REVOLADE
NOVARTIS
eltrombopague
Trat. de púrpura trombocitopênica idiopática.
Apresentações.
Comprimidos revestidos de 25 mg e 50 mg em cartuchos com 14 comprimidos.
VIA ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS (vide indicações)
Composição.
Cada comprimido revestido de Revolade® 25 mg contém 25 mg de eltrombopague como ácido livre equivalente a 31,9 mg de eltrombopague olamina1. Excipientes: celulose microcristalina, povidona k30, amidoglicolato de sódio, estearato de magnésio, manitol, hipromelose*, dióxido de titânio*, macrogol 400*, polissorbato 80*
Cada comprimido revestido de Revolade® 50 mg contém 50 mg de eltrombopague como ácido livre equivalente a 63,8 mg de eltrombopague olamina1. Excipientes: celulose microcristalina, povidona k30, amidoglicolato de sódio, estearato de magnésio, manitol, hipromelose*, dióxido de titânio*, macrogol 400*, óxido de ferro amarelo*, óxido de ferro vermelho*
1 - eltrombopague olamina é o sal bis-monoetanolamina de eltrombopague (ácido livre)
*Composição do revestimento
Informações técnicas.
1. INDICAÇÕES
Revolade® é um agonista do receptor de trombopoetina utilizado para o tratamento de plaquetopenia em pacientes adultos com púrpura trombocitopênica idiopática (PTI) de origem imune, os quais tiveram resposta insuficiente a corticosteroides, imunoglobulinas ou esplenectomia (retirada do baço).
Revolade® é utilizado em pacientes pediátricos acima de 6 anos com púrpura trombocitopênica idiopática (PTI) de origem imune, com duração de 6 meses ou mais desde o diagnóstico, os quais tiveram resposta insuficiente a corticosteroides, imunoglobulinas ou esplenectomia (retirada do baço).
Revolade® está indicado para pacientes com púrpura trombocitopênica idiopática que apresentam risco aumentado de sangramento e hemorragia. Revolade® não deve ser usado simplesmente para aumentar a contagem de plaquetas.
Revolade® é indicado em combinação com terapia imunossupressora padrão para o tratamento de primeira linha de pacientes adultos e pediátricos acima de 6 anos com Anemia Aplásica Severa (AAS).
Revolade® está indicado também para o tratamento de pacientes adultos com Anemia Aplásica Severa (AAS) adquirida que foram refratários à terapia imunossupressora prévia ou que foram extensamente tratados previamente e não sejam elegíveis ao transplante de células tronco hematopoiéticas.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
Estudos de Púrpura Trombocitopênica Idiopática (PTI) de origem imune
A eficácia de Revolade® foi demonstrada em dois estudos fase III randomizados, duplo-cegos, controlados com placebo (TRA102537 RAISE e TRA100773B) e em dois estudos abertos (REPEAT TRA108057 e EXTEND TRA105325) em pacientes adultos com PTI crônica previamente tratados. Todos os estudos tinham pacientes com características demográficas das populações de pacientes com PTI crônica, no que tange a raça, cor e sexo, com a maioria dos pacientes sendo brancos e aproximadamente dois terços sendo mulheres.
Estudos duplo-cegos controlados com placebo
TRA102537: No estudo RAISE, o desfecho primário de eficácia foi a probabilidade de atingir uma contagem plaquetária ≥50.000/microL e ≤400.000/microL durante o período de tratamento de seis meses em pacientes que receberam Revolade® em comparação a placebo. Cento e noventa e sete pacientes foram randomizados na proporção de 2:1 de Revolade® (n=135) para placebo (n=62) e estratificados com base no status de esplenectomia, uso de medicação para PTI na avaliação basal e contagem plaquetária basal. Os pacientes receberam a medicação do estudo por até seis meses, durante os quais a dose de Revolade® podia ser ajustada com base nas contagens plaquetárias individuais. Todos os pacientes iniciaram o tratamento com Revolade® 50 mg. Do dia 29 até o final do tratamento, 15 a 28% dos pacientes tratados com Revolade® foram mantidos com ≤ 25 mg e 29 a 53% receberam 75 mg. Além disso, os pacientes podiam ter as doses das medicações concomitantes para a PTI reduzidas e receber tratamento de resgate, conforme as recomendações ditadas pelos padrões locais de cuidados médicos (standard of care). As contagens plaquetárias medianas basais foram 16.000/microL para ambos os grupos.
A probabilidade de atingir uma contagem plaquetária entre 50.000/microL e 400.000/microL durante o período de tratamento de seis meses foi oito vezes mais alta nos pacientes tratados com Revolade® do que nos que receberam placebo (odds ratio [OR] = 8,2; IC de 99%: 3,59-18,73; p < 0,001). As contagens plaquetárias medianas foram mantidas em mais de 50.000/microL em todas as visitas durante o tratamento, a começar pelo dia 15, no grupo de Revolade®. Em contraste, no grupo de placebo elas permaneceram abaixo de 30.000/microL ao longo do estudo.
Na avaliação basal, 77% dos pacientes do grupo de placebo e 73% dos pacientes do grupo de Revolade® relataram qualquer sangramento (de graus 1-4 da OMS). Sangramento clinicamente significativo (de graus 2-4 da OMS) na avaliação basal foi relatado em 28% e 22% dos pacientes nos grupos de placebo e Revolade®, respectivamente. A proporção de pacientes com qualquer sangramento (de graus 1-4) e sangramento clinicamente significativo (de graus 2-4) diminuiu em relação à avaliação basal em aproximadamente 50% ao longo de todo o período de tratamento de seis meses em pacientes que receberam Revolade®. Na comparação com o grupo de placebo, a probabilidade de qualquer sangramento (de graus 1-4) e a de sangramento clinicamente significativo (de graus 2-4) foram, respectivamente, 76% e 65% mais baixas em pacientes tratados com Revolade® em relação àqueles que receberam placebo (p < 0,001).
O tratamento com Revolade® permitiu que um número significativamente maior de pacientes reduzisse ou descontinuasse os tratamentos basais de PTI, em comparação com placebo (59% vs 32%; p < 0,016).
Um número significativamente menor de pacientes tratados com Revolade® necessitou tratamento de resgate, em comparação com pacientes que receberam placebo [18% vs 40%; p=0,001].
Quatro pacientes que receberam placebo e 14 tratados com Revolade® tiveram pelo menos um estímulo hemostático (definido como um procedimento invasivo de diagnóstico ou cirúrgico) durante o estudo. Um número menor de pacientes tratados com Revolade® (29%) necessitou tratamento de resgate para controlar o estímulo hemostático, em comparação com pacientes que receberam placebo (50%).
Em termos de melhorias da qualidade de vida relacionada à saúde, observaram-se melhoras significativas, em relação à avaliação basal, no grupo tratado com Revolade® para fadiga, incluindo o grau de impacto e severidade nas atividades diárias relacionadas à trombocitopenia [medidos pela subescala de vitalidade do SF36, o inventário de motivação e energia, e pelo extrato de seis itens da subescala de trombocitopenia do FACIT-Th]. Comparando-se o grupo de Revolade® com o de placebo, melhoras estatisticamente significativas foram observadas nas atividades diárias relacionadas à trombocitopenia, especificamente no que se refere à motivação, à energia e à fadiga, bem como às funções físicas e emocionais e à saúde mental de modo geral. A probabilidade de melhora na qualidade de vida relacionada à saúde durante o tratamento foi significativamente maior entre pacientes tratados com Revolade® do que com placebo.
TRA100773B: Nesse estudo, o desfecho primário de eficácia foi a proporção de respondedores, definidos como pacientes que tiveram um aumento das contagens plaquetárias ≥50.000/microL no Dia 43 em relação a um valor basal < 30.000/microL. Os pacientes que se retiraram prematuramente do estudo devido a uma contagem plaquetária > 200.000/microL foram considerados respondedores; aqueles que descontinuaram o tratamento por qualquer outra razão foram considerados não respondedores, independente da contagem plaquetária. Cento e catorze pacientes com PTI crônica previamente tratados foram randomizados na proporção de 2:1, 76 para Revolade® e 38 para placebo. Todos os pacientes iniciaram o tratamento com Revolade® 50 mg. Do dia 22 até o final do tratamento, 6 a 39% receberam até 75 mg. As contagens plaquetárias medianas basais foram 18.000/microL para o grupo tratado com eltrombopague e 17.000/microL para o grupo tratado com placebo, respectivamente.
Cinquenta e nove por cento dos pacientes tratados com Revolade® responderam ao tratamento, em comparação com 16% dos que receberam placebo. A probabilidade de resposta foi 9 vezes mais alta com Revolade® do que com placebo (OR=9,6; IC de 95%: 3,31-27,86; p < 0,001). Na avaliação basal, 61% dos pacientes do grupo de Revolade® e 66% dos pacientes do grupo de placebo relataram qualquer sangramento (de graus 1-4). No Dia 43, 39% dos pacientes do grupo tratado com Revolade® apresentaram sangramento, em comparação com 60% dos que receberam placebo. A análise durante o período de tratamento, usando-se um modelo de medições repetidas para dados binários, confirmou que uma proporção mais baixa de pacientes tratados com Revolade® teve sangramento (de graus 1-4) em qualquer ponto do tempo ao longo do tratamento (do Dia 8 até o Dia 43), em comparação com pacientes do grupo de placebo (OR=0,49; IC de 95%: 0,26-0,89; p=0,021). Dois pacientes que receberam placebo e um tratado com Revolade® tiveram pelo menos um estímulo hemostático durante o estudo.
Nos estudos RAISE e TRA100773B, independentemente do uso de medicação para PTI, do status de esplenectomia e da contagem plaquetária basal (≤15.000/microL; > 15.000/microL) na ocasião da randomização, a resposta a Revolade® foi similar à observada com placebo. Nos estudos RAISE e TRA100773B, no subgrupo de pacientes PTI com contagem plaquetária basal ≤15.000/microL, a contagem plaquetária mediana não atingiu o nível alvo ( > 50.000/microL), apesar de em ambos os estudos, 43 % desses pacientes tratados com Revolade® responderam após 6 semanas de tratamento. Adicionalmente, no estudo RAISE, 42% dos pacientes com contagem plaquetária basal ≤ 15.000/microL tratados com Revolade® responderam ao final do período de 6 meses de tratamento. Quarenta e dois a 60 % dos pacientes tratados com Revolade® no estudo RAISE estavam recebendo 75 mg do Dia 29 até o final do tratamento.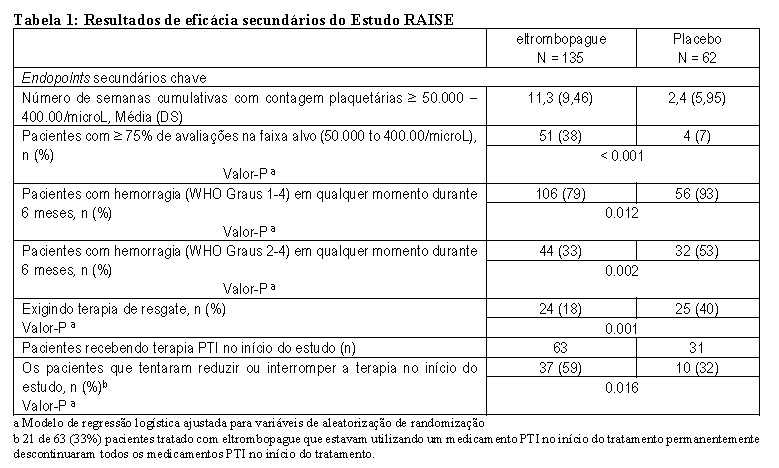

Estudos Abertos
TRA108057: O REPEAT foi um estudo aberto com doses repetidas que avaliou a eficácia, a segurança e a consistência da resposta após a administração repetida, intermitente e de curta duração de Revolade® durante três ciclos de tratamento de adultos com PTI crônica previamente tratados. O ciclo foi definido como um período de até seis semanas de tratamento, seguido de um período de quatro semanas sem tratamento. A duração dos períodos de terapia e pós terapia foi definida pela contagem de plaquetas do paciente. Os pacientes deveriam interromper o tratamento para que o ciclo atingisse uma contagem de plaquetas > 200/microL, ou quando atingiram a 6ª semana. Os pacientes começaram o próximo ciclo quando a contagem de plaquestas caiu abaixo de 20/microL, ou quando atingiu a 4ª semana de pós terapia. O ponto de corte primário nesse estudo foi a proporção de pacientes que atingiram uma contagem plaquetária ≥50.000/microL e pelo menos duas vezes o valor basal nos Ciclos 2 ou 3, considerando-se essa resposta no Ciclo 1.
Dos 52 pacientes que responderam ao tratamento no Ciclo 1, 33 (63%) atingiram contagem plaquetária ≥50.000/microL e pelo menos duas vezes o valor basal no Dia 8 do Ciclo 1. No Dia 15, 37 dos 47 pacientes avaliáveis (79%) atingiram esse nível de resposta.
Demonstrou-se redução de qualquer sangramento (de graus 1-4 da OMS) e do sangramento clinicamente significativo (de graus 2-4 da OMS) durante as fases de tratamento, em cada ciclo. Na visita basal do Ciclo 1, 50% dos pacientes relataram qualquer sangramento e 19% sangramento clinicamente significativo. Na visita do Dia 43 do mesmo ciclo, a proporção de pacientes com sangramento diminuiu: foi de 12% e 0%, respectivamente. Resultados similares foram observados durante os ciclos de tratamento subsequentes.
Oito pacientes controlaram com sucesso dez estímulos hemostáticos sem a necessidade de tratamento adicional para elevar as contagens plaquetárias e sem qualquer sangramento imprevisto.
TRA105325: O EXTEND foi um estudo de extensão, aberto que avaliou a segurança e a eficácia de Revolade® em pacientes com PTI crônica que haviam sido recrutados anteriormente em outro estudo com Revolade®. Neste estudo, os pacientes foram autorizados a modificar a dose da medicação em estudo, reduzi-la ou eliminar medicações concomitantes para PTI.
Revolade® foi administrado a 302 pacientes com PTI: 218 completaram um ano de tratamento, 180 completaram dois anos, 107 completaram 3 anos, 75 completaram 4 anos, 34 completaram 5 anos e 18 completaram 6 anos de terapia. A contagem plaquetária mediana basal foi de 19.000/microL antes da administração de Revolade®. As contagens medianas aos um, dois, três, quatro, cinco, seis e sete anos de estudo foram de 85.000/microL, 85.000/microL, 105,000/microL, 64,000/microL, 75,000/microL, 119,000/microL e 76,000/microL, respectivamente. A dose diária mediana de Revolade® global foi de 50,8 mg.
Na avaliação basal, 57% dos pacientes tinham qualquer sangramento (de graus 1-4 da OMS) e 17% apresentavam sangramento clinicamente significativo. A proporção relativa a ambos os tipos de sangramento diminuiu em aproximadamente 50% na maioria das avaliações até um ano.
Sessenta e um por cento dos pacientes que receberam a dose de uma medicação basal para PTI descontinuaram permanentemente ou reduziram de forma prolongada pelo menos uma medicação para PTI sem necessidade de tratamento de resgate subsequente. Oitenta e três por cento desses pacientes mantiveram essa descontinuação ou redução por pelo menos 24 semanas. Quarenta e seis por cento desses pacientes descontinuaram completamente pelo menos uma medicação basal para PTI sem necessidade de tratamento de resgate subsequente e 29% interromperam permanentemente todos os medicamentos de PTI sem nunca receber terapia de resgate no tratamento.
Pacientes pediátricos acima de 6 anos
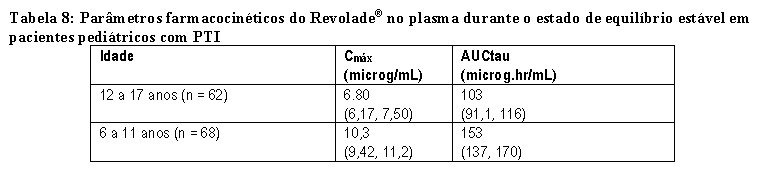
A segurança e a eficácia de Revolade® em pacientes pediátricos com PTI crônica previamente tratada foram demonstradas em dois estudos.
TRA115450 (PETIT2): O desfecho primário foi uma resposta plaquetária mantida, definida como a proporção de pacientes recebendo Revolade®, em comparação com o placebo, que atingiu contagens de plaquetas ≥ 50.000/microL por pelo menos 6 das 8 semanas (na ausência de terapia de resgate) entre as semanas 5 e 12 durante o período randomizado duplo-cego. Os pacientes foram diagnosticados com PTI crônica há pelo menos 1ano e eram refratários ou recidivantes a pelo menos uma terapia prévia para PTI ou incapaz de continuarem outros tratamentos por razão médica e tiveram contagem de plaquetas ≤ 30.000/microL. Noventa e dois pacientes foram randomizados em três faixas etárias estratificadas (proporção de 2:1) para Revolade® (n = 63) ou placebo (n = 29). A dose de Revolade® poderia ser ajustada com base na contagem individual de plaquetas.
No geral, uma proporção significativamente maior de pacientes do grupo de Revolade® (40%) em comparação com o grupo placebo (3%) alcançou o desfecho primário (taxa de probabilidade: 18,0 [IC de 95%: 2,3; 140,9] p < 0,001) que foi semelhante nas três faixas etárias (Tabela 3).
Uma proporção significativamente maior de pacientes tratados com Revolade® (75%) em comparação com placebo (21%) teve uma resposta plaquetária (pelo menos uma contagem de plaquetas ≥ 50.000/microL durante as primeiras 12 semanas de tratamento randomizado na ausência de terapia de resgate) (taxa de probabilidade: 11,7, [IC de 95%: 4,0; 34,5], p < 0,001). A proporção de pacientes que responderam ao Revolade® no período aberto de 24 semanas (80%) foi semelhante à observada durante a parte randomizada do estudo.
Estatisticamente menos pacientes tratados com Revolade® exigiram tratamento de resgate durante o período randomizado, em comparação com os pacientes do grupo placebo (19% [12/63] vs. 24% [7/29], p = 0,032).
Os pacientes foram autorizados a reduzir ou descontinuar a terapia basal para PTI, apenas durante a fase aberta do estudo e 53% (8/15) dos pacientes foram capazes de reduzir (n = 1) ou descontinuar (n = 7) a terapia basal para PTI, principalmente corticosteroides, sem a necessidade de terapia de resgate.
No início do estudo, 71% dos pacientes no grupo de Revolade® e 69% no grupo placebo relataram qualquer sangramento (graus 1 a 4, conforme a OMS). Na semana 12, a proporção de pacientes do grupo de Revolade® que relatou qualquer sangramento foi reduzida para metade do basal (36%). Em comparação, na semana 12, 55% dos pacientes do grupo placebo relataram qualquer sangramento.
TRA108062 (PETIT): O desfecho primário foi a proporção de pacientes que alcançou contagens de plaquetas ≥ 50.000/microL pelo menos uma vez entre as semanas 1 e 6 do período randomizado. Os pacientes eram refratários ou recidivantes a pelo menos uma terapia prévia para PTI com a contagem de plaquetas ≤ 30.000/microL (n= 67). Durante o período randomizado do estudo, os pacientes foram randomizados em três faixas etárias estratificadas (proporção de 2:1) para Revolade® (n = 45) ou placebo (n = 22). A dose de Revolade® poderia ser ajustada com base na contagem individual de plaquetas.
No geral, uma proporção significativamente maior de pacientes do grupo de Revolade® (62%) em comparação com os pacientes do grupo placebo (32%) alcançou o desfecho primário (taxa de probabilidade: 4,3 [IC de 95%: 1,4; 13,3] p = 0,011). A Tabela 4 mostra a resposta plaquetária nas três faixas etárias.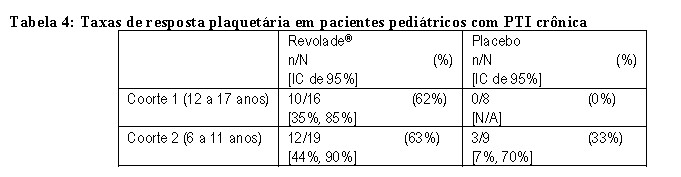
Uma proporção significativamente maior de pacientes tratados com Revolade® (36%) em comparação com placebo (0%) teve uma resposta plaquetária (contagens de plaquetas ? 50.000/microL por pelo menos 60% de avaliação entre as semanas 2 e 6) (taxa de probabilidade: 5,8, [IC de 95%: 1,2; 28,9], p = 0,002).
Estatisticamente menos pacientes tratados com Revolade® exigiram tratamento de resgate durante o período randomizado, em comparação com os pacientes do grupo placebo (13% [6/45] vs. 50% [11/22], p = 0,002).
Os pacientes foram autorizados a reduzir ou descontinuar a terapia basal para PTI apenas durante a fase aberta do estudo e 46% (6/13) dos pacientes foram capazes de reduzir (n = 3) ou descontinuar (n = 3) a terapia basal para PTI, principalmente corticosteroides, sem a necessidade de terapia de resgate.
No início do estudo, 78% dos pacientes no grupo de Revolade® e 82% no grupo placebo relataram qualquer sangramento (graus 1 a 4, conforme a OMS). A proporção de pacientes do grupo de Revolade® que relatou qualquer sangramento reduziu para 22% na semana 6. Em comparação, 73% dos pacientes do grupo placebo relataram qualquer sangramento na semana 6.
Estudos de Anemia Aplásica Severa (AAS) sem terapia imunossupressora definitiva prévia
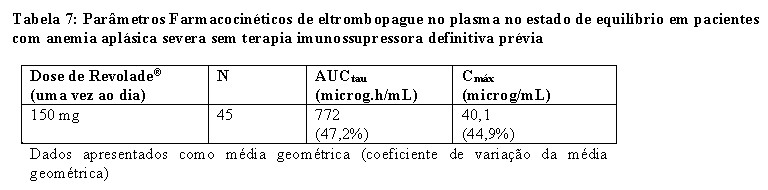
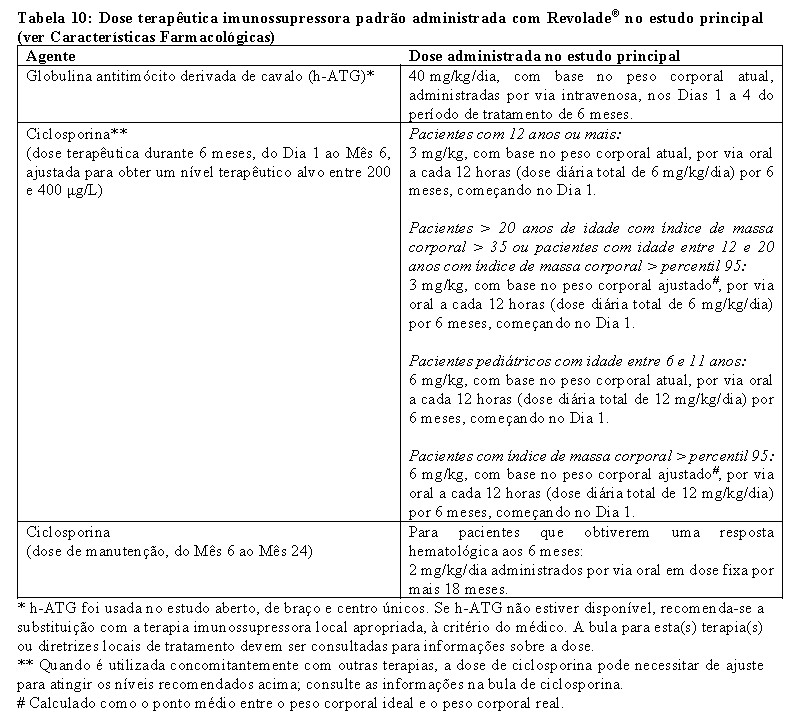
Eltrombopague em combinação com globulina antitimócito derivada de cavalo (h-ATG) e ciclosporina foi investigado em um estudo de coorte sequencial aberto, de braço e centro únicos, em pacientes com anemia aplásica severa que não receberam terapia imunossupressora definitiva prévia (isto é, terapia com ATG, alentuzumabe ou ciclofosfamida em altas doses). As múltiplas coortes diferiram entre o início do tratamento e a duração do eltrombopague e o início da dose baixa de ciclosporina (dose de manutenção) em pacientes que atingiram uma resposta hematológica aos 6 meses. Um total de 153 pacientes receberam eltrombopague em coortes sequenciais:
• eltrombopague no Dia 14 ao Mês 6 (D14-M6) mais h-ATG e ciclosporina (regime da Coorte 1 do estudo, n = 30), • eltrombopague no Dia 14 ao Mês 3 (D14-M3) mais h-ATG e ciclosporina (regime da Coorte 2 do estudo, n = 31), com metade dos pacientes elegíveis para receber baixas doses de ciclosporina (dose de manutenção) se obtiveram uma resposta hematológica aos 6 meses, • eltrombopague no Dia 1 ao Mês 6 (D1-M6) mais h-ATG e ciclosporina (regime da Coorte 3 do estudo, n = 92), com todos os pacientes elegíveis para receber doses baixas de ciclosporina (dose de manutenção) se obtiveram uma resposta hematológica aos 6 meses.
A dose inicial de eltrombopague foi de 150 mg, uma vez por dia, para adultos e adolescentes com idades entre os 12 e os 17 anos (dose reduzida de 75 mg para asiáticos do leste e sudeste), e 75 mg uma vez por dia para pacientes com 6 a 11 anos de idade (dose reduzida de 37,5 mg para asiáticos do leste e sudeste). A dose de eltrombopague foi reduzida quando a contagem de plaquetas excedeu 200.000/mL e interrompida e reduzida quando excedeu 400.000/mL.
Todos os pacientes receberam h-ATG 40 mg/kg/dia nos Dias 1 a 4 do período de tratamento de 6 meses e uma dose diária total de ciclosporina de 6 mg/kg/dia durante 6 meses em pacientes com 12 anos ou mais ou 12 mg/kg/dia por 6 meses em pacientes com 6 a 11 anos de idade. Uma dose de manutenção de 2 mg/kg/dia de ciclosporina foi administrada por mais 18 meses em 15 pacientes que obtiveram uma resposta hematológica aos 6 meses na Coorte D14-M3 de eltrombopague e todos os pacientes que obtiveram uma resposta hematológica aos 6 meses na Coorte D1-M6 de eltrombopague.
Os dados do esquema recomendado de eltrombopague do Dia 1 ao Mês 6 em combinação com h-ATG e ciclosporina (regime da Coorte 3 do estudo) são apresentados abaixo. Esta coorte teve as maiores taxas de resposta completas.
Na Coorte D1-M6 de eltrombopague, a idade mediana foi de 28 anos (variando de 5 a 82 anos) com 16,3% e 28,3% dos pacientes ≥ 65 anos de idade e < 18 anos de idade, respectivamente. 45,7% dos pacientes eram do sexo masculino e a maioria dos pacientes era branca (62,0%).
A eficácia de eltrombopague em combinação com h-ATG e ciclosporina foi estabelecida com base na resposta hematológica completa aos 6 meses. Uma resposta completa foi definida como parâmetros hematológicos que atendem a todos os 3 dos seguintes valores em duas medições consecutivas do hemograma, com pelo menos uma semana de intervalo: contagem absoluta de neutrófilos (ANC) > 1.000/mL, contagem de plaquetas > 100.000/mL e hemoglobina > 10 g/dL. Uma resposta parcial foi definida como melhora da contagem sanguínea que não atende aos critérios padrão para pancitopenia grave em anemia aplásica severa equivalente a 2 dos seguintes valores em duas medições consecutivas de hemograma, com intervalo mínimo de uma semana: ANC > 500/mL, contagem de plaquetas > 20.000/mL ou contagem de reticulócitos > 60.000/mL.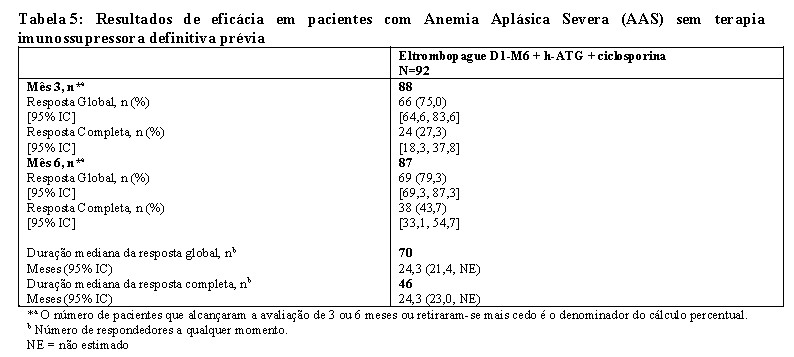
As taxas de resposta hematológica global e completa no Ano 1 (N = 78) são de 56,4% e 38,5% e no Ano 2 (N = 62) são 38,7% e 30,6%, respectivamente.
População pediátrica
Trinta e sete pacientes com idades entre 2 e 17 anos foram incluídos no estudo de coorte sequencial de braço único. Dos 36 pacientes que atingiram o ponto de avaliação de 6 meses ou retiraram-se mais cedo, a taxa de resposta completa aos 6 meses foi de 30,6% (0/2 nos pacientes com 2 a 5 anos, 1/12 nos pacientes com 6 a 11 anos e 10/22 em pacientes com idade entre 12 e 17 anos) e a taxa de resposta global aos 6 meses foi de 72,2% (2/2 em pacientes com 2 a 5 anos, 7/12 em pacientes de 6 a 11 anos e 17/22 em pacientes de 12 a 17 anos). Dos 25 pacientes avaliáveis na Coorte D1-M6 de eltrombopague, a taxa de resposta completa aos 6 meses foi de 28% (7/25) e a taxa de resposta global aos 6 meses foi de 68,0%.
Estudos de Anemia Aplásica Severa (AAS) refratária
Revolade® foi avaliado em um estudo de fase II não randomizado, de braço único, aberto, realizado em um centro em 43 pacientes com AAS, que tiveram uma resposta insuficiente a pelo menos uma terapia imunossupressora prévia e que apresentavam uma contagem de plaquetas ≤ 30.000 / microL.
Foi considerado que a maioria dos pacientes, 33 (77%), apresentava "doença refratária primária", definida como ausência de resposta prévia adequada à terapia imunossupressora (IST) em qualquer linhagem. Os 10 pacientes restantes apresentaram resposta plaquetária insuficiente às terapias anteriores. Todos os 10 pacientes tinham recebido pelo menos 2 regimes de IST prévios e 50% tinham recebido pelo menos 3 regimes de IST prévios. Os pacientes com diagnóstico de anemia de Fanconi, infecção não responsiva à terapia apropriada, tamanho de clone de PNH em neutrófilos ≥ 50%, foram excluídos da participação.
A população tratada tinha idade mediana de 45 anos (variação de 17 a 77 anos) e 56% dos pacientes eram do sexo masculino. Inicialmente a contagem de plaquetas média foi de 20.000 / microL, a hemoglobina foi de 8,4 g/dL, e CAN foi de 0,58 x 109/L e a contagem absoluta de reticulócitos foi de 24,3 x 109/L. Oitenta e seis por cento dos pacientes eram dependentes da transfusão de glóbulos vermelhos, e 91% eram dependentes da transfusão de plaquetas. A maioria dos pacientes (84%) tinha recebido pelo menos 2 terapias imunossupressoras anteriores. Três pacientes apresentaram anomalias citogenéticas no início do estudo.
Revolade® foi administrado a uma dose inicial de 50 mg uma vez por dia (25 mg para pacientes de ascendência asiática) durante 2 semanas. O aumento gradual da dose em 25 mg ocorreu a cada 2 semanas, com base nos níveis de plaquetas, até no máximo 150 mg uma vez ao dia (75 mg para pacientes da Ásia).
O desfecho primário foi a resposta hematológica avaliada após 12 semanas de tratamento com Revolade®. A resposta hematológica foi definida como sendo um ou mais dos seguintes critérios: 1) aumento na contagem de plaquetas para 20.000/microL acima da linha de base ou a contagem de plaquetas estável com independência de transfusão para um mínimo de 8 semanas; 2) aumento de hemoglobina de > 1,5 g/dL, ou uma redução ≥ 4 unidades de transfusões de glóbulos vermelhos (RBC) durante 8 semanas consecutivas; 3) aumento na contagem absoluta de neutrófilos (CAN) de 100% ou um aumento CAN > 0,5 x 109 / L.
A taxa de resposta hematológica foi de 40% (17/43 pacientes; 95% CI: 25, 56),a maioria das respostas obtidas foi unilinhagem (13/17,76%), mas houve 3 respostas bilinhagem e 1 resposta trilinhagem na avaliação da semana 12. Revolade® foi interrompido após 16 semanas quando não foi observada nenhuma resposta hematológica ou independência de transfusão. Os pacientes que responderam continuaram o tratamento em uma fase de extensão do estudo. Um total de 14 pacientes entrou na fase de extensão do estudo. Nove desses pacientes atingiram uma resposta multilinhagem, 4 dos 9 permanecem em tratamento e 5 reduziram gradualmente a dose do Revolade® e mantiveram a resposta (mediana de acompanhamento: 20,6 meses, intervalo: 5,7 a 22,5 meses). Os 5 pacientes restantes descontinuaram o tratamento, três devido à recidiva na visita de extensão do mês 3.
Durante o tratamento com Revolade®, 59% (23/39) tornaram-se independentes de transfusão plaquetária (28 dias sem transfusão de plaquetas) e 27% (10/37) tornaram-se independentes de transfusão de glóbulos vermelhos (RBC) (56 dias sem transfusão de RBC). O período mais longo sem transfusão de plaquetas em pacientes não respondedores foi de 27 dias (mediana). O período mais longo sem transfusão de plaquetas nos pacientes respondedores foi de 287 dias (mediana). O período mais longo sem transfusão de glóbulos vermelhos (RBC) nos pacientes não respondedores foi de 29 dias (mediana). O período mais longo sem transfusão de glóbulos vermelhos (RBC) nos pacientes respondedores foi de 266 dias (mediana).
Mais de 50% dos pacientes respondedores que eram dependentes de transfusão, no início do tratamento, apresentaram redução > 80% na necessidade de transfusão de plaquetas e de RBC em comparação ao início do estudo.
Os resultados preliminares de um estudo de suporte em andamento (Estudo ELT116826) fase II, não randomizado, aberto, de braço único, em pacientes com AAS refratários, apresentaram resultados consistentes. Os resultados são limitados a 21 dos 60 pacientes planejados com respostas hematológicas reportadas por 52% dos pacientes aos 6 meses. Foram notificadas respostas multilinhagem em 45% dos pacientes.
CETB115J2411 (TAPER)
CETB115J2411 foi um estudo de fase II de braço único que incluiu pacientes com PTI tratados com Revolade® após falha na resposta na administração de corticosteroides de primeira linha, independentemente do tempo desde o diagnóstico. Um total de 105 pacientes foram incluídos no estudo e iniciaram o tratamento com Revolade® 50 mg uma vez por dia (25 mg uma vez ao dia para pacientes de descendência oriental/sudeste asiático, e para pacientes de descendência japonesa, estes receberam 12,5 mg uma vez ao dia). A dose de Revolade® foi ajustada durante o período de tratamento com base nas contagens individuais de plaquetas, com o objetivo de atingir uma contagem plaquetária ≥100.000/microL.
Dos 126 pacientes que foram selecionados para inclusão no estudo TAPER, 105 pacientes receberam pelo menos uma dose de Revolade®, 70 pacientes (66,7%) completaram o tratamento e 35 pacientes (33,3%) interromperam o tratamento precocemente.
Resultados da análise primária da resposta sustentada ao tratamento
Os pacientes que atingiram uma contagem de plaquetas de ≥100.000/microL e mantiveram a contagem de plaquetas durante 2 meses de ≥70.000/microL foram elegíveis para a redução gradual do Revolade® e a descontinuação do tratamento. Para ser considerado como tendo alcançado uma resposta sustentada fora do tratamento, os pacientes deveriam manter contagens de plaquetas ≥30.000/microL, na ausência de eventos adversos hemorrágicos ou qualquer terapia de resgate, tanto durante o período de redução gradual do tratamento quanto após a descontinuação do tratamento até o Mês 12.
O esquema de redução gradual recomendou reduções de dose de 25 mg a cada 2 semanas, se as contagens de plaquetas fossem estáveis, seguidas de doses de 25 mg em dias alternados por 2 semanas até a interrupção do tratamento.
A duração da redução gradual foi individualizada em função da dose inicial e da resposta do paciente. A redução gradual foi feita em decréscimos menores da droga de 12,5 mg a cada duas semanas para pacientes de ascendência do Leste e Sudeste Asiático. Se uma recaída (definida como contagem de plaquetas < 30,000/microL) ocorresse durante o período de tratamento de 12 meses, foi oferecido aos pacientes um novo ciclo de Revolade® na dose inicial adequada.
O estudo atingiu o objetivo primário ao demonstrar que Revolade® foi capaz de induzir resposta sustentada fora do tratamento, na ausência de eventos hemorrágicos ou do uso de terapia de resgate, até o mês 12 em 32 pacientes dos 105 pacientes incluídos (30,5%; p < 0.0001; 95% CI: 21.9, 40.2).
Oitenta e nove pacientes (84,8%) obtiveram resposta completa (contagem de plaquetas ≥100.000/microL) e 65 pacientes (61,9%) mantiveram a resposta completa por pelo menos 2 meses sem contagem de plaquetas < 70,000/microL. Quarenta e quatro pacientes (41,9%) foram capazes de atingir a redução gradual do Revolade® através da descontinuação do tratamento, mantendo a contagem de plaquetas ≥30.000/microL na ausência de eventos adversos hemorrágicos ou qualquer terapia de resgate (Tabela 12-2)
A duração média da resposta sustentada após a descontinuação do tratamento até o Mês 12 foi de 33,3 semanas (mín-máx: 4-51).
A análise global de segurança é consistente com dados anteriormente comunicados e a avaliação risco-benefício permaneceu inalterada para a utilização de Revolade® em pacientes com PTI.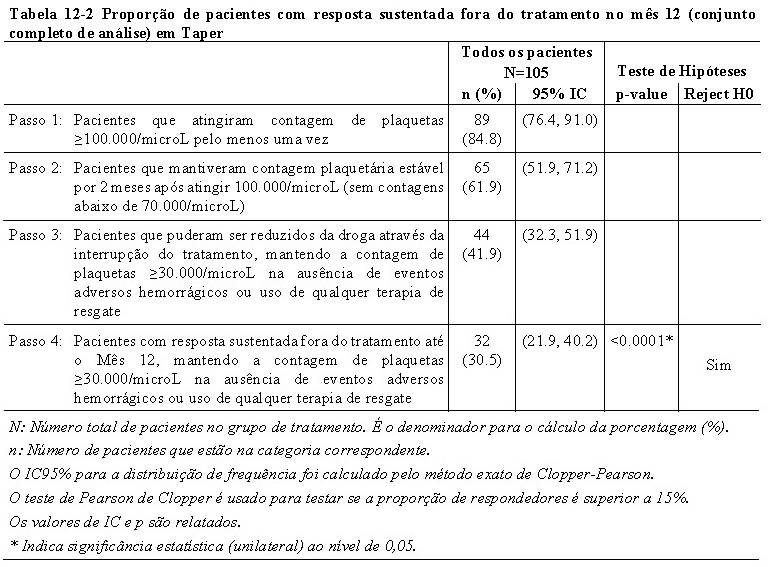
Resultados de uma resposta precoce no tratamento: análise pelo tempo de diagnóstico da PTI
Foi realizada uma análise ad-hoc nos n=105 pacientes por tempo desde o diagnóstico de PTI para avaliar a resposta precoce ao tratamento com Revolade® em quatro categorias diferentes de duração da PTI (PTI recém-diagnosticada < 3 meses, PTI persistente de 3 a < 6 meses, PTI persistente de 6 a ≤12 meses e PTI crônica > 12 meses).
49% dos pacientes (n=51) tiveram uma duração PTI de < 3 meses, 20% (n=21) de 3 a < 6 meses, 17% (n=18) de 6 a ≤12 meses e 14% (n=12 meses) =15) de > 12 meses.
Revolade® foi utilizado por uma duração mediana (25° a 75° percentil) de 6,2 meses (2,3 a 12,0). A mediana (25° a 75° percentil) da contagem de plaquetas no início do estudo foi de 16.000/microL (7.800 a 28.000/microL).
A resposta da contagem de plaquetas, definida como uma contagem de plaquetas ≥50.000/microL pelo menos uma vez até a semana 9 sem terapia de resgate, foi alcançada em 84% (IC de 95%: 71%, 93%) dos pacientes recém-diagnosticados (duração da PTI < 3 meses), 91% (95% IC: 70%, 99%) e 94% (95% IC: 73%, 100%) de pacientes com PTI persistente (ou seja, com diagnóstico de PTI de 3 a < 6 meses e 6 a ≤12 meses, respectivamente) e em 87% (IC 95%: 60%, 98%) dos pacientes com PTI crônica.
A taxa de resposta completa, definida como contagem de plaquetas ≥100.000/microL pelo menos uma vez até a semana 9 sem terapia de resgate, foi de 75% (IC de 95%: 60%, 86%) em pacientes recém-diagnosticados (duração da PTI < 3 meses), 76% (95% IC: 53%, 92%) e 72% (95% IC: 47%, 90%) em pacientes com PTI persistente (duração da PTI de 3 a < 6 meses e 6 a ≤12 meses, respectivamente) e 87% (95% IC: 60%, 98%) em pacientes com PTI crônica.
A taxa de resposta duradoura da contagem de plaquetas, definida como uma contagem de plaquetas ≥50.000/microL em pelo menos 6 de 8 avaliações consecutivas sem terapia de resgate durante os primeiros 6 meses do estudo, foi de 71% (IC de 95%: 56%, 83%) em pacientes com PTI recém-diagnosticada, 81% (IC de 95%: 58%, 95%) e 72% (IC de 95%: 47%, 90%) em pacientes com PTI persistente (duração da PTI de 3 a < 6 meses e 6 a ≤ 12 meses, respectivamente) e 80% (95% IC: 52%, 96%) em pacientes com PTI crônica.
Quando avaliada com a Escala de gravidade de Sangramento da OMS, a proporção de pacientes com PTI recentemente diagnosticados e persistentes sem sangramento na Semana 4 variou de 88% a 95% em comparação com 37% a 57% no início do estudo. Para pacientes com PTI crônica, foi de 93% em comparação com 73% no início do estudo.
A segurança do Revolade® foi consistente em todas as categorias de ITP e de acordo com seu perfil de segurança conhecido.
Referências Bibliográficas
1. A randomized, double-blind, placebo-controlled phase III study, to evaluate the efficacy, safety and tolerability of eltrombopag olamine (SB-497115-GR), a thrombopoietin receptor agonist, administered for 6 months as oral tablets once daily in adult subjects with previously treated chronic idiopathic thrombocytopenic purpura (ITP). Study TRA102537. Report UM2008/00026/00, 2008.
2. A double-blind, randomized, placebo-controlled, parallel group study to investigate the efficacy, safety, tolerability, pharmacokinetics and pharmacodynamics of SB-497115-GR, a thrombopoietin receptor agonist, administered at 30, 50 and 75 mg as oral tablets once-daily for 6 weeks to adult male and female subjects with refractory, chronic immune thrombocytopenic purpura. Study TRA100773B. Report RM2006/00266/01
3. An open-label repeat dosing study of eltrombopag olamine (SB-497115-GR) in adult subjects, with chronic idiopathic thrombocytopenic purpura (ITP) REPEAT. Repeated ExPosure To Eltrombopag in Adults with Idiopathic Thrombocytopenic Púrpura. Study TRA108057. Report UM2008/00028/0, 2008.
4. EXTEND (Eltrombopag Extended Dosing Study): An extension study of eltrombopag olamine (SB-497115-GR) in adults with idiopathic thrombocytopenic purpura (ITP) previously enrolled in an eltrombopag study. Study TRA10532. Report UM2008/00050/00, 2008.
5. PETIT (Pediatric patients with Thrombocytopenia from ITP): A three part, staggered cohort, open-label and double blind, randomized, placebo controlled study to investigate the efficacy, safety, tolerability and pharmacokinetics of eltrombopag, a thrombopoietin receptor agonist, in previously treated pediatric patients with chronic idiopathic thrombocytopenic purpura (ITP). Study TRA108062. Report 2013N179281_00, 2014.
6. PETIT2 (Pediatric patients with Thrombocytopenia from ITP): A two-part, double-blind, randomized, placebo-controlled and open-label study to investigate the efficacy, safety and tolerability of eltrombopag, a thrombopoietin receptor agonist, in pediatric patients with previously treated chronic immune (idiopathic) thrombocytopenic purpura (ITP). Study TRA115450. Report 2013N178554_01, 2014.
7. A Pilot Study of a Thrombopoietin-receptor Agonist (TPO-R agonist), Eltrombopag, in Aplastic Anemia Patients with Immunosuppressive-therapy Refractory Thrombocytopenia. Study ELT112523. Report 2013N170687_01, 2014.
8. CETB115J2411: A phase II, open-label, prospective, single-arm, study to assess ability of eltrombopag to induce sustained remission in subjects with ITP who are refractory or relapsed after first-line steroids (TAPER).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: agonista do receptor de trombopoetina. Código ATC: B02BX 05
Farmacodinâmica
Mecanismo de ação
A trombopoietina (TPO) é a principal citocina envolvida na regulação da megacariopoiese e produção de plaquetas e o principal ligante de receptor de trombopoietina (TPO-R). O eltrombopague interage com o domínio transmembrana do TPO-R e inicia cascatas de sinalização similares, porém não idênticas, às da trombopoietina (TPO) endógena, induzindo a proliferação e a diferenciação de megacariócitos provenientes das células progenitoras da medula óssea.
Efeitos farmacodinâmicos
Revolade® difere da TPO quanto aos efeitos sobre a agregação plaquetária, no sentido de que o tratamento com Revolade® de plaquetas humanas normais não aumenta a