RUKOBIA
GLAXOSMITHKLINE
trometamol cetorolaco
Analgésico. Antiinflamatório não-hormonal.
Apresentações.
Comprimidos revestidos de liberação prolongada de 600 mg em cartuchos com 60 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de liberação prolongada de Rukobia 600 mg contém: fostensavir 600 mg (equivalentes a 724,56 mg de fostensavir trometamol), excipientes* q.s.p. 1 comprimido revestido de liberação prolongada
*hiprolose, hipromelose, sílica coloidal hidrofóbica, estearato de magnésio, água purificada e Opadry® II 85F170022 Bege (álcool polivinílico, dióxido de titânio, macrogol, talco, óxido de ferro amarelo e óxido de ferro vermelho).
Informações técnicas.
1. INDICAÇÕES
Rukobia é indicado em combinação com outros agentes antirretrovirais para o tratamento de adultos altamente experimentados ao tratamento que apresentem infecção pelo vírus da imunodeficiência humana-1 (HIV-1) resistente a vários medicamentos para os quais não é possível construir um regime antiviral supressor devido a considerações de resistência, intolerância ou segurança (ver Resultados de Eficácia).
2. RESULTADOS DE EFICÁCIA
A eficácia de fostensavir em indivíduos adultos vivendo com HIV altamente experimentados ao tratamento para HIV se baseia em dados de um estudo de fase III, parcialmente randomizado, internacional, duplo-cego e controlado por placebo (BRIGHTE [205888]).
O estudo BRIGHTE foi realizado em 371 indivíduos vivendo com HIV-1 altamente experimentados com resistência a várias classes. Todos os indivíduos deveriam apresentar uma carga viral maior ou igual a 400 cópias/mL e ≤2 classes de antirretrovirais (ARV) permanecendo como base devido à resistência, intolerância, contraindicação ou outras preocupações de segurança. No recrutamento, os indivíduos da Coorte Randomizada recebiam um, porém não mais que dois agentes antirretrovirais totalmente ativos e disponíveis, que poderiam ser combinados como parte de um regime de base eficaz. Na Coorte Randomizada, 272 indivíduos receberam cegamente fostensavir 600 mg duas vezes ao dia (n = 203) ou placebo (n = 69), além de seu regime corrente em falha, por 8 dias de monoterapia funcional. Após o Dia 8, os indivíduos randomizados receberam abertamente fostensavir 600 mg duas vezes ao dia, mais uma terapia de base otimizada (TBO) selecionada pelo Investigador Principal. A Coorte Randomizada fornece evidência primária de eficácia de fostensavir. Na Coorte não Randomizada, 99 indivíduos sem agentes antirretrovirais totalmente ativos e aprovados disponíveis no recrutamento foram tratados abertamente com fostensavir 600 mg duas vezes ao dia, mais TBO a partir do Dia 1. O uso de medicamento(s) em investigação como um componente da TBO foi permitido na Coorte não Randomizada.
De modo geral, a maior parte dos indivíduos foi do gênero masculino (78%), brancos (70%), e a idade mediana foi de 49,0 anos (variação: 17- 73 anos). Na visita basal, o RNA do HIV-1 mediano foi de 4,6 log10 cópias/mL e a contagem mediana de células CD4+ foi de 80,0 células/mm3 (100 e 41 células/mm3 para indivíduos Randomizados e não Randomizados, respectivamente). Setenta e cinco por cento (75%) de todos os indivíduos tratados apresentaram uma contagem de células T CD4+ de < 200 células/mm3 na visita basal (com 30% < 20 células/mm3). Em geral, 86% apresentaram um histórico de Síndrome de ImunoDeficiência Adquirida (AIDS), e 8% apresentaram um histórico de coinfecção por vírus da hepatite B e/ou C. Setenta e um por cento (71%) dos indivíduos haviam sido tratados para HIV por > 15 anos, 85% haviam sido expostos a ≥5 regimes de tratamento diferentes para HIV após a admissão ao estudo.
Cinquenta e dois por cento dos indivíduos na Coorte Randomizada receberam um agente totalmente ativo em sua TBO ativa, 42% receberam dois e 6% não receberam nenhum. Na Coorte não Randomizada, 81% dos indivíduos não receberam agentes totalmente ativos em sua TBO original e 19% receberam um, incluindo 15% (n=15) que receberam ibalizumabe, que foi um agente em investigação no momento do início do estudo BRIGHTE.
A análise do desfecho primário, com base na redução média ajustada no RNA do HIV-1 nos Dias 1 a 8 na Coorte Randomizada, demonstrou superioridade do fostensavir em relação ao placebo (redução de 0,79 vs. 0,17 log10, respectivamente; p < 0,0001, população exposta de intenção de tratar [ITT-E]) (Tabela 1).
No Dia 8, 65% (131/203) e 46% (93/203) dos indivíduos apresentaram uma redução na carga viral desde o valor basal > 0,5 log10 c/mL e > 1 log10 c/mL, respectivamente, no grupo fostensavir, em comparação a 19% (13/69) e 10% (7/69) dos indivíduos, respectivamente, no grupo placebo.
Por análises de subgrupo, indivíduos randomizados tratados com fostensavir com RNA do HIV-1 basal > 1.000 c/mL atingiram uma redução mediana na carga viral de 1,02 log10 c/mL no Dia 8, em comparação a redução de 0,00 log10 c/mL em indivíduos tratados cegamente com placebo.
Os resultados virológicos por Análise de Snapshot ITT-E nas Semanas 24, 48 e 96 no estudo BRIGHTE (incluindo resultados pelas principais covariáveis basais) são apresentados na Tabela 2 para a Coorte Randomizada.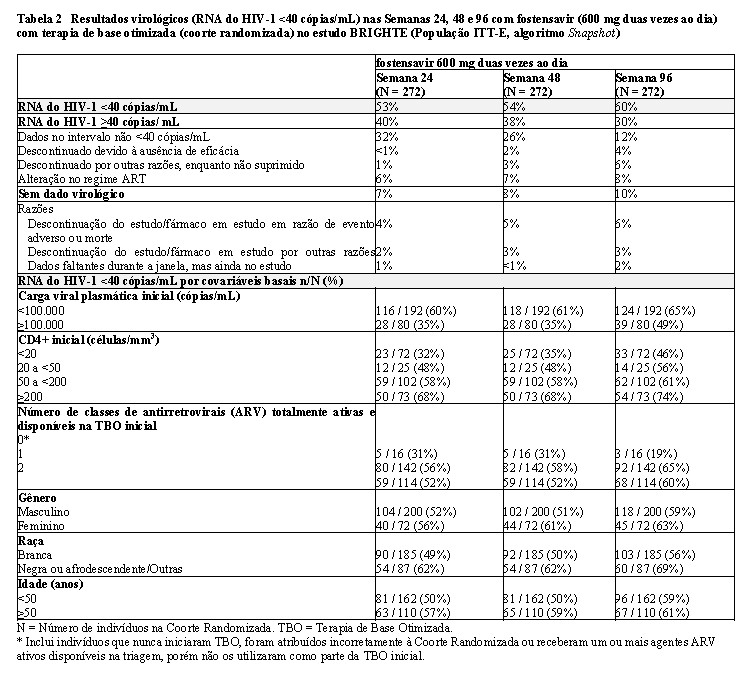
Na Coorte Randomizada, foi atingida carga viral < 200 cópias de RNA do HIV-1/mL em 68%, 69% e 64% dos indivíduos nas Semanas 24, 48 e 96, respectivamente. Nestes períodos, a proporção de indivíduos com carga viral < 400 cópias de RNA do HIV-1/mL foi de 75%, 70% e 64%, respectivamente (ITT-E, algoritmo Snapshot). As alterações médias na contagem de células T CD4+ desde o valor basal continuaram a aumentar com o tempo (isto é, 90 células/mm3 na Semana 24, 139 células/mm3 na Semana 48 e 205 células/mm3 na Semana 96). Com base em uma subanálise na Coorte Randomizada, os indivíduos nas menores contagens basais de células T CD4+ ( < 20 células/mm3) apresentaram um aumento similar na contagem de CD4+ com o tempo em comparação a indivíduos com contagem basal mais elevada de células T CD4+ ( > 50, > 100, > 200 células/mm3).
Na Coorte não Randomizada (indivíduos sem agentes antirretrovirais totalmente ativos e disponíveis no recrutamento), RNA do HIV-1 < 40 cópias/mL foi atingido em 37%, 38% e 37% dos indivíduos nas Semanas 24, 48 e 96, respectivamente. Nestes períodos, a proporção de indivíduos com RNA do HIV-1 < 200 cópias/mL foi de 42%, 43% e 39%, e a proporção de indivíduos com RNA do HIV-1 < 400 cópias/mL foi de 44%, 44% e 40%, respectivamente (ITT-E, algoritmo Snapshot). As alterações médias na contagem de células CD4+ desde o valor basal aumentaram com o tempo: 41 células/mm3 na Semana 24, 64 células/mm3 na Semana 48 e 119 células/mm3 na Semana 96.
Referências Bibliográficas:
1. Kozal M, Aberg J, Pialoux G, Cahn P, Thompson M, Molina JM, Grinsztejn B, Diaz R, Castagna A, Kumar P, Latiff G, DeJesus E, Gummel M, Gartland M, Pierce A, Ackerman P, Llamoso C, Lataillade M, for the BRIGHTE Trial Team. Fostemsavir in adults with multidrug-resistant HIV-1 infection. N Engl J Med. 2020;382(13):1232-1243.
2. Lataillade M, Lalezari JP, Kozal M, Aberg JA, Pialoux G, Cahn P, Thompson M, Molina J-M, Moreno S, Grinsztejn B, Diaz RS, Castagna A, Kumar PN, Latiff GH, De Jesus E, Wang M, Chabria S, Gartland M, Pierce A, Ackerman P, Llamoso C. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced individuals: week 96 results of the phase 3 BRIGHTE study. Lancet HIV. 2020;7(11):E740-E751.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Código ATC
Grupo farmacoterapêutico: antiviral para uso sistêmico, outros antivirais.
Código ATC: J05AX29.
Mecanismo de ação
O fostensavir é um pró-fármaco sem atividade bioquímica significativa ou antiviral, hidrolisado no componente ativo, tensavir, mediante clivagem de um grupo fosfonooximetil in vivo. O tensavir se liga diretamente à subunidade gp120 na glicoproteína gp160 do envelope de HIV-1 e inibe seletivamente a interação entre o vírus e receptores celulares CD4, prevenindo assim a entrada do vírus e infecção das células hospedeiras. O tensavir inibiu a ligação de CD4 solúvel a gp120 imobilizada de superfície com uma IC50 de 14 a 30 nM com uso de um ensaio de imunoabsorção enzimática (ELISA).
Efeitos farmacodinâmicos
Atividade antiviral em cultura celular
O tensavir apresentou potente atividade antiviral contra oito de nove cepas laboratoriais do HIV-1 subtipo B CCR5, CXCR4 e trópico duplo, com valores de EC50 que variam de 0,4 a 58 nM. Apenas a cepa RF de HIV-1 CXCR4-trópica apresentou redução da suscetibilidade ao tensavir (EC50 > 2.000 nM).
Em um estudo, um total de 103 isolados clínicos foi examinado quanto à suscetibilidade ao tensavir com uso de PBMCs como a célula hospedeira. Estes vírus abrangeram vários subtipos do Grupo M, incluindo A, B, B', C, D, F, CRF01_AE e G. Além disso, 2 vírus do Grupo O e 1 HIV-2 foram testados quanto à suscetibilidade ao tensavir. A coorte conteve principalmente vírus CCR5 trópicos, porém também houve algumas cepas CXCR4 trópicas e de tropismo duplo. Para a maioria dos subtipos, tensavir apresentou atividade potente, porém variável, independentemente do tropismo.
Contudo, todos os nove vírus examinados a partir do subtipo CRF01_AE, ambos os vírus examinados a partir do Grupo O e um HIV-2 apresentaram redução da suscetibilidade ao tensavir na concentração mais elevada testada.
Em outro estudo, um total de 1337 isolados foi examinado até o momento no Ensaio de Entrada PhenoSense. Estes incluem vírus de todos os indivíduos nos estudos de fase IIa (206267), fase IIb (205889) e fase III (205888), bem como outras amostras obtidas de plasma de indivíduos infectados. Um total de 881 destas amostras foi de indivíduos infectados pelo subtipo B, 156 amostras do subtipo C, 43 amostras do subtipo A, 17 amostras do subtipo A1, 48 amostras do subtipo F1, 29 amostras do subtipo BF1 e 19 amostras do subtipos BF. Além disso, houve 5 amostras CRF01_AE: quatro destas amostras apresentaram valores de IC50 acima da concentração máxima dos ensaios utilizados (100 nM ou 5 mM), ao passo que uma amostra apresentou uma IC50 de ~223 nM. De todos os isolados testados, 53,8% e 80,1% apresentaram IC50s < 1 nM e < 10 nM, respectivamente, para todos os subtipos. Cada um dos subtipos apresentou variações amplas de suscetibilidade ao tensavir. Para os vírus do subtipo B, as IC50s variaram de pM baixo a > 10 mM. Os demais subtipos apresentaram variações similares. As médias geométricas de IC50s variaram de 1,15 nM para vírus subtipo B a 34,91 nM para o subtipo BF1. Estes resultados demonstram que há uma grande variação de suscetibilidade intrínseca ao tensavir em envelopes pré-tratamento na população.
Atividade antiviral em combinação com outros agentes antivirais
Nenhum fármaco com atividade anti-HIV inerente teve ação antagonista do tensavir (as avaliações in vitro foram realizadas em combinação com abacavir, didanosina, zalcitabina, entricitabina, lamivudina, estavudina, fumarato de tenofovir desoproxila, zidovudina, efavirenz, nevirapina, amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, enfuvirtida, maraviroque, ibalizumabe, delavirdina, rilpivirina, darunavir, dolutegravir ou raltegravir). Além disso, antivirais sem atividade anti-HIV inerente (entecavir, ribavirina) não tiveram efeito aparente sobre a atividade de tensavir.
Efeito do soro humano e das proteínas séricas
Estudos in vitro não apresentaram efeito significativo do soro. A infecção de células PM1 ou MT-2 com cepas laboratoriais de HIV-1 LAI e HIV-1 NL4-3 demonstrou que a presença de 40% de soro humano reduziu a potência anti-HIV de tensavir em 1,5-2,1 vezes.
Resistência in vitro
Variantes de HIV-1 com suscetibilidade reduzida ao tensavir foram selecionadas em cultura celular após passagem dos vírus NL4-3, LAI e BaL em uma linhagem de células T. Foram identificados aminoácidos emergentes em gp120 que reduziram a suscetibilidade e incluíram L116P/Q, A204D, M426L, M434I e M475I (substituições S375I/N foram identificadas com base em dados in vivo com um inibidor de ligação relacionado).
Vírus recombinantes de substituição única foram modificados na base viral do HIV-1 LAI, e recombinantes resultantes foram examinados em relação ao tensavir. A substituição de M426L foi associada a uma redução de 81 vezes na sensibilidade ao tensavir para o vírus recombinante, enquanto uma alteração de M434I ou M475I apresentou um efeito moderado na sensibilidade ao inibidor (redução de 11 e 4,8 vezes, respectivamente). Duas outras substituições de aminoácido, L116P e A204D, localizadas distalmente ao bolso de ligação CD4 da gp120, conferiram níveis elevados de resistência ao tensavir em uma base de LAI (redução de > 340 vezes). Contudo, ambos os aminoácidos são rigorosamente preservados no gene do envelope clínico, e estes polimorfismos específicos nestas posições não foram observados clinicamente durante o tratamento com fostensavir.
O tensavir permaneceu ativo contra vírus CD4-independentes derivados de laboratório. O tratamento com fostensavir, portanto, provavelmente não promoverá resistência ao tensavir via geração de vírus independente de CD4.
Não houve evidência de resistência cruzada a agentes representativos de outras classes de ARVs, incluindo ITRNs (inibidores da transcriptase reversa análogos de nucleosídeos), ITRNNs (inibidores da transcriptase reversa não análogos de nucleosídeos), IPs (inibidores da protease) e INSTIs (inibidores de integrase). O tensavir manteve atividade do tipo selvagem contra vírus resistentes a tenofovir, abacavir, zidovudina, lamivudina, rilpivirina, efavirenz, atazanavir, darunavir ou raltegravir. Além disso, ITRNs (abacavir, tenofovir), ITRNNs (rilpivirina, efavirenz), IPs (atazanavir, darunavir) e INSTI raltegravir, mantiveram atividade contra mutantes direcionados ao sítio com redução da suscetibilidade ao tensavir (S375M, M426L, ou M426L mais M475I).
Não foi observada resistência cruzada entre tensavir e maraviroque ou enfuvirtida. O tensavir foi ativo contra vírus com resistência à enfuvirtida. Alguns vírus com tropismo CCR5 e resistentes ao maraviroque apresentaram suscetibilidade reduzida ao tensavir; no entanto, não houve correlação absoluta entre a resistência ao maraviroque e a sensibilidade reduzida ao tensavir. O maraviroque e a enfuvirtida mantiveram a atividade contra envelopes clínicos que tinham suscetibilidade reduzida ao tensavir e continham substituições S375H, M426L ou M426L mais M475I.
O tensavir foi ativo contra vários vírus resistentes ao ibalizumabe. O ibalizumabe manteve atividade contra mutantes sítio-dirigidos que tinham suscetibilidade reduzida ao tensavir (S375M, M426L ou M426L mais M475I). A gp120 E202 do HIV-1 foi identificada como uma substituição rara emergente do tratamento no BRIGHTE que pode reduzir a suscetibilidade ao tensavir e, dependendo do contexto da sequência do envelope, também pode resultar numa suscetibilidade reduzida ao ibalizumabe.
Resistência in vivo
Os resultados do estudo de fase III (BRIGHTE [205888]) em indivíduos adultos altamente experimentados ao tratamento demonstraram que, de modo geral, a resposta virológica no Dia 8 e períodos subsequentes (Semanas 24, 48 e 96) na Coorte Randomizada não foi um valor de fold-change (FC) da IC50 basal do tensavir ou presença de uma substituição da gp160 de interesse previstos de modo confiável.
A FC IC50 do tensavir > 100 vezes foi associada a uma alteração mediana no RNA do HIV-1 nos Dias 1 a 8 de < 0,5 log10 c/mL. De modo semelhante, a presença de substituições predefinidas da gp160 na visita basal, identificadas como possivelmente importantes para determinação de suscetibilidade fenotípica ao tensavir (S375H/I/M/N/T, M426L/P, M434I/K e M475I), foi associada a uma menor redução no RNA do HIV-1. No entanto, o aumento da FC IC50 basal de tensavir ou a presença de substituições predefinidas da gp160 não impediu os indivíduos de atingir uma resposta de > 1 log10 c/mL no Dia 8. Na realidade, 8 de 21 (38%) indivíduos com FC IC50 > 100 vezes não atingiram uma resposta no Dia 8 > 0,5 log10 c/mL e 7/21 (33%) indivíduos atingiram uma redução de > 1 log10 c/mL na carga viral. Indivíduos sem substituições predefinidas da gp160 presentes na visita basal atingiram uma alteração mediana no RNA do HIV-1 de -1,032 log10 c/mL no Dia 8, em comparação a uma alteração de -0,652 log10 c/mL na carga viral em indivíduos com substituições predefinidas da gp160 presentes. As substituições basais da gp160 mais associadas à resposta < 0,5 log10 c/mL no Dia 8 foram S375H/M/N e M426L.
Com a adição de uma terapia de base otimizada, o aumento da FC IC50 basal de tensavir ou a presença de substituições predefinidas da gp160 não influenciaram a durabilidade de resposta (RNA do HIV-1 < 40 c/mL) até a Semana 96. Estes resultados demonstram que a resposta a fostensavir, conforme determinada por fatores virológicos basais, depende altamente do contexto.
A porcentagem de indivíduos randomizados que apresentaram falha virológica definida pelo protocolo (PDVF) foi de 11% (31/272) até a Semana 24, 18% (49/272) até a Semana 48 e 23% (63/272) até a Semana 96. Os critérios para PDVF foram os seguintes: RNA do HIV-1 confirmado ou último disponível antes da descontinuação, ≥400 c/mL em qualquer momento após supressão anteriormente confirmada para < 400 c/mL, ou aumento confirmado ou último disponível de > 1 log10 c/mL no RNA do HIV-1 a qualquer momento acima do nível nadir, onde o nadir é ≥40 c/mL (antes da Semana 24); RNA do HIV-1 confirmado ou último disponível ≥400 c/mL (na Semana 24 ou posteriormente). Na população PDVF até a Semana 96, 48% (24/50) dos indivíduos randomizados avaliáveis apresentaram substituições genotípicas da gp160 de interesse decorrentes do tratamento. Mais frequentemente, houve surgimento de M426L (32%), S375N (24%), M475I (12%) e M434I (10%).
O aumento mediano na FC IC50 do tensavir entre indivíduos randomizados que atenderam aos critérios PDVF foi de 1,67 vez; 37% (19/51) dos indivíduos avaliáveis apresentaram FC IC50 do tensavir ≤10 vezes e 49% (25/51) apresentaram FC IC50 do tensavir ≤100 vezes no momento da falha virológica, indicando que uma proporção de indivíduos provavelmente manteve suscetibilidade fenotípica ao tensavir, apesar de atender aos critérios de falha virológica (FC IC50 do tensavir é normalizada a um vírus de referência de 1 nM e, portanto, é aproximadamente igual à IC50 de tensavir expressa em nM). Aproximadamente 30% (17/63) dos indivíduos randomizados que atenderam a PDVF foram subsequentemente capazes de atingir supressão virológica para < 40 c/mL.
Houve apenas dois indivíduos com vírus subtipo AE na triagem na Coorte Randomizada. Um indivíduo (FC IC50 > 4747 vezes e substituições da gp160 em S375H e M475I na visita basal) não respondeu ao fostensavir no Dia 8. Um segundo indivíduo (FC IC50 de 298 vezes e substituição da gp160 em S375N na visita basal) recebeu placebo durante a monoterapia funcional. Ambos os indivíduos foram suprimidos virologicamente na Semana 96, enquanto recebiam fostensavir mais terapia de base otimizada.
Efeitos no eletrocardiograma
Em um estudo duplo-cego, cruzado (cross-over) para QT, randomizado, controlado por placebo e ativo, 60 indivíduos saudáveis receberam administração oral de placebo, fostensavir 1200 mg uma vez ao dia, Fostensavir 2400 mg duas vezes ao dia e moxifloxacino 400 mg (controle ativo) em sequência randomizada. O fostensavir administrado na dose de 1200 mg uma vez ao dia não causou um efeito clinicamente significativo no intervalo QTc, uma vez que a alteração máxima média de correspondência temporal (limite de confiança superior bicaudal de 90%) de QTc ajustada ao placebo desde o valor basal com base no método de correção de Fridericia (QTcF) foi de 4,3 (6,3) milissegundos (abaixo do limiar clinicamente importante de 10 milissegundos). Contudo, fostensavir administrado na dose de 2400 mg duas vezes ao dia por 7 dias foi associado a um prolongamento clinicamente significativo do intervalo QTc, uma vez que a alteração média máxima de correspondência temporal desde o valor basal (limite de confiança superior bicaudal de 90%) ajustada por placebo no intervalo QTcF foi de 11,2 (13,3) milissegundos. A administração de fostensavir 600 mg duas vezes ao dia em estado de equilíbrio resultou em uma Cmáx média de tensavir aproximadamente 4,2 vezes menor que a concentração de tensavir prevista para aumentar o intervalo QTcF em 10 milissegundos (ver Advertências e Precauções).
Propriedades Farmacocinéticas
A farmacocinética de tensavir após administração de fostensavir é semelhante em indivíduos saudáveis e vivendo com HIV. A variabilidade entre indivíduos (%CV) na Cmáx e AUC plasmáticas do tensavir variou de 22 a 50% e Ct de 50 a 127% entre os estudos de fase I em indivíduos saudáveis. A magnitude de variabilidade foi similar em indivíduos vivendo com HIV (%CV na Cmáx e AUC plasmáticas de tensavir variou de 20,5 a 63% e a Ct de 20 a 165%). A variabilidade entre indivíduos na depuração oral e volume de distribuição oral central calculados a partir de análise de farmacocinética populacional de indivíduos saudáveis de estudos selecionados de fase I e pacientes vivendo com HIV-1 foi de 43% e 48%, respectivamente.
Absorção
O fostensavir é um pró-fármaco altamente solúvel metabolizado em tensavir pela fosfatase alcalina na superfície luminal do intestino delgado, e geralmente não é detectável no plasma após administração oral. O componente ativo, tensavir, é prontamente absorvido com o tempo mediano até concentrações plasmáticas máximas (Tmáx) em 2 horas após a dose (em jejum). Após a administração oral, os aumentos na exposição plasmática ao tensavir (Cmáx e AUC) pareceram ser proporcionais à dose ou discretamente acima do proporcional à dose, na variação de 600 mg a 1.800 mg de fostensavir. O tensavir é absorvido pelo intestino delgado e ceco/cólon ascendente proximal.
Os parâmetros farmacocinéticos após doses múltiplas orais de fostensavir 600 mg duas vezes ao dia e indivíduos saudáveis e indivíduos adultos vivendo com HIV-1 e altamente experimentados são ilustrados na Tabela 3.
A biodisponibilidade absoluta de tensavir foi de 26,9% após administração oral de uma dose única de 600 mg de fostensavir.
Efeito de Alimentos
O fostensavir pode ser administrado com ou sem alimentos. A biodisponibilidade de tensavir (AUC) não foi afetada por uma refeição padrão (aproximadamente 423 kcal, 36% de gordura), porém aumentou 81% com uma refeição hiperlipídica (aproximadamente 985 kcal, 60% de gordura) e não é considerada clinicamente significativa. Independentemente do teor calórico e lipídico, o alimento não afetou a Cmáx plasmática de tensavir.
Distribuição
De acordo com dados in vivo, o tensavir liga-se (cerca de 88%) às proteínas plasmáticas humanas. Albumina sérica humana é o principal contribuinte para a ligação às proteínas plasmáticas de tensavir em humanos. O volume de distribuição de tensavir em estado de equilíbrio (Vss) após administração intravenosa é calculado em 29,5 L. A proporção sangue-plasma de Cmáx total de radiocarbono foi de aproximadamente 0,74, indicando associação mínima de tensavir ou seus metabólitos aos eritrócitos. Ex vivo, a proporção sangue-plasma (determinada em 300, 1000 e 10.000 nanogramas/mL) variou de 0,785 a 0,963 [média (DP) geral de 0,869 ± 0,0599] sem dependência aparente da concentração na variação de concentração testada.
A fração livre de tensavir no plasma foi de aproximadamente 12 a 18% em indivíduos saudáveis, 23% em indivíduos com comprometimento hepático grave e 19% em indivíduos com comprometimento renal grave, e 12% em pacientes vivendo com HIV-1.
Metabolismo
In vivo, tensavir é metabolizado primariamente via hidrólise de esterase (36,1% da dose administrada) e secundariamente por vias oxidantes mediadas por CYP3A4 (21,2% da dose administrada). Outros metabólitos diferentes de CYP3A4 totalizam 7,2% da dose administrada. Glicuronidação é uma via metabólica menor ( < 1% da dose administrada).
O tensavir é extensamente metabolizado, representando o fato de que apenas 3% da dose administrada são recuperados na urina e fezes humanas. O tensavir é biotransformado em dois metabólitos inativos circulantes predominantes, BMS-646915 (um produto de hidrólise) e BMS-930644 (um produto de N-dealquilação). Fármacos indutores potentes da CYP3A são contraindicados com fostensavir (ver Contraindicações e Interações Medicamentosas).
Eliminação
O tensavir tem uma meia-vida terminal de aproximadamente 11 horas. A depuração plasmática do tensavir após administração intravenosa foi de 17,9 L/h, e a depuração aparente (CL/F) após administração oral foi de 66,4 L/h. Após a administração oral de uma dose única de 300 mg de 14C-fostensavir em um estudo de balanço de massa em humanos, 51% e 33% da radioatividade foram recuperados na urina e fezes, respectivamente. Com base na limitada coleta de bile neste estudo (3 a 8 horas após a dose), a eliminação biliar representou 5% da dose radioativa, sugerindo que uma fração de excreção fecal é de excreção biliar.
Populações especiais de pacientes
Crianças
A farmacocinética de tensavir não foi avaliada em crianças menores de 18 anos.
Idosos
A análise de farmacocinética populacional de tensavir com uso de dados em adultos vivendo com HIV-1 demonstrou que a idade não teve efeito clinicamente relevante na exposição ao tensavir. Os dados farmacocinéticos para tensavir em indivíduos de 65 anos ou mais são limitados. Pacientes idosos poderão ser mais suscetíveis ao prolongamento do intervalo QT induzido pela droga (ver Advertências e Precauções).
Disfunção renal
Não é necessário ajuste de dose de fostensavir em pacientes com disfunção renal leve, moderada ou grave e para pacientes com nefropatia em estágio terminal (ESRD). O efeito do comprometimento renal na exposição ao tensavir após uma dose única de 600 mg de fostensavir foi avaliado em um estudo aberto em 30 indivíduos adultos com função renal normal, comprometimento renal leve, moderado e grave e indivíduos com ESRD em hemodiálise (n=6 por grupo). A classificação de função renal se baseou na taxa de filtração glomerular estimada (TFGe), como segue: 60 ≤ TFGe ≤89 (leve), 30 ≤ TFGe < 60 (moderada), TFGe < 30 (grave) e ESRD em hemodiálise) mL/min/1,73 m2. Não houve efeito do comprometimento renal nos parâmetros de exposição farmacocinética (Cmáx e AUCs) de tensavir (total e não ligada) com base na TFGe e depuração de creatinina (CLcr). O fostensavir poderá ser administrado a pacientes com ESRD sem relação ao momento de hemodiálise, uma vez que tensavir não foi prontamente eliminado por hemodiálise, com aproximadamente 12,3% da dose administrada removidos durante a sessão de 4 horas de hemodiálise. A hemodiálise iniciada 4 horas após a administração de tensavir foi associada a um aumento médio de 46% na Cmáx plasmática total de tensavir e redução média de 11% na AUC em relação à farmacocinética sem hemodiálise.
Disfunção hepática
Não é necessário ajuste de dose de fostensavir em pacientes com disfunção hepática leve, moderada e grave. O efeito do comprometimento hepático na exposição a tensavir após uma dose única de 600 mg de fostensavir foi avaliado em um estudo aberto em 30 indivíduos adultos com função hepática normal (n=12), comprometimento hepático leve (classe A de Child-Pugh, n=6), moderado (classe B de Child-Pugh, n=6) e grave (classe C de Child-Pugh, n=6). As exposições total e não ligada a tensavir aumentaram conforme o aumento da gravidade de comprometimento hepático classificado por classes de Child-Pugh. Em pacientes com comprometimento hepático leve a grave, o aumento da exposição a Cmáx e AUC não ligadas e totais foi de 1,2 a 2,2 vezes; contudo, os limites superiores do IC de 90% bicaudal para o impacto do comprometimento hepático na Cmáx plasmática total e não ligada de tensavir são menores que o limiar de Cmáx estabelecidos com base na exposição-resposta a tensavir (ver Efeitos no Eletrocardiograma, acima).
Gênero
As análises farmacocinéticas populacionais não indicaram efeito clinicamente relevante do gênero na exposição ao tensavir. Dos 764 indivíduos incluídos na análise, 216 (28%) eram do gênero feminino.
Raça
As análises farmacocinéticas populacionais não indicaram efeito clinicamente relevante da raça na exposição ao tensavir. Dos 764 indivíduos incluídos na análise, 490 (64%) eram brancos, 177 (23%) negros/afrodescendentes, 5 (1%) asiáticos e 92 (12%) de outra raça.
Coinfecção por hepatite B ou C
Os dados farmacocinéticos para tensavir em pacientes com coinfecção por vírus de hepatite B e/ou C são limitados.
4. CONTRAINDICAÇÕES
Rukobia é contraindicado para pacientes que demonstraram hipersensibilidade a fostensavir ou a quaisquer componentes da formulação do produto.
Rukobia é contraindicado em combinação com fortes indutores potentes da CYP3A4, incluindo, mas não se limitando a carbamazepina, fenitoína (anticonvulsivantes), mitotano (antineoplásico), enzalutamida (inibidor de receptor de andrógeno), rifampicina (antimicobacteriano) e Erva de São João (Hypericum perforatum, suplemento fitoterápico), já que podem ocorrer diminuições significativas nas concentrações plasmáticas de temsavir (porção ativa do fostemsavir) o que pode resultar na perda de resposta virológica. (ver Interações Medicamentosas).
5. ADVERTÊNCIAS E PRECAUÇÕES
Síndrome de Reconstituição Imune
Pacientes vivendo com HIV que têm imunodeficiência acentuada por ocasião do início da terapia antirretroviral (TARV) podem apresentar uma reação inflamatória a infecções oportunistas assintomáticas ou residuais, que causa distúrbios clínicos graves ou agravamento dos sintomas. Em geral, essas reações foram observadas nas primeiras semanas ou meses após o início da TARV. Alguns exemplos pertinentes são a retinite por citomegalovírus, as infecções micobacterianas generalizadas e/ou focais e a pneumonia por Pneumocystis jiroveci (P. carinii). É preciso avaliar sem demora quaisquer sintomas inflamatórios e iniciar o tratamento, quando necessário. Também foi relatada a ocorrência de distúrbios autoimunes (como doença de Graves, polimiosite e síndrome de Guillain-Barré) nos casos de reconstituição imune, porém o tempo até que se iniciem é mais variável e esses distúrbios podem ocorrer muitos meses depois de iniciado o tratamento e, às vezes, têm apresentação atípica.
Prolongamento do Intervalo QTc
Em participantes saudáveis do estudo, uma dose supraterapêutica de Rukobia (2400 mg duas vezes ao dia) demonstrou prolongar substancialmente o intervalo QTc do eletrocardiograma (ver Características Farmacológicas). Rukobia deverá ser utilizado com cautela em pacientes com um histórico de prolongamento do intervalo QT, quando administrado concomitantemente com um medicamento com risco conhecido de Torsade de Pointes (por exemplo, amiodarona, disopiramida, dofetilida, ibutilida, procainamida, quinidina ou sotalol) ou em pacientes com cardiopatia preexistente relevante. Pacientes idosos poderão ser mais suscetíveis ao prolongamento do intervalo QT induzido por medicamento.
Pacientes coinfectados pelo vírus da Hepatite B ou C
O monitoramento da bioquímica hepática é recomendado em pacientes com coinfecção por hepatite B e/ou C. Deve ser aplicada especial diligência em iniciar ou manter uma terapia eficaz para hepatite B (consultando as diretrizes de tratamento) quando se inicia uma terapia com Rukobia em pacientes coinfectados pelo vírus da hepatite B.
Elevações nas transaminases hepáticas foram observadas em uma proporção maior de indivíduos com coinfecção pelo vírus da hepatite B e/ou C comparado com aqueles com monoinfecção por HIV. Algumas dessas elevações nas transaminases foram consistentes com a reativação da hepatite B, particularmente no ambiente onde a terapia com anti-hepatite foi suspensa.
Infecções oportunistas
Pacientes tratados com Rukobia ou qualquer outra terapia antirretroviral ainda podem ter infecções oportunistas e outras complicações da infecção por HIV. Portanto, os pacientes devem permanecer sob rigorosa observação clínica por médicos com experiência no tratamento dessas doenças associadas ao HIV.
Interações Medicamentosas
É necessário ter cuidado com a coadministração de medicamentos (prescritos ou de venda livre) que possam modificar a exposição ao tensavir, componente ativo de fostensavir, ou que possam ter sua exposição modificada pelo tensavir (ver Contraindicações e Interações Medicamentosas). O aumento da exposição ao tensavir poderá aumentar o risco de prolongamento do Intervalo QTc (ver Prolongamento do Intervalo QTc, acima e Características Farmacológicas), bem como a redução da exposição ao temsavir pode levar à perda do efeito terapêutico de Rukobia e possível desenvolvimento de resistência.
A coadministração de Rukobia com elbasvir/grazoprevir não é recomendada, uma vez que as concentrações aumentadas de grazoprevir podem aumentar o risco de elevações de ALT (ver Interações Medicamentosas).
São recomendadas modificações de dose e/ou titulação cuidadosa para determinadas estatinas que são substratos de OATP1B1/3 ou BCRP (rosuvastatina, atorvastatina, pitavastatina, sinvastatina e fluvastatina) quando coadministradas com Rukobia (ver Interações Medicamentosas).
Quando Rukobia foi coadministrado com contraceptivos orais, o tensavir aumentou as concentrações de etinilestradiol, e recomenda-se cautela particularmente em pacientes com fatores de risco adicionais para eventos tromboembólicos. Doses de terapias à base de estrógeno, incluindo contraceptivos orais, não deverão conter mais de 30 mg de etinilestradiol ao dia em pacientes que estão recebendo Rukobia (ver Interações Medicamentosas).
Gravidez e Lactação
Fertilidade
Não há dados sobre os efeitos de Rukobia na fertilidade masculina nem feminina. Estudos em animais não indicam efeitos de fostensavir na fertilidade masculina ou feminina em doses clinicamente relevantes (ver Informações Não Clínicas).
Gravidez
Não há estudos adequados e bem controlados de fostensavir em mulheres grávidas. O efeito de Rukobia na gestação humana é desconhecido.
Estudos em animais não indicam efeitos de fostensavir no desenvolvimento embriofetal em exposições clinicamente relevantes. O fostensavir foi associado a achados de toxicidade do desenvolvimento em estudos de reprodução em animais em exposições substancialmente mais elevadas que a dose terapêutica na presença de toxicidade materna. O tensavir demonstrou atravessar a placenta em um estudo de distribuição em animais após administração de fostensavir radiomarcado (ver Informações Não Clínicas).
Rukobia deverá ser utilizado durante a gestação apenas se o benefício esperado justificar o possível risco ao feto.
Lactação
Os especialistas da área de saúde recomendam que, sempre que possível, as mulheres vivendo com HIV não amamentem seus bebês para evitar a transmissão do HIV. Em situações em que o uso de fórmulas infantis não é viável e o aleitamento materno durante o tratamento antirretroviral for considerado, devem ser seguidos os guias locais para amamentação e tratamento.
De acordo com dados obtidos em animais, presume-se que o tensavir seja secretado no leite humano, embora não haja confirmação disso em seres humanos. O fostensavir foi associado à redução da sobrevida de filhotes durante o período máximo de lactação em um estudo em animais em exposições substancialmente mais elevadas que a dose terapêutica (ver Informações Não Clínicas). Portanto, recomenda-se que, quando possível, mulheres vivendo com HIV não amamentem enquanto estiverem fazendo uso de Rukobia.
Categoria C de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não houve estudos para investigar o efeito de Rukobia sobre a capacidade de dirigir ou operar máquinas. É preciso levar em conta o estado clínico do paciente e o perfil de eventos adversos de Rukobia ao avaliar essa capacidade.
Informações Não-Clínicas
Carcinogênese/Mutagênese
Fostensavir e tensavir não foram mutagênicos nem clastogênicos com uso de testes in vitro em bactérias e cultura de células de mamíferos, e nem em teste do micronúcleo in vivo em ratos. O fostensavir não foi carcinogênico em estudos de longo prazo em camundongos e ratos após administração por gavagem oral por até 26 e 100 semanas, respectivamente.
Toxicologia Reprodutiva
Fertilidade
A administração oral de fostensavir não causou efeitos adversos na fertilidade masculina ou feminina em ratos em doses de até 300 mg/kg/dia em machos e 600 mg/kg/dia em fêmeas ( > 100 vezes a exposição clínica humana com base na AUC a 600 mg duas vezes ao dia). Os efeitos em machos incluíram achados patológicos microscópicos dose-dependentes nos testículos e epidídimo, reduções nos pesos da próstata/vesícula seminal e redução da densidade do esperma (em > 85 vezes a exposição clínica humana com base na AUC a 600 mg duas vezes ao dia), com redução da motilidade e aumento do esperma anormal (em > 95 vezes a exposição clínica humana com base na AUC a 600 mg duas vezes ao dia). Estes achados não foram considerados clinicamente relevantes.
Gravidez
Não foram observadas anormalidades fetais após administração oral de fostensavir a ratas prenhes durante organogênese na dose de 600 mg/kg/dia [ > 100 vezes a exposição prevista humana à dose máxima recomendada em humanos (MRHD)]. Não foram observados efeitos adversos na gestação, parto ou desenvolvimento fetal e inicial da prole quando fostensavir foi administrado em doses orais de até 300 mg/kg/dia até a gestação e lactação ( > 100 vezes a exposição humana na MRHD).
Contudo, a administração oral de fostensavir a ratas prenhes resultou em anormalidades fetais (fenda palatina, olhos abertos, focinho encurtado, microstomia, boca/mandíbula mal alinhada e protrusão da língua) e reduções nos pesos corporais fetais na presença de maternotoxicidade (reduções nos pesos corporais e consumo de alimento) quando administrado na dose de 1000 mg/kg/dia ( > 200 vezes a exposição prevista humana na MRHD).
Não foram evidentes efeitos adversos na sobrevida embrionária e pesos fetais após administração oral de fostensavir a coelhas prenhes durante a organogênese na dose de 50 mg/kg/dia ( > 30 vezes a exposição prevista humana na MRHD). As reduções nos pesos corporais fetais e mortes embrionárias foram evidentes em > 65 vezes a exposição na MRHD. Em doses iguais ou maiores que 250 mg/kg/dia ( > 100 vezes as exposições na MRHD), a administração oral de fostensavir a coelhas prenhes resultou em