RYBREVANT
JANSSEN-CILAG
amivantamabe
Tratamento de câncer de pulmão cel. não peq..
Apresentações.
Solução para diluição para infusão de 50 mg/mL de amivantamabe em embalagem com 1 frasco-ampola de 7 mL.
USO INTRAVENOSO
USO ADULTO
Composição.
Cada frasco contém 350 mg de amivantamabe em 7 mL de solução.
Excipientes: histidina, cloridrato de L-histidina monoidratado, sacarose, polissorbato 80, levometionina, edetato dissódico di-hidratado e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Rybrevant® é indicado para o tratamento de pacientes com câncer de pulmão de não pequenas células (CPNPC) localmente avançado ou metastático com mutações de inserção do éxon 20 ativadoras do receptor do fator de crescimento epidérmico (EGFR), cuja doença apresentou progressão durante ou após quimioterapia à base de platina.
2. RESULTADOS DE EFICÁCIA
CPNPC Localmente Avançado ou Metastático com Mutações de Inserção do Éxon 20
EDI1001 (CHRYSALIS) é um estudo multicêntrico, aberto, de múltiplas coortes conduzido para avaliar a segurança e a eficácia de Rybrevant® em pacientes com CPNPC metastático ou locamente avançado. A eficácia foi avaliada em 81 pacientes com CPNPC localmente avançado ou metastático que apresentaram mutações de inserção do éxon 20 no EGFR, conforme determinado por teste prévio ao tratamento padrão local, cuja doença apresentou progressão durante ou após quimioterapia à base de platina, e que apresentaram acompanhamento mediano de 9,7 meses. Rybrevant® foi administrado por via intravenosa a 1050 mg em pacientes < 80 Kg ou 1400 mg em pacientes ≥ 80 Kg uma vez por semana durante 4 semanas, e então a cada 2 semanas subsequentes até a progressão da doença ou toxicidade inaceitável.
A idade mediana foi de 62 (faixa: 42-84) anos, sendo que 9% dos pacientes ≥ 75 anos de idade; 59% eram do sexo feminino; e 49% eram asiáticos e 37% eram caucasianos. O número mediano de terapias anteriores foi de 2 (faixa: 1 a 7 terapias). No período basal, 99% apresentaram status de desempenho do Grupo de Oncologia Cooperativo do Leste (ECOG) de 0 ou 1 (99%); 53% nunca fumaram; 75% apresentaram câncer em Estágio IV; e 22% tiveram tratamento anterior para metástases cerebrais. As inserções do éxon 20 foram observadas em 8 resíduos diferentes; os resíduos mais comuns foram A767 (24%), S768 (16%), D770 (11%) e N771 (11%).
Os resultados de eficácia são resumidos na Tabela 1.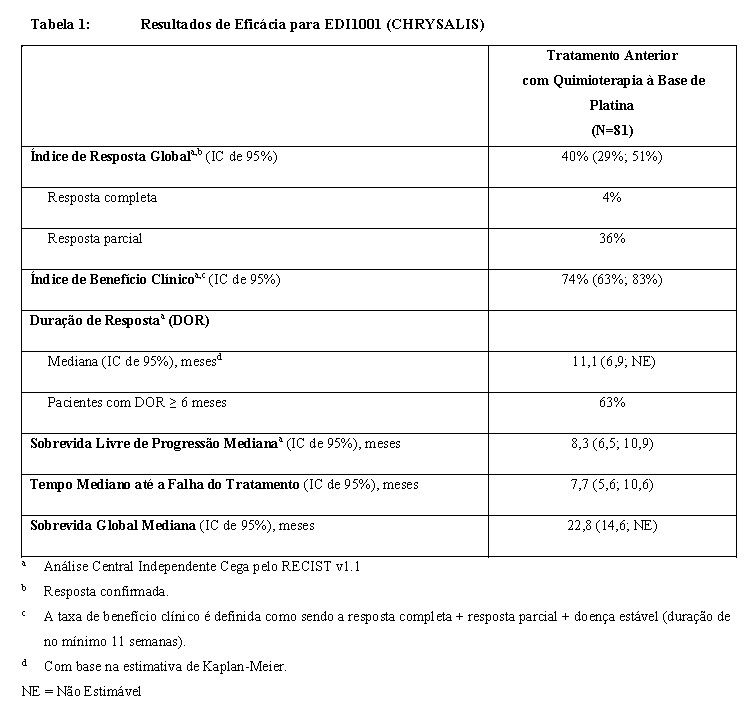
Referências bibliográficas:
1. PARK, K.; HAURA, E.; LEIGHL, N. et al. Amivantamab in EGFR Exon 20 Insertion – Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. In: Journal of Clinical Oncology, 2021. http://ascopubs.org/doi/full/10.1200/JCO.21.00662.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
O amivantamabe é um anticorpo biespecífico EGFR-MET completamente humano à base de IgG1 e de baixa fucose com atividade imunológica direcionada às células que tem como alvos tumores com mutações em EGFR resistentes e ativadores e mutações e amplificações em MET. Amivantamabe liga-se aos domínios extracelulares de EGFR e MET. Amivantamabe interrompe as funções sinalizadas por EGFR e MET através do bloqueio da ligação destes ligantes e amplificação da degradação de EGFR e MET, dessa forma, prevenindo o crescimento e progressão tumoral. A presença de EGFR e MET na superfície de células tumorais também possibilita o direcionamento dessas células para destruição por células imunológicas efetoras, tais como células exterminadoras naturais e macrófagos, através de citotoxicidade celular dependente de anticorpos (CCDA) e mecanismos de trogocitose, respectivamente.
Efeitos farmacodinâmicos
Albumina
O amivantamabe reduziu a concentração de albumina, um efeito farmacodinâmico da inibição do MET, tipicamente durante as 8 primeiras semanas. Subsequentemente, a concentração de albumina se estabilizou pelo restante do tratamento com amivantamabe.
Imunogenicidade
Assim como com todas as proteínas terapêuticas, há o potencial de imunogenicidade. Em um estudo clínico de pacientes com CPNPC localmente avançado ou metastático tratado com Rybrevant®, 3 (1%) dos 286 pacientes avaliáveis testaram positivo para anticorpos anti-amivantamabe.
Propriedades Farmacocinéticas
A área sob a curva de concentração-tempo (AUCsemana1) de amivantamabe aumenta proporcionalmente em relação a uma faixa de dose de 350 a 1750 mg (0,33 a 1,67 vezes a dose recomendada para pacientes < 80 Kg e 0,25 a 1,25 vezes a dose recomendada para pacientes ≥ 80 Kg).
Após a administração de Rybrevant® na dose e esquema recomendados, a concentração máxima (Cmáx) sérica ± DP média foi de 836 ± 264 mg/mL a 1050 mg para pacientes < 80 Kg e 655 ± 109 mg/mL a 1400 mg para pacientes < 80 Kg no término da dosagem semanal após a quinta dose. A AUC semana1 ± DP média após a quinta dose foi de 94,946 ± 35,440 mg.h/mL a 1050 mg para pacientes < 80 Kg e 76,946 ± 14,557 mg.h/mL a 1400 mg para pacientes ≥ 80 Kg. A AUCsemana1 sérica média foi de aproximadamente 2,9 vezes mais alta após a quinta dose depois da dosagem semanal em comparação à primeira dose.
Quando Rybrevant® foi administrado, o estado de equilíbrio de amivantamabe foi alcançado em aproximadamente 2 meses em cada período de dosagem de 2 semanas (até a nona infusão) a 1050 mg, e a proporção ± DP média de amivantamabe de AUCsemana1 no estado de equilíbrio até a AUCsemana1 após a primeira dose foi de 2,44 ± 0,54.
Distribuição
O volume de distribuição ± DP médio de amivantamabe estimado a partir de parâmetros PK populacionais foi de 5,13 ± 1,78 L após a administração da dose recomendada de Rybrevant® .
Eliminação
O clearance de amivantamabe diminuiu com a dose crescente e com dosagem múltipla. Estimou-se que o clearance linear ± DP médio foi de 360 ± 144 mL/dia e a meia-vida terminal estimada ± DP média associada ao clearance linear estimado a partir de estimativas de parâmetros PK populacionais foi de 11,3 ± 4,53 dias após a administração da dose recomendada de Rybrevant® .
Populações especiais
Pacientes Pediátricos (17 anos de idade ou menos)
A farmacocinética de Rybrevant® em pacientes pediátricos não foi investigada.
Idosos (65 anos de idade ou mais)
Não foram observadas diferenças clinicamente significativas na farmacocinética de amivantamabe com base na idade (32-87 anos).
Insuficiência renal
Nenhum efeito clinicamente significativo sobre a farmacocinética de amivantamabe foi observado em pacientes com insuficiência renal leve (60≤ clearance de creatinina [CrCl] < 90 mL/min) e moderada (29 ≤ CrCl < 60 mL/min). O efeito da insuficiência renal severa (15≤ CrCl < 29mL/min) sobre a farmacocinética de amivantamabe é desconhecido.
Insuficiência hepática
É improvável que alterações na função hepática apresentem um efeito sobre a eliminação de amivantamabe, uma vez que moléculas à base de IgG1 como amivantamabe não são metabolizadas através de vias hepáticas.
Não foi observado um efeito clinicamente significativo na farmacocinética de amivantamabe com base no insuficiência hepática leve [(bilirrubina total ≤ LSN e AST > LSN) ou (LSN < bilirrubina total ≤ 1,5 x LSN)]. O efeito da insuficiência hepática moderada (bilirrubina total de 1,5 a 3 vezes a LSN) e severa (bilirrubina total > 3 vezes a LSN) sobre a farmacocinética de amivantamabe é desconhecido.
Gênero
O clearance de amivantamabe foi 24% mais alto em homens do que em mulheres; entretanto, não foram observadas diferenças clinicamente significativas na farmacocinética de amivantamabe com base em gênero.
Peso
O volume central de distribuição e clearance de amivantamabe aumentou com o peso corporal crescente. Exposições similares de amivantamabe foram alcançadas na dose recomendada de Rybrevant® em pacientes com peso corporal < 80 Kg que receberam 1050 mg e pacientes com peso corporal ≥ 80 Kg que receberam 1400 mg.
Informação não clínica
Em estudos de toxicidade de doses repetidas em macacos cynomolgus, amivantamabe foi bem tolerado em doses semanais de até 120 mg/Kg por via intravenosa durante 3 meses e até 125 mg/Kg por via subcutânea durante 2 semanas. Não houve efeitos sobre a função dos sistemas cardiovascular, respiratório e nervoso. A patologia clínica demonstrou elevações não adversas na alanina aminotransferase (ALT), no aspartato aminotransferase (AST) e nas globulinas séricas, e reduções não adversas na albumina em comparação ao grupo de controle. Todos esses valores retornaram às faixas normais em grupos de recuperação.
Carcinogenicidade e Mutagenicidade
Nenhum estudo animal foi realizado para estabelecer o potencial carcinogênico de amivantamabe. Estudos de rotina de genotoxicidade e carcinogenicidade geralmente não são aplicáveis a medicamentos biológicos, pois grandes proteínas não podem se difundir em células e não podem interagir com DNA ou material cromossômico.
Toxicologia Reprodutiva
Nenhum estudo toxicológico foi realizado para avaliar os possíveis efeitos de amivantamabe.
4. CONTRAINDICAÇÕES
Rybrevant® é contraindicado em pacientes com hipersensibilidade conhecida a amivantamabe ou a qualquer um dos componentes da formulação.
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações Relacionadas à Infusão
Rybrevant® pode causar reações relacionadas à infusão (RRI); os sinais e sintomas da RRI incluem dispneia, rubor, febre, calafrios, náuseas, desconforto no peito, hipotensão e vômitos. Com base na população de segurança (vide "Reações Adversas"), a RRI ocorreu em 66% dos pacientes tratados com Rybrevant®. Entre os pacientes que receberam tratamento na Semana 1, Dia 1, 65% experimentaram uma RRI, enquanto a incidência de RRI foi de 3,4% com a infusão do Dia 2, 0,4% com a infusão da Semana 2 e cumulativamente 1,1% com as infusões subsequentes. Das RRIs relatadas, 97% eram de Grau 1-2, 2,2% eram de Grau 3 e 0,4% eram de Grau 4. O tempo médio para o início foi de 1 hora (intervalo de 0,1 a 18 horas) após o início da infusão. A incidência de modificações na infusão devido a RRI foi de 62% e 1,3% dos pacientes interromperam definitivamente o Rybrevant® devido à IRR. Pré-medique com anti-histamínicos, antipiréticos e glicocorticoides e faça a infusão de Rybrevant® conforme recomendado (vide "Posologia e Modo de Usar"). Administre Rybrevant® por via periférica na Semana 1 e na Semana 2 (vide "Posologia e Modo de Usar"). Monitore os pacientes para quaisquer sinais e sintomas de reações à infusão durante a infusão de Rybrevant® em um ambiente onde medicamentos e equipamentos de ressuscitação cardiopulmonar estejam disponíveis. Interrompa a infusão se houver suspeita de RRI. Reduza a taxa de infusão ou descontinue definitivamente Rybrevant® com base na gravidade (vide "Posologia e Modo de Usar").
Doença Pulmonar Intersticial/Pneumonite Rybrevant® pode causar doença pulmonar intersticial (DPI)/pneumonite. Com base na população de segurança (vide "Reações Adversas"), DPI/pneumonite ocorreu em 3,3% dos pacientes tratados com Rybrevant®, com 0,7% dos pacientes apresentando DPI/pneumonite de Grau 3. Três pacientes (1%) interromperam o uso de Rybrevant® devido a DPI / pneumonite. Monitore os pacientes para novos ou agravamento dos sintomas indicativos de DPI/pneumonite (por exemplo, dispneia, tosse, febre). Suspenda imediatamente o Rybrevant® em pacientes com suspeita de DPI/pneumonite e descontinue permanentemente se DPI/pneumonite for confirmada (vide "Posologia e Modo de Usar").
Reações Dermatológicas
Rybrevant® pode causar erupção na pele (incluindo dermatite acneiforme), prurido e pele seca. Com base na população de segurança (vide "Reações Adversas"), a erupção cutânea ocorreu em 74% dos pacientes tratados com Rybrevant® , incluindo erupção cutânea de Grau 3 em 3,3% dos pacientes. O tempo médio para o início da erupção foi de 14 dias (variação: 1 a 276 dias). Erupção cutânea que levou à redução da dose ocorreu em 5% dos pacientes, e Rybrevant® foi descontinuado permanentemente devido a erupção cutânea em 0,7% dos pacientes (vide "Reações Adversas"). A necrólise epidérmica tóxica (NET) ocorreu em um paciente (0,3%) tratado com Rybrevant® . Instrua os pacientes a limitar a exposição ao sol durante e por 2 meses após o tratamento com Rybrevant®. Aconselhe os pacientes a usar roupas de proteção e usar protetor solar de amplo espectro UVA/UVB. O creme emoliente sem álcool é recomendado para pele seca. Se ocorrerem reações cutâneas, inicie corticosteroides tópicos e antibióticos tópicos e/ou orais. Para reações de Grau 3, adicione esteróides orais e considere uma consulta dermatológica. Encaminhar prontamente os pacientes que apresentam erupção cutânea grave, aparência ou distribuição atípica ou ausência de melhora em 2 semanas a um dermatologista. Pausar, reduzir a dose ou descontinuar permanentemente Rybrevant® com base na gravidade (vide "Posologia e Modo de Usar").
Toxicidade Ocular
Rybrevant® pode causar toxicidade ocular, incluindo ceratite, sintomas de olho seco, hiperemia conjuntival, visão turva, alteração na acuidade visual, prurido ocular e uveíte. Com base na população de segurança (vide "Reações Adversas"), ceratite ocorreu em 0,7% e uveíte em 0,3% dos pacientes tratados com Rybrevant®. Todos os eventos foram de Grau 1-2. Encaminhar prontamente os pacientes que apresentam sintomas oculares a um oftalmologista. Interrompa, reduza a dose ou descontinue permanentemente Rybrevant® com
fetal Com base em seu mecanismo de ação e achados em modelos ani
mais, Rybrevant® pode causar danos fetais quando administrado a mulheres grávidas. A administração de outras moléculas inibidoras de EGFR a animais grávidas resultou em um aumento da incidência de comprometimento do dbase na gravidade (vide "Posologia e Modo de Usar").
Toxicidade embriofetal
Com base em seu mecanismo de ação e achados em modelos animais, Rybrevant® pode causar danos fetais quando administrado a mulheres grávidas. A administração de outras moléculas inibidoras de EGFR a animais grávidas resultou em um aumento da incidência de comprometimento do desenvolvimento embriofetal, embrioletalidade e aborto. Aconselhe as mulheres com potencial reprodutivo do risco potencial para o feto. Aconselhe as pacientes do sexo feminino com potencial reprodutivo a usar anticoncepcionais eficazes durante o tratamento e por 3 meses após a dose final de Rybrevant®. (vide "Gravidez e Lactação").
Efeitos sobre a Capacidade de Dirigir e Usar Máquinas
Nenhum estudo foi realizado sobre os efeitos na capacidade de dirigir e usar máquinas. Se pacientes apresentarem sintomas relacionados ao tratamento que afetam a sua capacidade de concentrar e reagir, recomenda-se que não dirijam ou usem máquinas até que o efeito cesse.
Gravidez, Lactação e Fertilidade Gravidez (Categoria C)
Resumo do Risco Com base no mecanismo de ação e achados em modelos animais, Rybrevant® pode causar dano fetal quando administrado a mulheres grávidas. Não há dados disponíveis sobre o uso de Rybrevant® em mulheres grávidas ou dados de animais para avaliar o risco de Rybrevant® na gravidez. O bloqueio ou degradação de EGFR em modelos animais resultou no comprometimento do desenvolvimento embriofetal, incluindo efeitos no desenvolvimento placentário, pulmonar, cardíaco, cutâneo e neural. A ausência de sinalização de EGFR ou MET resultou em embrioletalidade, malformações e morte pósnatal em animais (vide "Dados"). Avise as mulheres grávidas sobre o risco potencial para o feto.
Dados
Dados Animais Não foram realizados estudos em animais para avaliar os efeitos do amivantamabe na reprodução e no desenvolvimento fetal; no entanto, com base em seu mecanismo de ação, Rybrevant® pode causar danos fetais ou anomalias de desenvolvimento. Em camundongos, o EGFR é criticamente importante nos processos reprodutivos e de desenvolvimento, incluindo implantação de blastocisto, desenvolvimento placentário e sobrevivência e desenvolvimento embriofetal/pósnatal. A redução ou eliminação da sinalização de EGFR embriofetal ou materna pode prevenir a implantação, pode causar perda embriofetal durante vários estágios da gestação (por meio de efeitos no desenvolvimento da placenta) e pode causar anomalias de desenvolvimento e morte precoce em fetos sobreviventes. Resultados de desenvolvimento adversos foram observados em vários órgãos em embriões/neonatos de camundongos com interrupção da sinalização de EGFR. Da mesma forma, a supressão de MET ou seu ligante HGF foi embrionariamente letal devido a graves defeitos no desenvolvimento da placenta e os fetos exibiram defeitos no desenvolvimento muscular em vários órgãos. Sabe-se que a IgG1 humana atravessa a placenta; portanto, o amivantamabe tem o potencial de ser transmitido da mãe para o feto em desenvolvimento.
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Resumo do Risco Não existem dados sobre a presença de amivantamabe no leite humano na produção de leite, ou seus efeitos na criança amamentada. Devido ao potencial de reações adversas graves de Rybrevant® em bebês amamentados, aconselhe as mulheres a não amamentar durante o tratamento com Rybrevant® e por 3 meses após a dose final.
Contracepção
Devido ao risco de que Rybrevant® pode provocar dano fetal quando administrado a mulheres grávidas, oriente as pacientes com potencial reprodutivo de utilizar uma contracepção eficaz durante o tratamento e por 3 meses após a última dose de Rybrevant®. Pacientes do sexo masculino devem usar contracepção eficaz (p. ex. preservativo) e não devem doar ou armazenar sêmen durante o tratamento e por 3 meses após a última dose de Rybrevant® .
Atenção: Este medicamento contém açúcar, portanto, deve ser usado com cautela em pessoas com diabetes.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação medicamentosa.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Rybrevant® deve ser armazenado sob refrigeração entre 2°C a 8°C. Não congele. Armazene na embalagem original para proteger da luz. A validade de Rybrevant® é de 18 meses a partir da data de sua fabricação.
Após diluição:
Como as soluções de amivantamabe não contêm conservantes, salvo se o método de abertura/diluição prevenir o risco de contaminação microbiana, o produto deve ser usado imediatamente. Administre as soluções diluídas dentro do período de 10 horas (incluindo o tempo de infusão) em temperatura de 15°C a 25°C e em iluminação ambiente.
Aspecto físico
A solução para diluição para infusão é incolor a amarela pálida e livre de conservantes.
Número de lote, datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original. Antes de usar, observe o aspecto do medicamento antes do uso. Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
Rybrevant® deve ser administrado por um profissional de saúde com auxílio médico apropriado para tratar reações relacionadas à infusão (RRIs) caso ocorram (vide "Advertências e Precauções"). Administre os medicamentos pré-infusão (vide "Posologia -Medicamentos Pré-infusão"). Ao considerar o uso de Rybrevant®, a presença de mutações de inserção no éxon 20 do EGFR deve ser estabelecida usando um teste validado (vide "Resultados de Eficácia").
Dosagem - Adultos (≥18 anos)
A dose recomendada de Rybrevant® é fornecida na Tabela 2, e o esquema de dose está disposto na Tabela 3 (vide "Taxas de Infusão -Tabela 5"). 

Medicamentos pré-infusão Antes da infusão inicial de Rybrevant® (Dias 1 e 2 da Semana 1), administre anti-histamínicos, antipiréticos e glicocorticoides para reduzir o risco de RRIs. Para doses subsequentes, administre anti-histamínicos e antipiréticos. Administre antieméticos conforme necessário. 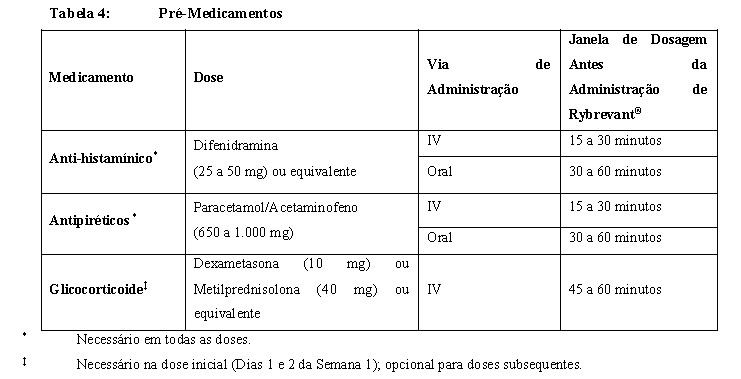
Taxas de Infusão Administre a infusão de Rybrevant® por via intravenosa de acordo com as taxas de infusão na Tabela 5. Devido à frequência de RRIs na primeira dose, a infusão via veia periférica na Semana 1 e na Semana 2 deve ser considerada para minimizar a exposição ao medicamento em caso de RRI; uma infusão por cateter de linha central pode ser administrada durante as semanas subsequentes. Recomenda-se a diluição da primeira dose o mais próximo da administração quanto possível, permitindo flexibilidade máxima no tratamento da RRI. 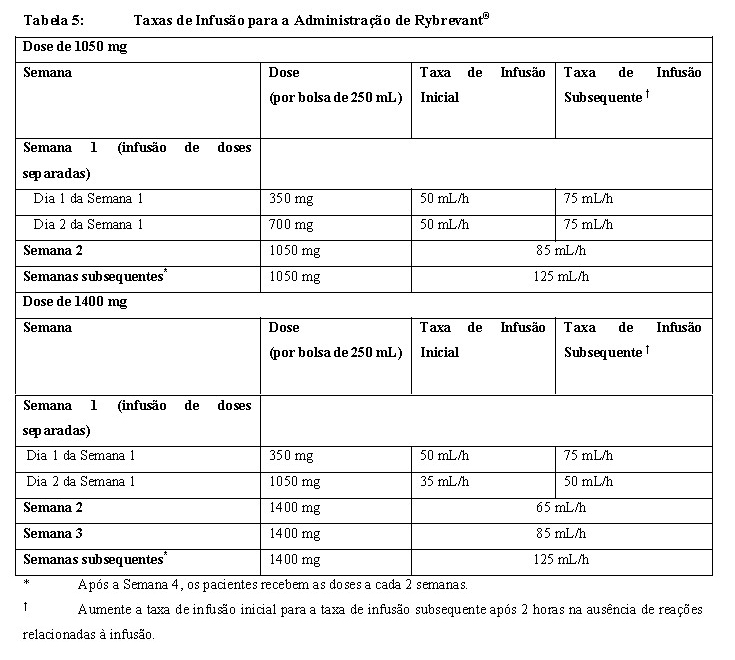
Tratamento de Reações Relacionadas à Infusão
• Interrompa infusão no primeiro sinal de RRIs. Administre medicamentos adjuvantes adicionais (p. ex. glicocorticoides, anti-histamínicos, antipiréticos e antieméticos adicionais), conforme clinicamente indicado.
• Grau 1-3 (leve-severo): Após a recuperação dos sintomas, retome a infusão a 50% da taxa anterior. Se não houver sintomas adicionais, a taxa pode ser aumentada de acordo com a taxa de infusão recomendada (veja a Tabela 5). Administre medicamentos pré-infusão na próxima dose (veja a Tabela 4).
• Grau 3 ou Grau 4 Recorrentes (risco à vida): Descontinue Rybrevant® permanentemente (vide "Advertências e Precauções").
Dose(s) Perdida(s) Se uma dose planejada de Rybrevant® for perdida, a dose deve ser administrada o quanto antes e o esquema de dosagem deve ser devidamente ajustado, mantendo o intervalo de tratamento.
Modificações da Dose Interrompa o Rybrevant® na presença de reações adversas de Grau 3 ou 4 até que a reação adversa seja resolvida*. Se uma interrupção durar 7 dias ou menos, retome a administração de Rybrevant® na dose atual. Se uma interrupção for maior do que 7 dias, considere retomar a administração de Rybrevant® na dose reduzida, conforme descrito na Tabela 6.
Reações na Pele e nas Unhas
Se o paciente desenvolver reações na pele e nas unhas de Grau 2 insatisfatoriamente toleradas ou de Grau 3, considere a interrupção de Rybrevant® até a reação adversa melhorar (vide "Advertências e Precauções").
Doença Pulmonar Intersticial
Se o paciente desenvolver doença pulmonar intersticial (DPI) ou pneumonite, descontinue o Rybrevant® permanentemente (vide "Advertências e Precauções").
Populações especiais
Pacientes Pediátricos (17 anos de idade e mais novos) A segurança e a eficácia de Rybrevant® não foram estabelecidas em pacientes pediátricos.
Idosos (65 anos de idade ou mais) Dos 362 pacientes tratados com Rybrevant® em EDI1001, 41% tinham 65 anos de idade ou mais, e 12% tinham 75 anos de idade ou mais. Não foram observadas diferenças na segurança ou na eficácia entre estes pacientes e os mais jovens. Nenhum ajuste de dose é necessário (vide "Propriedades Farmacocinéticas").
Insuficiência renal
Não foram conduzidos estudos formais de amivantamabe em pacientes com insuficiência renal. Com base nas análises farmacocinéticas (PK) populacionais, nenhum ajuste de doseé necessário para pacientes com insuficiência renal leve ou moderado. Não há dados disponíveis em pacientes com insuficiência renal grave (vide "Propriedades Farmacocinéticas").
Insuficiência hepática
Não foram conduzidos estudos formais de amivantamabe em pacientes com insuficiência hepática. Com base em análises PK populacionais, nenhum ajuste de doseé necessário para pacientes com insuficiência hepática leve. Não há dados disponíveis em pacientes com insuficiência hepática moderada ou severa (vide "Propriedades Farmacocinéticas").
Administração
Preparação para Administração
A solução de Rybrevant® deve ser diluída e preparada para infusão intravenosa por um profissional de saúde usando técnica asséptica.
1. Determine a dose necessária (1050 mg ou 1400 mg) e o número de frascos-ampola Rybrevant® necessários com base no peso do paciente no período basal (vide "Dosagem"). Cada frasco-ampola de Rybrevant® contém 350 mg de amivantamabe.
2. Verifique se a solução de Rybrevant® está incolor a amarela pálida. Não o use se houver descoloração ou houver presença de partículas visíveis.
3. Retire e então descarte um volume de solução de dextrose [glicose] a 5% ou de solução de cloreto de sódio a 0,9% da bolsa de infusão igual ao volume de Rybrevant® a ser incluída (ou seja, descarte 7 mL de diluente da bolsa de infusão para cada frasco-ampola de Rybrevant®). As bolsas de infusão devem ser feitas de policloreto de vinila (PVC), polipropileno (PP), polietileno (PE) ou mistura de poliolefina (PP+PE).
4. Retire 7 mL de Rybrevant® de cada frasco-ampola e adicione à bolsa de infusão. O volume final na bolsa de infusão deve ser de 250 mL. Cada frasco-ampola contém um excedente de 0,5 mL para garantir um volume extraível suficiente. Descarte qualquer porção não usada restante no frasco-ampola.
5. Inverta a bolsa levemente para misturar a solução. Não agite.
6. Inspecione a solução diluída visualmente antes da administração. Não use se descoloração ou partículas visíveis forem observadas.
7. Soluções diluídas devem ser administradas dentro do período de 10 horas (incluindo o tempo de infusão) em temperatura ambiente (15°C a 25°C) e em iluminação ambiente.
Administração
1 Administre a solução diluída por infusão intravenosa usando um conjunto de infusão equipado com um regulador de fluxo e com um filtro de polietersulfona (PES) de baixa ligação proteica, não pirogênica, estéril e com acesso em linha (tamanho de poros de 0,2 micrômetros). Os conjuntos de administração devem ser feitos de poliuretano (PU), polibutadieno (PBD), PVC, PP ou PE.
2 Não realize a infusão com Rybrevant® concomitantemente no mesmo acesso em linha intravenoso com outros agentes.
3 Este medicamento destina-se apenas para uso único. Qualquer sobra de medicamento não usada deve ser descartada de acordo com as exigências locais.
Este medicamento não pode ser misturado com outros produtos, exceto aqueles mencionados na seção "Posologia e Modo de Usar".
9. REAÇÕES ADVERSAS
As seguintes reações adversas são discutidas nas outras seções desta bula:
• Reações Relacionadas à Infusão (vide "Advertências e Precauções")
• Doença Pulmonar Intersticial/Pneumonite (vide "Advertências e Precauções")
• Reações Adversas Dermatológicas (vide "Advertências e Precauções")
• Toxicidade Ocular (vide "Advertências e Precauções")
Experiência dos Estudos Clínicos
Como os estudos clínicos são conduzidos em condições amplamente variáveis, as taxas de reações adversas observadas nos estudos clínicos de um medicamento não podem ser comparadas diretamente às taxas dos estudos clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
A população de segurança descrita em "Advertências e Precauções" reflete a exposição ao Rybrevant® como agente único no estudo CHRYSALIS em 302 pacientes com CPNPC localmente avançado ou metastático que receberam uma dose de 1050 mg (para pacientes < 80 kg) ou 1400 mg (para pacientes ≥80 kg) uma vez por semana durante 4 semanas e, em seguida, a cada 2 semanas. Entre os 302 pacientes que receberam Rybrevant®, 36% foram expostos por 6 meses ou mais e 12% foram expostos por mais de um ano. Na população de segurança, as reações adversas mais frequentes (≥ 20%) foram erupção cutânea, reação relacionada à infusão, paroníquia, dor musculoesquelética, dispneia, náusea, edema, tosse, fadiga, estomatite, constipação, vômitos e prurido. As anormalidades laboratoriais de Grau 3 a 4 mais comuns (≥ 2%) foram diminuição dos linfócitos, diminuição do fosfato, diminuição da albumina, aumento da glicose, aumento da gama glutamil transferase, diminuição do sódio, diminuição do potássio e aumento da fosfatase alcalina.
Os dados descritos abaixo refletem a exposição ao Rybrevant® na dosagem recomendada em 129 pacientes com CPNPC localmente avançado ou metastático com mutações de inserção do Exon 20 do EGFR cuja doença progrediu durante ou após a quimioterapia à base de platina. Entre os pacientes que receberam Rybrevant®, 44% foram expostos por 6 meses ou mais e 12% foram expostos por mais de um ano.
A mediana de idade foi de 62 anos (variação: 36 a 84 anos); 61% eram mulheres; 55% eram asiáticos, 35% eram brancos e 2,3% eram negros; e 82% tinham peso corporal basal < 80 kg.
Reações adversas graves ocorreram em 30% dos pacientes que receberam Rybrevant®. As reações adversas graves em ≥ 2% dos pacientes incluíram embolia pulmonar, pneumonite/DPI, dispneia, dor musculoesquelética, pneumonia e fraqueza muscular. Reações adversas fatais ocorreram em 2 pacientes (1,5%) devido a pneumonia e 1 paciente (0,8%) devido a morte súbita.
A descontinuação permanente de Rybrevant® devido à uma reação adversa ocorreu em 11% dos pacientes. As reações adversas que resultaram na descontinuação permanente de Rybrevant® em ≥1% dos pacientes foram pneumonia, RRI, pneumonite/DPI, dispneia, derrame pleural e erupção cutânea.
As interrupções da dose de Rybrevant® devido a uma reação adversa ocorreram em 78% dos pacientes. Reações relacionadas à infusão (RRI) que necessitaram interrupções da infusão ocorreram em 59% dos pacientes. As reações adversas que exigiram interrupção da dose em ≥5% dos pacientes incluíram dispneia, náusea, erupção cutânea, vômito, fadiga e diarreia.
Reduções de dose de Rybrevant® devido a uma reação adversa ocorreram em 15% dos pacientes. As reações adversas que necessitaram redução da dose em ≥ 2% dos pacientes incluíram erupção cutânea e paroníquia.
As reações adversas mais comuns (≥ 20%) foram erupção cutânea, RRI, paroníquia, dor musculoesquelética, dispneia, náusea, fadiga, edema, estomatite, tosse, constipação e vômitos. As anormalidades laboratoriais de Grau 3 a 4 mais comuns (≥ 2%) foram diminuição dos linfócitos, diminuição da albumina, diminuição do fosfato, diminuição do potássio, aumento da glicose, aumento da fosfatase alcalina, aumento da gama-glutamil transferase e diminuição do sódio.
A Tabela 7 resume as reações adversas no CHRYSALIS.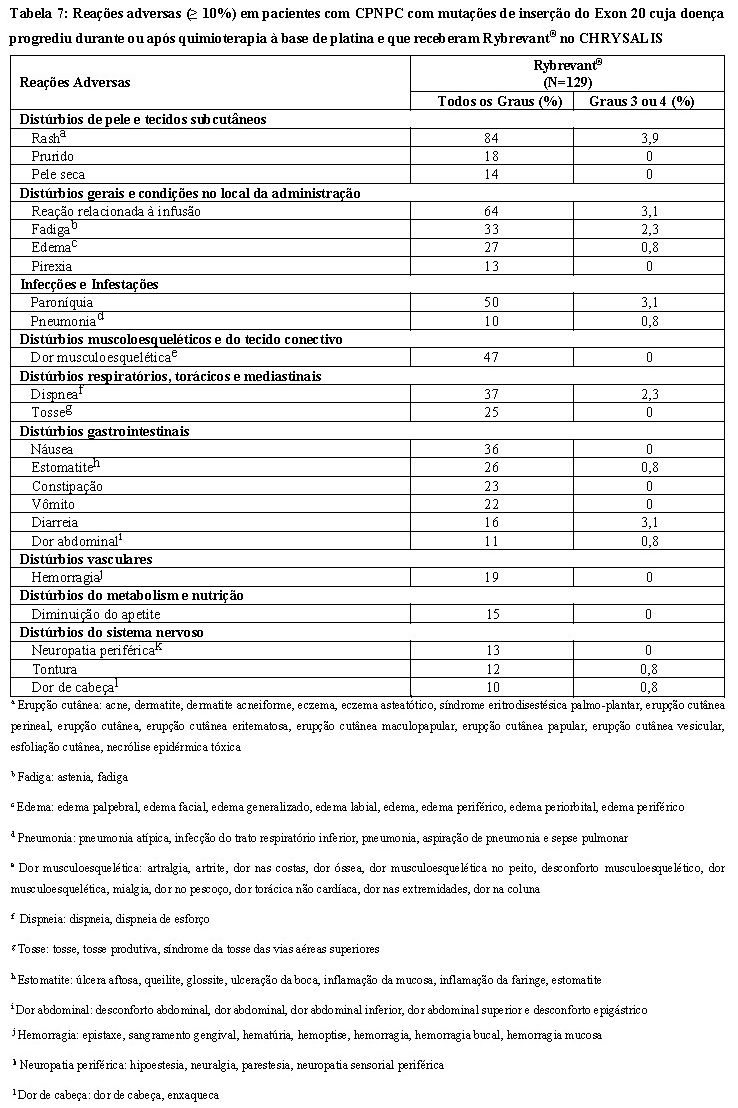
As reações adversas clinicamente relevantes em < 10% dos pacientes que receberam Rybrevant® incluíram toxicidade ocular, DPI/pneumonite e necrólise epidérmica tóxica (NET).
A Tabela 8 resume as anormalidades laboratoriais no CHRYSALIS.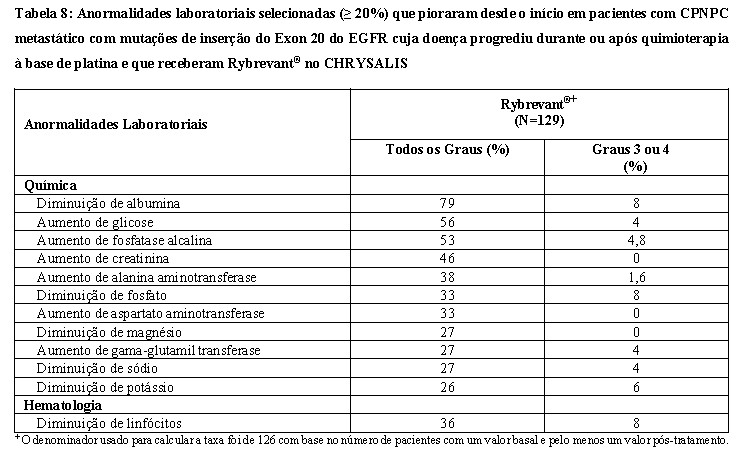
Imunogenicidade
Tal como acontece com todas as proteínas terapêuticas, existe potencial para imunogenicidade. A detecção da formação de anticorpos é altamente dependente da sensibilidade e especificidade do ensaio. Além disso, a incidência observada de positividade de anticorpos (incluindo anticorpos neutralizantes) em um ensaio pode ser influenciada por vários fatores, incluindo metodologia do ensaio, manuseio da amostra, tempo de coleta da amostra, medicamentos concomitantes e doença subjacente. Por estas razões, a comparação da incidência de anticorpos nos estudos descritos abaixo com a incidência de anticorpos em outros estudos ou com outros medicamentos com amivantamabe pode ser errônea.
No CHRYSALIS, 3 dos 286 (1%) pacientes que foram tratados com Rybrevant® e avaliados quanto à presença de anticorpos antidroga (AAD), testaram positivo para anticorpos anti-amivantamabe emergentes do tratamento (um em 27 dias, um em 59 dias e um em 168 dias após a primeira dose) com títulos de 1:40 ou menos. Não há dados suficientes para avaliar o efeito de AAD na farmacocinética, segurança ou eficácia do Rybrevant® .
Atenção: este medicamento é novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos através do VigiMed, disponível no site da ANVISA.
10. SUPERDOSE
Sinais e sintomas
Não há informações sobre a superdosagem de Rybrevant® .
Tratamento
Não há um antídoto específico conhecido para a superdosagem de Rybrevant®. Na hipótese de superdosagem, interrompa a administração de Rybrevant®, e adote medidas adjuvantes gerais até que a toxicidade clínica tenha sido diminuída ou resolvida.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS-1.1236.3436
VENDA SOB PRESCRIÇÃO MÉDICA
USO RESTRITO A HOSPITAIS Esta bula foi aprovada pela ANVISA em 23/12/2021.