 a 3 vezes o LSN e qualquer AST). Insuficiência hepática leve não teve efeito clinicamente significativo na farmacocinética de SARCLISA. O efeito de insuficiência hepática moderada (bilirrubina total > 1,5 vezes a 3 vezes LSN e qualquer AST) e comprometimento hepático grave (bilirrubina total > 3 vezes LSN e qualquer AST) na farmacocinética de SARCLISA é desconhecida. No entanto, como o isatuximabe é um anticorpo monoclonal, não se espera que seja eliminado por meio do metabolismo mediado por enzimas hepáticas e, assim, não se espera que a variação na função hepática afete a eliminação de isatuximabe (vide item 8. Posologia e modo de usar - Populações especiais).
Insuficiência renal
Não foram realizados estudos formais de isatuximabe em pacientes com insuficiência renal. A análise farmacocinética da população em 476 pacientes incluiu 192 pacientes com insuficiência renal leve (60 mL/min/1,73 m2 ≤ taxa estimada de filtração glomerular (eTFG) < 90 mL/min/1,73 m2), 163 pacientes com insuficiência renal moderada (30 mL/min/1,73 m2≤ eTFG < 60 mL/min/1,73 m2) e 12 pacientes com insuficiência renal grave (TFG < 30 mL/min/1,73 m2). As análises sugeriram que não há efeito clinicamente significativo do comprometimento renal leve a grave, na farmacocinética de SARCLISA em comparação com o comprometimento normal da função renal (bilirrubina total > 3 vezes o LSN e qualquer AST) na farmacocinética de SARCLISA. Uma análise farmacocinética em 22 pacientes com doença renal em estágio terminal (DRET), incluindo pacientes em diálise (TFGe < 15 ml/min/1,73 m2), não mostrou efeitos clinicamente significativos da DRET na farmacocinética de SARCLISA em comparação com aqueles de função renal normal, ou com comprometimento leve a moderado da função renal. Nenhum ajuste de dose de SARCLISA é necessário em pacientes com comprometimento renal leve, moderado, grave ou em estágio terminal.
Dados de segurança pré-clínica
Toxicidade por dose repetida
Nenhuma informação essencial.
Genotoxicidade e carcinogenicidade
Nenhum estudo de genotoxicidade e carcinogenicidade foi realizado com SARCLISA.
Toxicidade reprodutiva e de desenvolvimento
Os estudos de toxicidade reprodutiva, desenvolvimento e toxicidade embrionária não foram realizados com SARCLISA.
Outros estudos de toxicidade
Nenhuma informação essencial.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em pacientes com hipersensibilidade ao isatuximabe ou a qualquer excipiente da formulação.
5. ADVERTÊNCIAS E PRECAUÇÕES
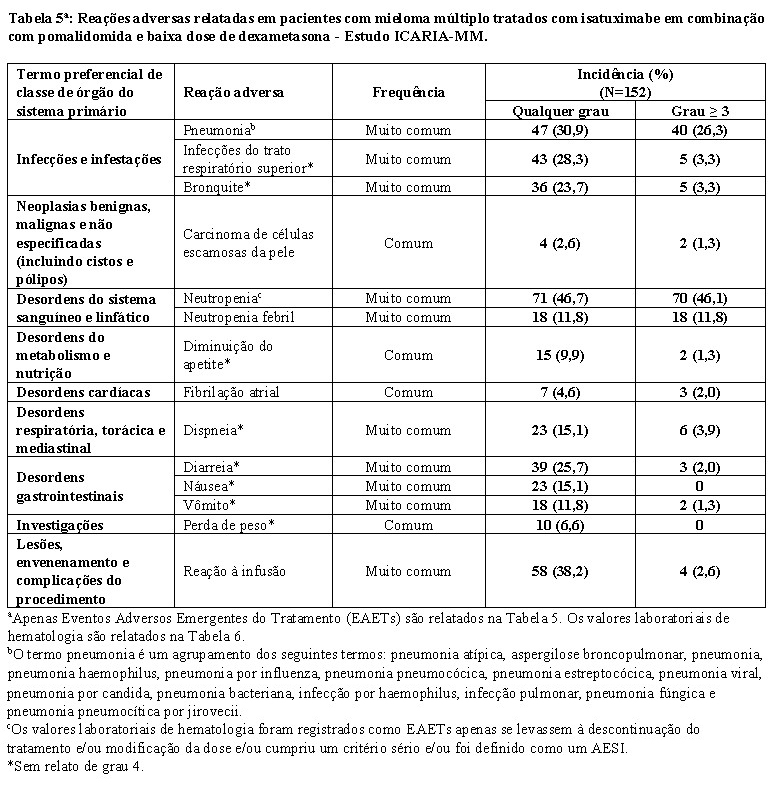
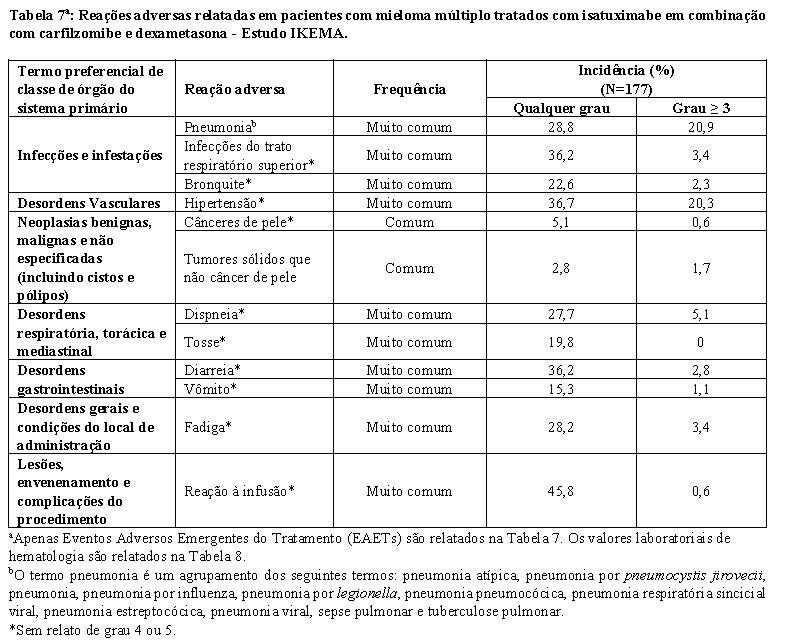
Reações à infusão
Reações à infusão (RIs), principalmente leves ou moderadas, foram observadas em 38,2% dos pacientes tratados com SARCLISA no ICARIA-MM, e em 45,8% dos pacientes tratados com Isa-Kd no IKEMA (vide item 9. Reações Adversas). No ICARIA-MM, todas as RIs começaram durante a primeira infusão SARCLISA e foram resolvidas no mesmo dia na maioria dos pacientes. Os sintomas mais comuns de uma RI incluem dispneia, tosse, calafrios e náusea. Os sinais e sintomas graves mais comuns incluem hipertensão, dispneia e broncoespasmo (vide item 9. Reações Adversas). No IKEMA, as RIs ocorreram no dia da infusão em 99,2% dos episódios. Em pacientes tratados com Isa-Kd, 94,4% daqueles que tiveram uma RI, esta ocorreu durante o primeiro ciclo de tratamento. Todas as RIs foram resolvidas. Os sintomas mais comuns de uma RI incluíram tosse, dispneia, congestão nasal, vômito e náusea. Os sinais e sintomas graves mais comuns incluíam hipertensão e dispneia (vide item 9. Reações Adversas). SARCLISA pode causar reações graves à infusão, incluindo reações anafiláticas (vide item 9. Reações Adversas). Para diminuir o risco e a gravidade das RIs, os pacientes devem ser pré-medicados antes da infusão de SARCLISA com paracetamol, difenidramina ou equivalente; a dexametasona deve ser usada como tratamento pré-medicação e anti-mieloma (vide item 8. Posologia e modo de usar -Pré-medicação). Os sinais vitais devem ser monitorados com frequência durante toda a infusão com SARCLISA. Quando necessário, interrompa a infusão de SARCLISA e forneça assistência médica apropriada e medidas de suporte (vide item 8. Posologia e modo de usar -Administração e Ajuste de Dose). No caso dos sintomas não melhorarem para grau ≤1 após a interrupção da infusão de SARCLISA, persistirem ou piorarem apesar do uso de medicamentos apropriados, se houver necessidade de hospitalização ou risco de vida descontinue permanentemente SARCLISA e institua manobra apropriada.
Interferência no teste sorológico (teste indireto de antiglobulina)
SARCLISA se liga ao CD38 nas células vermelhas do sangue (CVS) e pode resultar em um teste de antiglobulina indireta falso positivo (teste indireto de Coombs). Essa interferência com o teste indireto de Coombs pode persistir por aproximadamente 6 meses após a última infusão de SARCLISA. O teste indireto de antiglobulina foi positivo durante o tratamento com Isa-Pd em 67,7% dos pacientes testados e 63,3% durante o tratamento com Isa-Kd. Nos pacientes com teste positivo de antiglobulina indireta, as transfusões de sangue foram administradas sem evidência de hemólise. Tipagem ABO/RhD não foi afetado pelo tratamento com SARCLISA. Para evitar possíveis problemas com a transfusão de hemácias, os pacientes que estão sendo tratados com SARCLISA devem realizar exames de tipo sanguíneo e de triagem antes da primeira infusão com SARCLISA. A fenotipagem pode ser considerada antes do início do tratamento com SARCLISA, conforme a prática local. Se o tratamento com SARCLISA já tiver sido iniciado, o banco de sangue deve ser informado de que o paciente está recebendo SARCLISA e a interferência de SARCLISA nos testes de compatibilidade sanguínea pode ser resolvida usando ditiotreitol (DTT) nas células vermelhas tratadas para interromper a ligação do isatuximabe. Uma vez que o sistema de grupo sanguíneo Kell também é sensível ao tratamento com DTT, unidades Kell-negativas devem ser fornecidas após descartar ou identificar aloanticorpos usando células vermelhas tratadas com DTT. Se uma transfusão de emergência for necessária, hemácias compatíveis com ABO/RhD não compatíveis podem ser administradas de acordo com as práticas locais do banco de sangue (vide item Interferência com testes laboratorial e diagnóstico). Atualmente, não há informações disponíveis sobre por quanto tempo a interferência com o teste indireto de Coombs pode persistir após a última infusão de SARCLISA. Com base na meia-vida de isatuximabe, prevê-se que o teste indireto de Coombs positivo mediado por isatuximabe pode persistir por aproximadamente 6 meses após a última infusão.
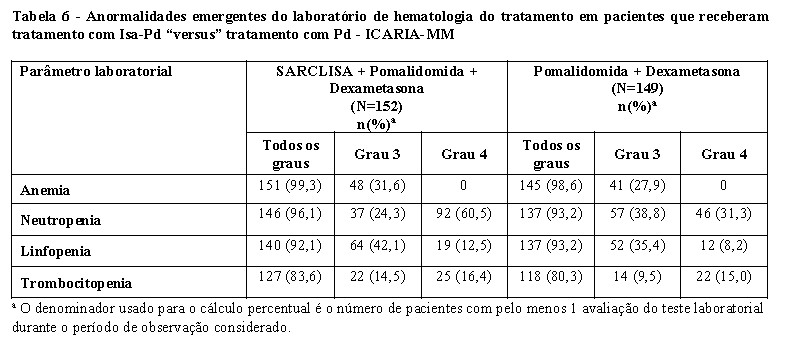
Neutropenia
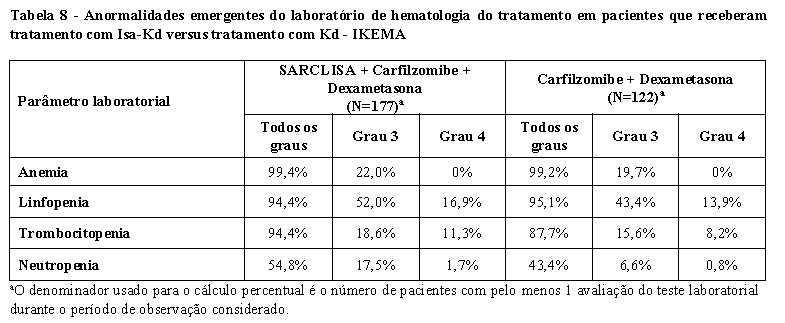
Em pacientes tratados com Isa-Pd, neutropenia ocorreu como uma anormalidade laboratorial em 96,1% dos pacientes e como uma reação adversa em 46,7%, destes foi relatada neutropenia Grau 3-4 como uma anormalidade laboratorial em 84,9% dos pacientes e como reações adversas em 45,4% dos pacientes. Complicações neutropênicas foram observadas em 30,3% dos pacientes, incluindo 11,8% de neutropenia febril e 25,0% de infecções neutropênicas. Em pacientes tratados com Isa-Kd, a neutropenia ocorreu como anormalidade laboratorial em 54,8% dos pacientes e como uma reação adversa em 4,5% dos pacientes, destes foi relatada neutropenia de Grau 3-4 como uma anormalidade laboratorial em 19,2% dos pacientes (com 17,5% de Grau 3 e 1,7% Grau 4) e como uma reação adversa em 4,0% dos pacientes. Complicações neutropênicas foram observadas em 2,8% dos pacientes, incluindo 1,1% de neutropenia febril e 1,7% de infecções neutropênicas (vide item 9. Reações adversas).
Monitore a contagem completa de células sanguíneas periodicamente durante o tratamento. Antibacterianos e profilaxia antiviral (como a profilaxia de herpes zoster) podem ser consideradas durante o tratamento. Monitore os pacientes com neutropenia quanto a sinais de infecção. Não são recomendadas reduções de dose de SARCLISA. Atrasos na dose de SARCLISA e o uso de fatores estimuladores de colônias (por exemplo, G-CSF) podem ser necessários para permitir a melhoria da contagem de neutrófilos (vide item 8. Posologia e modo de usar -Ajuste de dose).
Segunda malignidade primária
Segundas malignidades primárias (SMPs) foram relatadas com isatuximabe em combinação com pomalidomida e dexametasona (ICARIA-MM) e com isatuximabe em combinação com carfilzomibe e dexametasona (IKEMA). A incidência geral de SPMs em todos os pacientes expostos a SARCLISA é de 4,3%. Os médicos devem monitorar cuidadosamente os pacientes antes e durante o tratamento de acordo com Diretrizes do International Myeloma Working Group (IMWG) para a ocorrência de SPM e iniciar tratamento conforme indicado (vide item 9. Reações adversas).
Síndrome da lise tumoral
Casos de síndrome da lise tumoral (SLT) foram relatados em participantes que receberam regimes contendo isatuximabe. Os pacientes devem ser monitorados atentamente e as precauções apropriadas devem ser tomadas.
Interferência na avaliação de respostas
SARCLISA é um anticorpo monoclonal IgG kappa que pode ser detectado eventualmente em ambas as dosagens de eletroforese de proteínas séricas (EPS) e imunofixação (EFI) utilizados para o monitoramento clínico de proteína M endógena (vide item Interferência com testes laboratorial e diagnóstico). Essa interferência pode afetar a precisão da determinação da resposta completa em alguns pacientes com proteína IgG kappa mieloma. Vinte e dois pacientes no braço Isa-Pd que atendiam aos critérios RPMB com apenas positividade residual à imunofixação foram testados quanto à interferência. As amostras de soro desses pacientes foram testadas por espectrometria de massa para separar o sinal de SARCLISA do sinal da proteína M do mieloma. Em 11 dos 22 pacientes, não houve proteína M do mieloma residual detectável no nível de sensibilidade do teste de imunofixação (25 mg/dL); 10 dos 11 pacientes tinham mieloma do subtipo IgG no início, mostrando interferência de SARCLISA com o ensaio de imunofixação. No braço Isa-Kd, dos 27 pacientes identificados com potencial interferência e testados por espectrometria de massa no nível de sensibilidade do teste de imunofixação (25 mg / dL), 15 pacientes tiveram resposta não completa (não-RC) de acordo com o Comitê de Resposta Independente (CRI) não demostraram proteína M do mieloma residual detectável. Entre esses 15 pacientes, 11 deles tiveram < 5% de células plasmáticas na medula óssea. Isso indica que 11 pacientes adicionais dos 179 do braço Isa-Kd (6,1%) poderiam ter RC como melhor resposta, levando a uma taxa potencial de RC de 45,8% (vide item Interferência com testes laboratorial e diagnóstico).
Interferência com testes laboratorial e diagnóstico
Interferência nos testes sorológicos
Como a proteína CD38 é expressa na superfície dos glóbulos vermelhos, SARCLISA, um anticorpo anti-CD38, pode interferir nos testes sorológicos do banco de sangue com possíveis reações falso positivas em testes indiretos de antiglobulina (testes indiretos de Coombs), testes de detecção (triagem) de anticorpos, painéis de identificação de anticorpos e combinações cruzadas de globulina anti-humano (AHG) em pacientes tratados com SARCLISA.
Interferência nos testes de eletroforese e imunofixação de proteínas séricas
SARCLISA pode ser detectada eventualmente por ensaios de eletroforese de proteínas séricas (EPS) e imunofixação (EFI) usados para o monitoramento da proteína M e pode interferir na classificação precisa da resposta, com base nos critérios do International Myeloma Working Group (IMWG). Em pacientes com resposta parcial muito boa persistente, onde há suspeita de interferência, considere o uso de um ensaio IFE isatuximabe-específico validado (Sebia Hydrashift) para remover a interferência de isatuximabe e visualizar especificamente qualquer proteína M sérica restante, para facilitar a determinação da resposta completa.
Gravidez e lactação
Não existem dados disponíveis sobre a utilização de SARCLISA em mulheres grávidas. Não foram realizados estudos de toxicidade na reprodução animal com SARCLISA. Não é possível concluir se SARCLISA é seguro para uso durante a gravidez.
Anticorpos monoclonais de imunoglobulina G1 são conhecidos por atravessar a placenta. Com base em seu mecanismo de ação, SARCLISA pode causar danos fetais quando administrado a uma mulher grávida. SARCLISA pode causar depleção de células imunes fetais (CD38-positiva) e diminuição da densidade óssea. O uso de SARCLISA em mulheres grávidas não é recomendado. Aconselhe as mulheres grávidas sobre o risco potencial para o feto.
A administração de vacinas vivas a recém-nascidos e bebês expostos ao SARCLISA no útero deve ser adiada até que uma avaliação hematológica seja realizada.
Mulheres com potencial para engravidar, tratadas com SARCLISA devem usar métodos contraceptivos eficazes durante o tratamento e por pelo menos 5 meses após a interrupção do tratamento com SARCLISA.
A combinação de SARCLISA com pomalidomida é contraindicada em mulheres grávidas, pois a pomalidomida pode causar defeitos congênitos e morte do feto. Consulte as informações de prescrição da pomalidomida sobre o uso durante a gravidez. Para outros medicamentos administrados com SARCLISA, consulte as respectivas informações de bulas atuais.
Não há dados disponíveis sobre a presença de SARCLISA no leite humano, na produção de leite ou nos efeitos sobre o lactente. No entanto, sabe-se que a imunoglobulina G humana está presente no leite humano. Os anticorpos podem ser secretados no leite humano. Não se pode tirar conclusões se SARCLISA é seguro para uso durante a amamentação. O uso de SARCLISA em mulheres que amamentam não é recomendado.
Fertilidade
Não há dados disponíveis em humanos e animais para determinar os efeitos potenciais de SARCLISA na fertilidade em homens e mulheres (vide item 3. Características Farmacológicas - Dados de segurança pré-clínica).
Pacientes idosos
Apesar de não terem sido observadas diferenças nessa população nos estudos clínicos, não é possível descartar a possibilidade de maior sensibilidade em pacientes idosos.
Alterações na capacidade de dirigir veículos e utilizar máquinas
Não foram realizados estudos sobre os efeitos na capacidade de dirigir e utilizar máquinas. Com base nas reações adversas relatadas, não se espera que SARCLISA influencie a capacidade de dirigir e utilizar máquinas (vide item 9. Reações Adversas). No entanto, foram relatadas fadiga e tontura em pacientes que tomam SARCLISA e isso deve ser levado em consideração ao dirigir ou utilizar máquinas. Para outros medicamentos administrados com SARCLISA, consulte as respectivas informações nas bulas atuais.
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Atenção diabéticos: este medicamento contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
SARCLISA não tem impacto sobre a farmacocinética de pomalidomida ou carfilzomibe, ou vice-versa.
Incompatibilidades/Compatibilidades
SARCLISA não deve ser misturado com outros medicamentos exceto aqueles mencionados no item 8. Posologia e modo de usar - Modo de preparo e administração.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Este medicamento deve ser mantido sob refrigeração (em temperatura entre 2 e 8°C). Proteger da luz. Não congelar. Não agitar.
Após diluição
A solução para infusão de SARCLISA deve ser preparada em cloreto de sódio a 0,9% ou glicose a 5%. A estabilidade microbiológica, química e física em uso da solução de infusão SARCLISA foi demonstrada por 48 horas entre 2°C e8°C, seguidas por 8 horas (incluindo o tempo de infusão) à temperatura ambiente. Nenhuma proteção contra a luz é necessária para o armazenamento na bolsa de infusão.
Prazo de validade: 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparado manter entre 2°C e 8°C por até 48 horas, seguidas por 8 horas (incluindo o tempo de infusão) à temperatura ambiente.
Características físicas e organolépticas
Este medicamento apresenta-se como uma solução incolor a ligeiramente amarelada, essencialmente livre de partículas visíveis.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
SARCLISA deve ser administrado por um profissional de saúde, em um ambiente em que instalações de ressuscitação estejam disponíveis.
A dose recomendada de SARCLISA é de 10 mg/kg de peso corpóreo administrada por infusão intravenosa em combinação com pomalidomida e dexametasona (Isa-Pd) ou em combinação com carfilzomibe e dexametasona (Isa-Kd), de acordo com
o esquema da Tabela 3.
Pré-medicação
A pré-medicação deve ser usada antes da infusão de SARCLISA com os seguintes medicamentos para reduzir o risco e a gravidade das reações à infusão (RIs):
• Dexametasona 40 mg via oral ou IV (ou 20 mg via oral ou IV em pacientes com idade ≥ 75 anos): quando administrado em combinação com SARCLISA e pomalidomida.
• Dexametasona 20 mg (IV nos dias de infusão de SARCLISA e/ou carfilzomibe, e via oral nos demais dias): quando administrado em combinação com SARCLISA e carfilzo