POMALYST
B-MS
pomalidomina
Antineoplásico. Imunomodulador.
Apresentações.
Cada embalagem contém 14 ou 21 cápsulas duras de 1 mg, 2 mg, 3 mg ou 4 mg.
USO ORAL
USO ADULTO ACIMA DE 18 ANOS
Composição.
Pomalyst® 1 mg:
Cada cápsula dura contém 1 mg de pomalidomida. Excipientes: manitol, amido pré-gelatinizado, estearil fumarato de sódio, gelatina, dióxido de titânio, óxido de ferro amarelo, indigotina.
Pomalyst® 2 mg:
Cada cápsula dura contém 2 mg de pomalidomida. Excipientes: manitol, amido pré-gelatinizado, estearil fumarato de sódio, gelatina, dióxido de titânio, óxido de ferro amarelo, vermelho de eritrosina, indigotina.
Pomalyst® 3 mg:
Cada cápsula dura contém 3 mg de pomalidomida. Excipientes: manitol, amido pré-gelatinizado, estearil fumarato de sódio, gelatina, dióxido de titânio, óxido de ferro amarelo, azul de indigotina, indigotina.
Pomalyst® 4 mg:
Cada cápsula dura contém 4 mg de pomalidomida. Excipientes: manitol, amido pré-gelatinizado, estearil fumarato de sódio, gelatina, dióxido de titânio, FCF azul brilhante, indigotina.
Proibido para mulheres grávidas.
Este medicamento pode causar o nascimento de crianças sem braços e sem pernas.
Este medicamento é somente seu. Não passe para ninguém.
Este medicamento não provoca aborto e não evita filhos.
Informações técnicas.
1 INDICAÇÕES
1.1 Em combinação com bortezomibe e dexametasona (PVd) - pacientes que apresentam mieloma múltiplo recidivado ou refratário após pelo menos uma terapia anterior, incluindo lenalidomida:
Pomalyst® em combinação com bortezomibe e dexametasona é indicado para o tratamento de pacientes com mieloma múltiplo recidivado ou refratário que receberam pelo menos um esquema de tratamento anterior, incluindo lenalidomida.
1.2 Em combinação com dexametasona (Pd) - pacientes que apresentam mieloma múltiplo recidivado e refratário:
Pomalyst® em combinação com dexametasona é indicado para o tratamento de pacientes com mieloma múltiplo recidivado e refratário que receberam pelo menos dois regimes de tratamento anteriores, incluindo lenalidomida e bortezomibe, e demonstraram progressão da doença na última terapia.
2 RESULTADOS DE EFICÁCIA
2.1 Resumo de eficácia
2.1.1 Esquema de PVd: Mieloma Múltiplo Recidivado ou Refratário
Um estudo clínico randomizado, aberto, Fase 3, de dois braços, multicêntrico (CC-4047-MM-007) avaliou a eficácia e segurança de pomalidomida em combinação com bortezomibe e baixa dose de dexametasona (PVd) versus bortezomibe e baixa dose de dexametasona (Vd) em pacientes adultos, anteriormente tratados, com mieloma múltiplo recidivado ou refratário.
Os pacientes receberam pelo menos um tratamento prévio, um dos quais deve ter sido o esquema contendo lenalidomida.
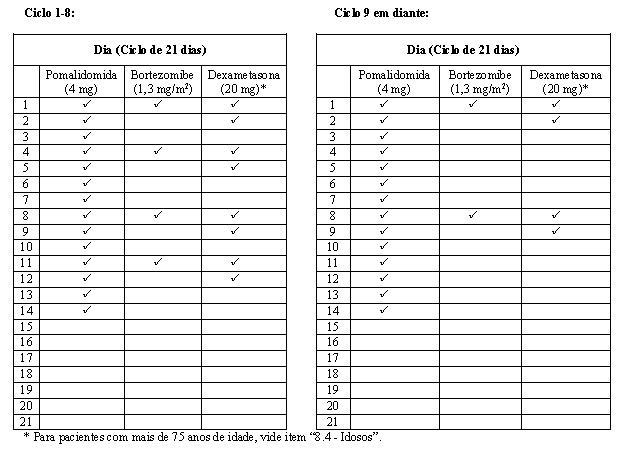
Os pacientes no braço PVd receberam 4 mg de pomalidomida via oral nos Dias 1 a 14 de cada ciclo de 21 dias. Baixa dose de dexametasona 20 mg foi administrada uma vez ao dia nos Dias 1, 2, 4, 5, 8, 9, 11 e 12 de um ciclo de 14 dias do Ciclo 1 ao 8, em seguida, uma vez ao dia nos Dias 1, 2, 8 e 9 de cada ciclo subsequente de 21 dias a partir do Ciclo 9 em diante.
Pacientes com > 75 anos de idade receberam dexametasona 10 mg usando o mesmo cronograma de tratamento que os pacientes mais jovens. O bortezomibe (1,3 mg/m2/dose) foi administrado nos Dias 1, 4, 8 e 11 do ciclo de 21 dias do Ciclo 1 ao 8, então na mesma dose nos Dias 1 e 8 do ciclo de 21 dias do Ciclo 9 em diante.
Para o braço Vd, bortezomibe e dexametasona seguiram a mesma dose e cronograma que o braço PVd. O tratamento continuou até a progressão da doença ou toxicidade inaceitável. As doses foram reduzidas, o tratamento foi temporariamente interrompido ou suspenso, conforme necessário para tratar a toxicidade.
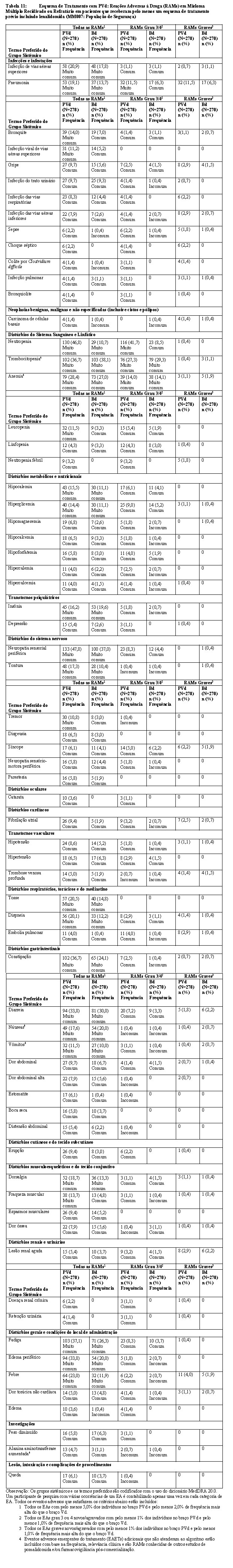
Um total de 559 pacientes foi randomizado no estudo: 281 no braço PVd e 278 no braço Vd. A maior parte dos indivíduos da pesquisa era branca (84%), 54% eram do sexo masculino e a idade mediana para a população geral era de 68 anos (mín, máx: 27, 89 anos). As características demográficas e da doença basal para a população com Intenção de Tratar (ITT) são resumidas nas tabelas a seguir. De modo geral, as características demográficas e relacionadas à doença foram geralmente compatíveis entre os braços de tratamento.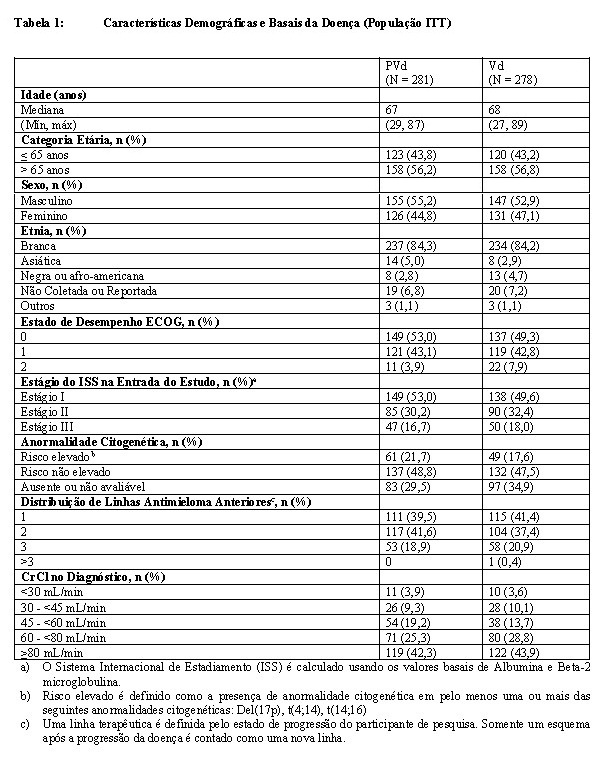
O desfecho primário de eficácia foi a sobrevida livre de progressão (SLP). A SLP foi definida como o tempo entre a randomização e a progressão da doença ou óbito. A resposta foi avaliada por um Comitê Independente de Revisão (IRAC) de acordo com os critérios do IMWG (Grupo de Trabalho Internacional do Mieloma) usando a população de intenção de tratar como a análise primária. A sobrevida global foi um desfecho secundário.
Os resultados de eficácia estão resumidos na tabela abaixo. A SLP foi significativamente mais longa com PVd do que com Vd: HR 0,61 (IC de 95%: 0,49, 0,77) indicando uma redução de 39% no risco de progressão da doença ou óbito para o braço PVd.
Qualidade de vida
A Qualidade de Vida Relacionada à Saúde (HRQoL) foi avaliada como o desfecho de eficácia exploratório usando o Módulo do Questionário de QoL da Organização Europeia para Pesquisa e Tratamento de Câncer para Pacientes com Mieloma Múltiplo (EORTC QLQ-MY20), Módulo Core 30 de Qualidade de vida da Organização Europeia para Pesquisa e Tratamento de Câncer (EORTC QLQ-C30) e o Questionário Europeu de Qualidade de Vida de Cinco Dimensões (EQ-5D). Com a QoL Global do EORTC QLQ-C30 como o domínio de interesse primário.
Os resultados das análises dos dados de HRQoL indicaram que não houve diferenças clinicamente significativas entre os grupos de tratamento. Melhoras nos sintomas da doença foram observadas em ambos os tratamentos. Os achados das análises sugeriram que o tratamento com Pomalyst (POM) + bortezomibe (BTZ) + baixa dose de dexametasona (LD-DEX), em comparação com BTZ +LD-DEX, em pacientes com mieloma múltiplo recidivado ou refratário participantes da pesquisa não comprometeu a HRQoL
2.1.2 Esquema de Pd: Mieloma Múltiplo Recidivado e Refratário
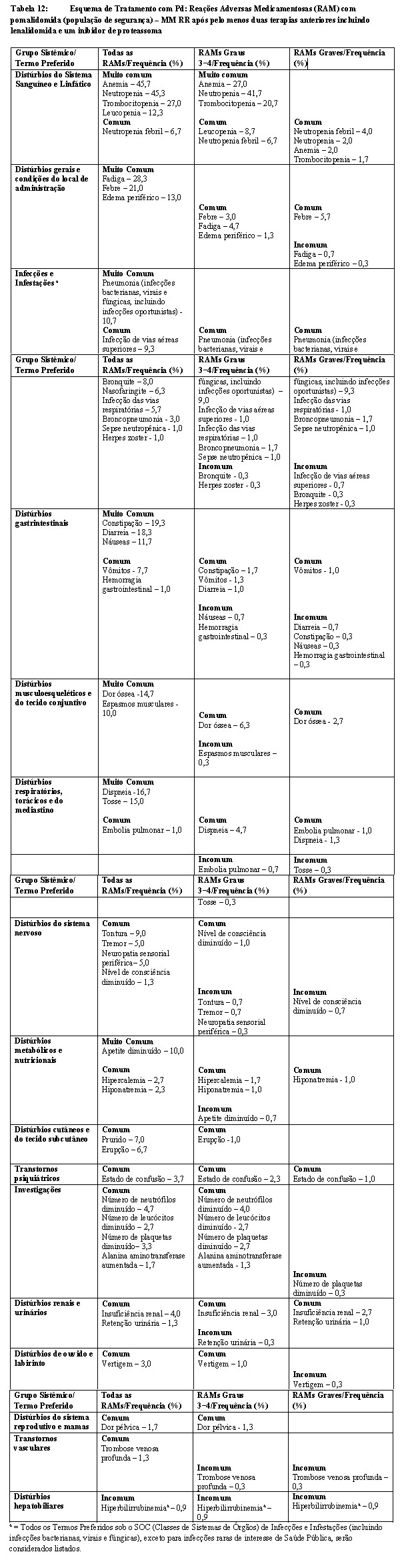
A eficácia e a segurança de pomalidomida em combinação com dexametasona foram avaliadas em um estudo Fase III, multicêntrico, randomizado e aberto (CC-4047-MM-003), em que a terapia de pomalidomida combinada com baixa dose de dexametasona (Pom+LD-dex) foi comparada com alta dose de dexametasona (HD-dex) isolada em pacientes adultos previamente tratados que apresentam mieloma múltiplo recidivado e refratário, que receberam pelo menos dois esquemas de tratamento anteriores e falharam com lenalidomida e bortezomibe, e demonstraram progressão da doença na última terapia. Um total de 455 indivíduos foi incluído no estudo: 302 no braço Pom+LD-dex e 153 no braço HD-dex. A maior parte dos indivíduos era do sexo masculino (59%) e branca (79%); a idade mediana para a população geral foi de 64 anos (mín, máx: 35, 87 anos).
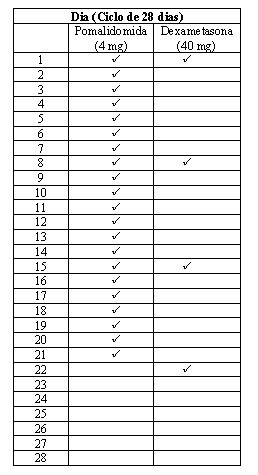
Os pacientes no braço Pom+LD-dex receberam 4 mg de pomalidomida via oral nos Dias 1 a 21 de cada ciclo de 28 dias. LD-dex (40 mg) foi administrado uma vez ao dia nos Dias 1, 8, 15 e 22 de um ciclo de 28 dias. Indivíduos > 75 anos de idade iniciaram o tratamento com 20 mg de dexametasona usando o mesmo cronograma. Para o braço HD-dex, dexametasona (40 mg) foi administrado uma vez ao dia, nos Dias 1 a 4, 9 a 12 e 17 a 20 de um ciclo de 28 dias. Indivíduos > 75 anos de idade iniciaram o tratamento com 20 mg de dexametasona usando o mesmo cronograma. O tratamento continuou até os indivíduos apresentarem progressão da doença. Os indivíduos no braço HD-dex após a progressão da doença tiveram a opção de receber Pomalyst® isoladamente em um estudo complementar (CC-4047-MM-003/C).
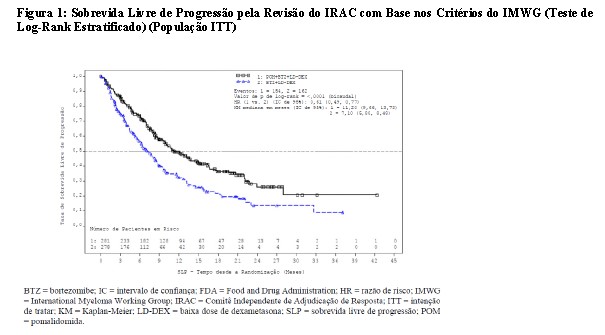
O desfecho primário de eficácia foi a sobrevida livre de progressão (SLP) de acordo com os critérios do IMWG. Para a população com Intenção de Tratar (ITT), o tempo de SLP mediana pela revisão do Comitê Independente de Revisão (IRAC) foi de 15,7 semanas (IC de 95%: 13,0, 20,1) no braço Pom + LD-dex; a taxa de sobrevida livre de eventos estimada de 26 semanas foi de 35,99% ±3,46. No braço HD-dex, o tempo de SLP mediana foi de 8,0 semanas (IC de 95%: 7,0, 9,0); a taxa de sobrevida livre de eventos estimada de 26 semanas foi de 12,15% ±3,63%. Resultados idênticos foram obtidos pela revisão do IRAC com base nos critérios do Grupo Europeu para Transplante de Sangue e Medula Óssea (EBMT). Os resultados da análise da população avaliável para eficácia com base nos critérios do Grupo de Trabalho Internacional do Mieloma (IMWG), bem como nos critérios do EBMT, foram compatíveis com os observados na população ITT. Independentemente do subgrupo avaliado, a SLP foi geralmente compatível com a observada na população ITT para ambos os grupos de tratamento.
A Sobrevida Livre de Progressão é resumida na Tabela 3 para a população ITT. Uma curva de Kaplan-Meier para a SLP para a população ITT é fornecida na Figura 2.
Corte de dados: 07 Set 2012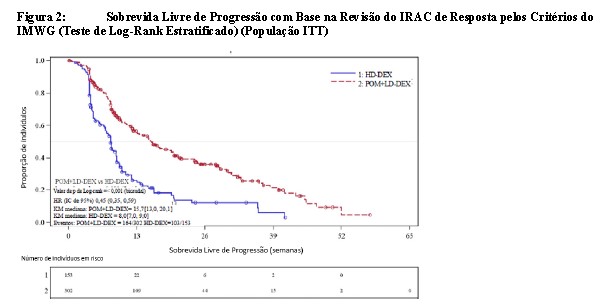
A Sobrevida Global (SG) foi um dos desfechos secundários do estudo. Um total de 226 (74,8%) dos indivíduos tratados com Pom + LD-dex e 95 (62,1%) dos indivíduos tratados com HD-dex estavam vivos até a data limite para corte de dados (07 de setembro de 2012). O tempo de SG mediana das estimativas de Kaplan-Meier não foi alcançado para Pom + LD-dex, mas seria esperado em pelo menos 48 semanas, o limite inferior do IC de 95%. O tempo de SG mediana para o braço HD-dex foi de 34 semanas (IC de 95%: 23,4, 39,9).
A taxa livre de evento de 1 ano foi de 52,6% (+ 5,72%) para o braço Pom + LD-dex e 28,4% (±7,51%) para o braço HD-dex. A diferença na SG entre os dois braços de tratamento foi estatisticamente significativa (p < 0,001).
Os resultados para a população avaliável para eficácia foram compatíveis com os observados na população ITT.
Uma curva de Kaplan-Meier para a SG para a população ITT é fornecida na Figura 3. 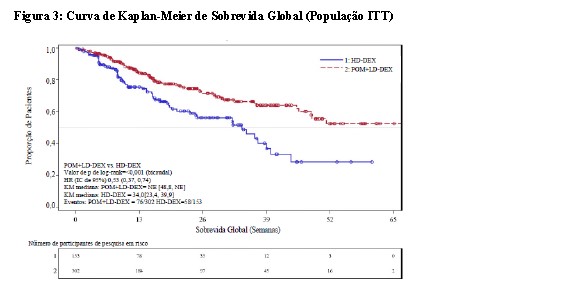
Dois estudos randomizados adicionais (MM-002 e IFM-2009-02) foram realizados para avaliar a eficácia e segurança de pomalidomida.
O Estudo 1 (MM-002) foi um estudo de duas fases, multinacional, multicêntrico, randomizado e aberto em pacientes que apresentam mieloma múltiplo recidivado e refratário que foram refratários à sua última terapia para mieloma e que receberam lenalidomida e bortezomibe. Na primeira fase do Estudo 1, a dose máxima tolerada (DMT) das cápsulas de pomalidomida foi determinada como 4 mg/dia, administrada em 21/28 dias até a progressão da doença. Na segunda fase do Estudo 1, a segurança e a eficácia de pomalidomida 4 mg, 21/28 dias até a progressão da doença foram avaliadas isoladamente e em combinação com baixa dose de dexametasona (40 mg/dia administrado nos dias 1, 8, 15 e 22 de cada ciclo de 28 dias). Os pacientes no braço de pomalidomida isolado foram autorizados a adicionar baixa dose de dexametasona após a progressão da doença.
O Estudo 2 (IFM-2009-02) foi um estudo multicêntrico, randomizado e aberto de pomalidomida em combinação com baixa dose de dexametasona em pacientes com mieloma múltiplo recidivado e refratário, que apresentaram doença progressiva e que receberam lenalidomida e bortezomibe. Neste estudo, os pacientes foram randomizados para pomalidomida 4 mg/dia em 21/28 dias até a progressão da doença (Braço A) ou pomalidomida 4 mg/dia em 28/28 dias até a progressão da doença. No braço A e no braço B, pomalidomida foi administrado em combinação com baixa dose de dexametasona (40 mg administrado nos dias 1, 8, 15 e 22 de cada ciclo de 28 dias).
A Tabela 4 resume as características basais do paciente e da doença nos dois estudos. Em ambos os estudos, as características basais demográficas e relacionadas à doença foram bem equilibradas e comparáveis entre os braços do estudo.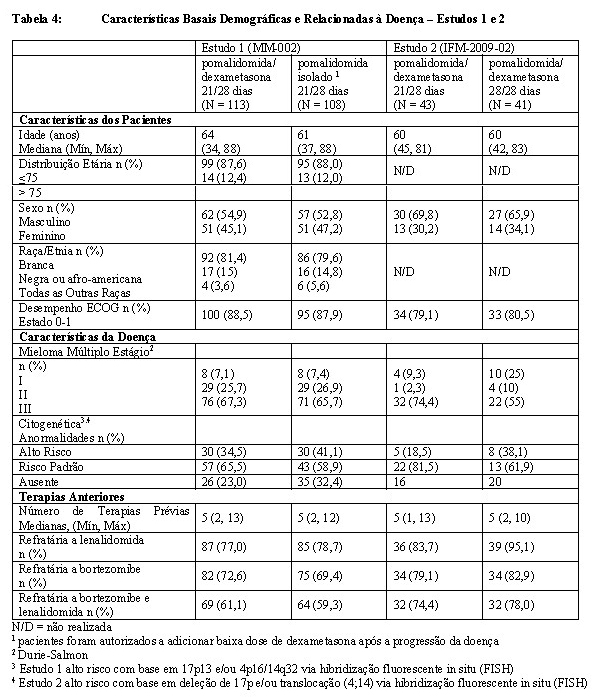
No Estudo 1, com base na avaliação pelo Comitê de Adjudicação de Revisão Independente (IRAC), a taxa de resposta global na população com intenção de tratar (ITT) foi de 30%. No geral, 34 pacientes (30%) apresentaram uma resposta a pomalidomida, incluindo 1 paciente (0,9%) com resposta completa e 33 pacientes (29,2%) com resposta parcial. A taxa de resposta global foi compatível, independentemente do tipo de terapia antimieloma prévia a que esses pacientes foram expostos durante suas histórias de tratamento ou do último tratamento imediato ao qual se tornaram refratários antes de receber pomalidomida. O desfecho de eficácia primário no Estudo 1 (fase 2) foi a sobrevida livre de progressão (SLP).
A duração da resposta mediana para os respondedores tratados com pomalidomida mais dexametasona foi de aproximadamente 32,1 semanas (IC de 95% = 22,1 a 39,9 semanas). A duração da resposta mediana para os indivíduos tratados com pomalidomida isolado ainda não foi alcançada (Tabela 5: Resultados da SLP nos Estudos 1 e 2).
O desfecho de eficácia primário do estudo 2 foi a taxa de resposta. Com base na avaliação pelo IRC (Comitê de Revisão Independente), a taxa de resposta global na população com Intenção de Tratar (ITT) foi de 35% (IC de 95%: 25%-46%); não foi observada diferença significativa entre as taxas de resposta no Braço A e no Braço B (p=0,943). No geral, 29 pacientes (35%) apresentaram uma resposta a pomalidomida, incluindo 3 pacientes (4%) com resposta completa e 26 pacientes (31%) com resposta parcial.
A SLP foi de 25,1 semanas em ambos os braços de tratamento. A duração da resposta mediana foi de 45,7 semanas (IC de 95%: 15,1 a 54,7 semanas) no Braço A, em comparação com 31,6 semanas no Braço B, uma diferença equivalente a aproximadamente 3 meses.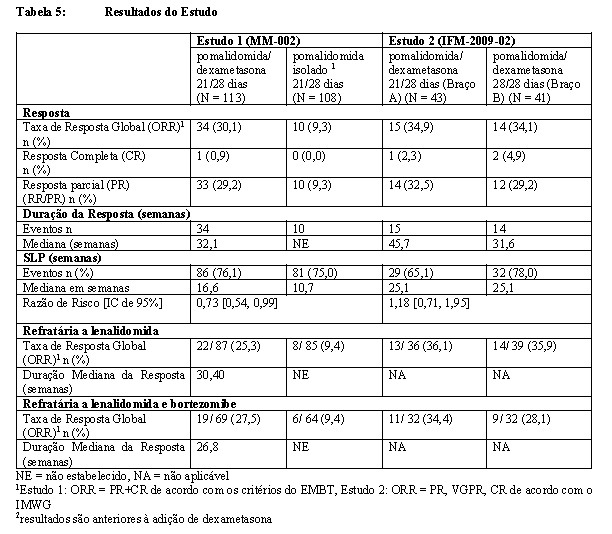
2.2 Dados de segurança pré-clínica
Genotoxicidade/Carcinogenicidade
A pomalidomida não foi mutagênica em ensaios de mutação bacteriana e mamífera e não induziu aberrações cromossômicas em linfócitos do sangue periférico humano ou formação de micronúcleos em eritrócitos policromáticos na medula óssea de ratos que receberam doses de até 2000 mg/kg/dia. Não foram conduzidos estudos de carcinogenicidade.
Fertilidade e Desenvolvimento Embrionário Precoce
Em um estudo de fertilidade e desenvolvimento embrionário inicial em ratos, pomalidomida foi administrada a machos e fêmeas em doses de 25, 250 e 1000 mg/kg/dia. Ratos machos foram tratados a partir de 28 dias antes da coabitação e continuamente até o dia antes da necropsia; ratas foram tratadas a partir de 14 dias antes da coabitação, durante o período de acasalamento e até o 7° dia de gestação. O exame uterino no Dia de Gestação 13 revelou uma redução no número médio de embriões viáveis e um aumento na perda pós-implantação em todos os níveis de dose. Portanto, o NOAEL para esses efeitos observados foi < 25 mg/kg/dia (AUC24h - 39960 ng·h/mL; exposição 99 vezes maior na dose mais baixa testada em relação a uma dose de 4 mg). Quando machos tratados neste estudo foram acasalados com fêmeas não tratadas, todos os parâmetros uterinos foram comparáveis aos controles. Com base nestes resultados, os efeitos observados foram atribuídos ao tratamento das fêmeas.
Desenvolvimento Embriofetal
A pomalidomida demonstrou ser teratogênica em ratos e coelhos, quando administrada durante o período de organogênese importante. No estudo de toxicidade do desenvolvimento embriofetal em ratos, foram observadas malformações de ausência de bexiga urinária, ausência de glândula tireoide e fusão e mal alinhamento dos elementos vertebrais lombares e torácicos (arcos centrais e/ou neurais) em todos os níveis de dose (25, 250 e 1000 mg/kg/dia). Não houve toxicidade materna observada neste estudo. Portanto, o NOAEL materno foi de 1000 mg/kg/dia e o NOAEL para toxicidade do desenvolvimento foi < 25 mg/kg/dia (AUC24h = 34340 ng h/mL no Dia de Gestação 17; exposição 85 vezes maior na menor dose testada em relação a uma dose clínica de 4 mg). Em coelhos, pomalidomida em doses de 10 a 250 mg/kg produziu malformações do desenvolvimento embriofetal. Foi observado aumento das anomalias cardíacas em todas as doses, com aumentos significativos na dose de 250 mg/kg/dia. Nas doses de 100 e 250 mg/kg/dia, houveram aumentos discretos na perda pós-implantação e discretas reduções nos pesos corporais fetais.
Na dose de 250 mg/kg/dia, as malformações fetais incluíam anomalias de membros (anteriores e/ou posteriores flexionados e/ou rotacionados, dígito não anexado ou ausente) e malformações esqueléticas associadas (metacarpo não ossificado, falange e metacarpo mal alinhados, dígito ausente, falange não ossificada e tíbia curta não ossificada ou dobrada), dilatação moderada do ventrículo lateral cerebral, inserção anormal da artéria subclávia direita, lobo intermediário ausente nos pulmões, rim de baixa localização, alteração da morfologia hepática, pélvis de ossificação incompleta ou ausente, aumento da média de costelas torácicas supranumerárias e redução da média de tarsos ossificados. Foram observadas redução discreta no ganho de peso corporal materno, redução significativa nos triglicerídeos e redução significativa nos pesos esplênicos absoluto e relativo com 100 e 250 mg/kg/dia. O NOAEL materno foi de 10 mg/kg/dia e o NOAEL para desenvolvimento foi < 10 mg/kg/dia (AUC 24h = 418 ng h/mL no Dia de Gestação 19; exposição 1 vez na menor dose testada em relação a uma dose clínica de 4 mg).
2.3 Referências bibliográficas
• San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol 2013; 14: 1055-66.
• Richardson PD, Oriol A, Beksac M, et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): a randomised, open-label, phase 3 trial Lancet Oncol. 2019 Jun;20(6):781-794.
3 CARACTERÍSTICAS FARMACOLÓGICAS
3.1 Propriedades farmacodinâmicas
3.1.1 Mecanismo de ação
Grupo farmacoterapêutico: agente imunomodulador (código ATC L04 AX06).
A pomalidomida apresenta atividade tumoricida direta antimieloma, atividades imunomoduladoras e inibe o suporte de células estromais para o crescimento de células tumorais de mieloma múltiplo. Especificamente, a pomalidomida inibe a proliferação e induz a apoptose de células tumorais hematopoiéticas. Além disso, a pomalidomida inibe a proliferação de linhagens celulares de mieloma múltiplo resistentes a lenalidomida e sinergiza com dexametasona em linhagens celulares sensíveis a lenalidomida e resistentes a lenalidomida para induzir apoptose de células tumorais. A pomalidomida aumenta a imunidade mediada por células T e por células natural killer (NK), e inibe a produção de citocinas pró-inflamatórias (p. ex., TNF-a e IL-6) pelos monócitos. A pomalidomida também inibe a angiogênese ao bloquear a migração e a adesão de células endoteliais.
A pomalidomida se liga diretamente à proteína cereblon (CRBN), que é parte de um complexo E3 ligase que inclui a proteína ligante ao dano do ácido desoxirribonucleico (DNA) 1 (DDB1), a culina 4 (CUL4) e a Roc1, e pode inibir a autoubiquitinação de CRBN dentro do complexo. As E3 ubiquitina ligases são responsáveis pela poliubiquitinação de uma variedade de proteínas substratos, e podem explicar em parte os efeitos celulares pleiotrópicos observados com o tratamento com pomalidomida.
Na presença de pomalidomida in vitro, os substrato de proteínas Aiolos e Ikaros são direcionadas para a ubiquitinação e subsequente degradação levando a efeitos citotóxicos e imunomoduladores diretos. In vivo, a terapia com pomalidomida levou à redução dos níveis de Ikaros em pacientes com mieloma múltiplo recidivado e refratário a lenalidomida.
As atividades pró-eritropoiéticas de pomalidomida foram demonstradas em células-tronco hematopoiéticas CD34+ induzidas a se diferenciarem em direção ao fenótipo eritroide. Essas atividades se manifestaram como uma maturação eritroide tardia, aumento da proliferação de células eritroides imaturas e indução da produção de hemoglobina fetal (HbF).
3.1.2 Eletrofisiologia cardíaca
Um estudo de QTc foi conduzido para avaliar os efeitos de pomalidomida no intervalo QT em doses únicas de 4 mg e 20 mg. Uma dose única de pomalidomida até 20 mg não foi associada ao prolongamento do intervalo QT em indivíduos saudáveis do sexo masculino. Não se espera que a pomalidomida resulte em prolongamento clinicamente significativo do intervalo QT em pacientes nas doses terapêuticas aprovadas.
3.2 Propriedades farmacocinéticas
3.2.1 Absorção
A pomalidomida é absorvida com uma Cmax que ocorre entre 2 e 3 horas e é > 70% absorvida após a administração de uma dose oral única. A exposição sistêmica (AUC) de pomalidomida aumenta de maneira proporcional à dose. Após doses múltiplas, a pomalidomida apresenta uma taxa de acúmulo de 27% a 31%.
Pacientes com mieloma múltiplo que receberam pomalidomida 4 mg/dia, em monoterapia ou combinação com dexametasona, a exposição no estado de equilíbrio foi caracterizada em AUC de 860 ng?h/mL (CV% = 37%) e Cmax de 75 ng/mL (CV% = 32%).
A administração concomitante com uma refeição rica em gorduras e calorias diminui a velocidade de absorção, diminuindo a Cmax média plasmática em ~27%, mas tem um efeito mínimo na extensão geral da absorção, com uma diminuição de 8% na AUC média. Portanto, a pomalidomida pode ser administrada independentemente da ingestão de alimentos.
3.2.2 Distribuição
A pomalidomida tem um volume de distribuição aparente médio (Vd/F) entre 62 e 138 L em estado de equilíbrio.
A pomalidomida é distribuída no sêmen de indivíduos saudáveis em uma concentração de aproximadamente 67% do nível plasmático em 4 horas após a dose (~Tmax) depois de 4 dias de administração, uma vez ao dia de 4 mg.
A ligação in vitro de enantiômeros de pomalidomida à proteínas no plasma humano varia de 12% a 44% e não é dependente da concentração.
3.2.3 Metabolismo
A pomalidomida é o principal componente circulante (aproximadamente 70% da radioatividade plasmática) in vivo em indivíduos saudáveis que receberam uma dose oral única de [14C] -pomalidomida (2 mg). Nenhum metabólito estava presente em > 10% em relação à radioatividade parental ou total no plasma.
A pomalidomida é eliminada em humanos por várias vias, incluindo metabolismo mediado por CYP, hidrólise não dependente de CYP e excreção de droga inalterada. As vias metabólicas predominantes de radioatividade excretada são a hidroxilação com subsequente glicuronidação ou hidrólise. In vitro, a CYP1A2 e CYP3A4 foram identificadas como as principais enzimas envolvidas na hidroxilação de pomalidomida mediada por CYP, com contribuições menores adicionais de CYP2C19 e CYP2D6.
A administração concomitante de pomalidomida com o potente CYP3A4/5 (e inibidor da P-gp), cetoconazol, ou com o potente indutor de CYP3A4/5, carbamazepina, não apresentou efeito clinicamente relevante na exposição a pomalidomida.
A administração concomitante do potente inibidor de CYP1A2 fluvoxamina com pomalidomida na presença de cetoconazol aumentou a exposição média a pomalidomida em 107% com um intervalo de confiança de 90% [91% a 124%] em comparação com pomalidomida mais cetoconazol. Em um segundo estudo para avaliar a contribuição de um inibidor de CYP1A2 isolado às alterações do metabolismo, a administração concomitante de fluvoxamina isolado com pomalidomida aumentou a exposição média a pomalidomida em 125% com um intervalo de confiança de 90% [98% a 157%] em comparação com pomalidomida isolado. Se inibidores potentes de CYP1A2 forem administrados concomitantemente com pomalidomida, reduza a dose de pomalidomida em 50%.
A administração concomitante de múltiplas doses de 4 mg de pomalidomida com 20 mg a 40 mg de dexametasona (um indutor fraco a moderado de várias enzimas CYP, incluindo CYP3A) a pacientes que apresentam mieloma múltiplo não apresentou efeito sobre a farmacocinética de pomalidomida em comparação com pomalidomida administrado isoladamente.
A pomalidomida é um substrato da P-glicoproteína in vitro, mas isso não parece limitar sua absorção em humanos, onde pelo menos 73% da droga foi absorvida. A administração concomitante de pomalidomida com o inibidor de P-gp, cetoconazol, não apresentou efeito clinicamente relevante na exposição a pomalidomida, portanto, não são previstas interações medicamentosas clinicamente relevantes quando pomalidomida é administrada concomitantemente com inibidores da P-glicoproteína.
Com base em dados in vitro, pomalidomida não é um inibidor ou indutor das isoenzimas do citocromo P-450 e não inibe a P-glicoproteína ou outros transportadores estudados. Não são previstas interações medicamentosas clinicamente relevantes quando pomalidomida é administrada concomitantemente com substratos dessas vias.
A administração de pomalidomida em fumantes, com inalação de tabaco conhecido por induzir a isoforma CYP1A2, não apresentou efeito clinicamente relevante na exposição a pomalidomida em relação à exposição a pomalidomida observada em não fumantes.
3.2.4 Excreção
A pomalidomida é eliminada com uma meia-vida plasmática mediana de aproximadamente 9,5 horas em indivíduos saudáveis e aproximadamente 7,5 horas em pacientes que apresentam mieloma múltiplo. A pomalidomida tem um clearance corporal total médio (CL/F) de 7-10 L/h.
Após uma administração oral única de [14C]-pomalidomida (2 mg) em indivíduos saudáveis, aproximadamente 73% e 15% da dose radioativa foram eliminados na urina e nas fezes, respectivamente, com aproximadamente 2% e 8% do radiocarbono dosado eliminado como pomalidomida na urina e nas fezes.
A pomalidomida é extensivamente metabolizada antes da excreção, com os metabólitos resultantes eliminados principalmente na urina. Os três metabólitos predominantes na urina (formados por hidrólise ou hidroxilação com subsequente glicuronidação) representam aproximadamente 23%, 17% e 12%, respectivamente, da dose na urina.
Os metabólitos dependentes de CYP representam aproximadamente 43% da radioatividade total excretada, ao passo que os metabólitos hidrolíticos não dependentes de CYP representaram 25%, e a excreção de pomalidomida inalterado representou 10% (2% na urina e 8% nas fezes).
3.2.5 Farmacocinética em crianças
Em geral, não há diferença significativa na farmacocinética da pomalidomida entre crianças e pacientes adultos.
3.2.6 Farmacocinética em idosos
Em indivíduos com idades entre 61 e 82 anos, os parâmetros farmacocinéticos médios da AUC (0-∞) e Cmax foram geralmente semelhantes aos indivíduos mais jovens.
3.2.7 Farmacocinética no comprometimento renal
As análises farmacocinéticas da população mostraram que os parâmetros farmacocinéticos de pomalidomida não foram notavelmente afetados em pacientes com insuficiência renal (definidos pelo clearance de creatinina ou ritmo de filtração glomerular estimado [eGFR]) em relação aos pacientes com função renal normal (CrCl ≥60 mL/minuto). A exposição média normalizada da AUC a pomalidomida foi de 98,2% com um intervalo de confiança de 90% [77,4% a 120,6%] em pacientes com insuficiência renal moderada (eGFR ≥30 a ≤45 mL/minuto/1,73 m2) em relação aos pacientes com função renal normal.
A exposição média normalizada da AUC de pomalidomida foi de 100,2% com um intervalo de confiança de 90%
[79,7% a 127,0%] em pacientes com insuficiência renal severa que não necessitam de diálise (CrCl < 30 ou eGFR < 30 mL/minuto/ 1,73 m
2) em relação aos pacientes com função renal normal. A exposição média normalizada da AUC à pomalidomida aumentou em 35,8% com um intervalo de confiança de 90% [7,5% a 70,0%] em pacientes com insuficiência renal severa que necessitam de diálise (CrCl < 30 mL/minuto com necessidade de diálise) em relação aos pacientes com função renal normal. As alterações médias na exposição à pomalidomida em cada um desses grupos de insuficiência renal não são de uma magnitude que exijam ajustes de dosagem.
3.2.8 Farmacocinética no comprometimento hepático
Os parâmetros farmacocinéticos foram modestamente alterados em pacientes com comprometimento hepático (definidos pelos critérios de Child-Pugh) em relação a indivíduos saudáveis. A exposição média à pomalidomida aumentou em 51% com um intervalo de confiança de 90% [9% a 110%] em pacientes com comprometimento hepático leve em relação aos indivíduos saudáveis. A exposição média à pomalidomida aumentou em 58% com um intervalo de confiança de 90% [13% a 119%] em pacientes com comprometimento hepático moderado em relação aos indivíduos saudáveis. A exposição média à pomalidomida aumentou em 72% com um intervalo de confiança de 90% [24% a 138%] em pacientes com comprometimento hepático severo em relação aos indivíduos saudáveis. Os aumentos médios na exposição à pomalidomida em cada um desses grupos de comprometimento hepático não têm magnitude para a qual são necessários ajustes no cronograma ou na dose.
4 CONTRAINDICAÇÕES
• Gravidez.
• Mulheres em idade fértil, exceto quando todas as condições de prevenção da gravidez forem atendidas (vide itens "5 - ADVERTÊNCIAS E PRECAUÇÕES" e "5.3 - Gravidez, lactação e fertilidade").
• Pacientes do sexo masculino incapazes de seguir ou cumprir as medidas contraceptivas necessárias (vide item "5 - ADVERTÊNCIAS E PRECAUÇÕES" e "5.3 - Gravidez, lactação e fertilidade").
• Hipersensibilidade à pomalidomida ou a qualquer um dos excipientes.
Para informações sobre os medicamentos utilizados em combinação com pomalidomida, consultar a bula do respectivo produto.
Categoria X: Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento, a menos que todas as condições do Programa de Prevenção de Gravidez sejam cumpridas.
5 ADVERTÊNCIAS E PRECAUÇÕES
5.1 Geral
Advertência de Gravidez
A pomalidomida é um análogo da talidomida.
A talidomida é um agente teratogênico humano conhecido que provoca defeitos congênitos severos potencialmente fatais. A pomalidomida demonstrou ser teratogênica em ratos e coelhos, quando administrada durante o período de organogênese importante (vide itens "5.3 - Gravidez, lactação e fertilidade" e "2.2 - Dados de Segurança Pré-Clínica"). Caso Pomalyst® seja administrado durante a gestação, um efeito teratogênico em humanos não pode ser descartado.
Eventos Tromboembólicos
Alguns pacientes que receberam pomalidomida em combinação com bortezomibe e dexametasona ou em combinação com dexametasona desenvolveram eventos tromboembólicos venosos (predominantemente trombose venosa profunda e embolia pulmonar) e eventos trombóticos arteriais (infarto do miocárdio e acidente vascular cerebral). Pacientes com fatores de risco conhecidos para tromboembolismo - incluindo trombose prévia - devem ser monitorados cuidadosamente. Ações devem ser tomadas para tentar minimizar todos os fatores de risco modificáveis (por exemplo, tabagismo, hipertensão e hiperlipidemia). Pacientes e médicos são aconselhados a observar os sinais e sintomas de tromboembolismo. Os pacientes devem ser instruídos a procurar atendimento médico se desenvolverem sintomas como falta de ar, dor no peito e inchaço nos braços ou pernas. Terapia anticoagulante (exceto se contraindicada) é recomendada (tal como ácido acetilsalicílico, varfarina, heparina ou clopidogrel). Uma decisão de adotar medidas profiláticas deve ser tomada cuidadosamente após uma avaliação dos fatores de risco subjacentes do paciente individualmente.
Em estudos clínicos, os pacientes receberam ácido acetilsalicílico profilático ou terapia antitrombótica alternativa. O uso de agentes eritropoiéticos acarreta o risco de eventos trombóticos, incluindo tromboembolismo. Portanto, agentes eritropoiéticos ou outros agentes que possam aumentar o risco de trombose, como a terapia de reposição hormonal, devem ser utilizados com precaução.
Distúrbios da tiroide
Foram descritos casos de hipotiroidismo. Antes do início do tratamento, recomenda-se o controle otimizado de comorbidades que influenciem a função tireoidiana. A monitorização inicial e contínua da função tiroideia é recomendada.
Neuropatia periférica
Os pacientes com neuropatia periférica de Grau ≥ 2 foram excluídos dos ensaios clínicos com a pomalidomida. Devem ser tomadas as devidas precauções quando se considerar o tratamento destes pacientes com pomalidomida.
Disfunção cardíaca significativa
Pacientes com disfunção cardíaca significativa [insuficiência cardíaca congestiva (Classe III ou IV da NY Heart Association); enfarte do miocárdio no prazo de 12 meses do início do tratamento; angina de peito instável ou pouco controlada] foram excluídos dos ensaios clínicos com a pomalidomida. Foram notificados eventos cardíacos, incluindo insuficiência cardíaca congestiva, edema pulmonar e fibrilação auricular (vide item "9 - REAÇÕES ADVERSAS"), principalmente em pacientes com doença cardíaca pré-existente ou com fatores de risco cardíaco. Devem ser tomadas as devidas precauções quando se considerar o tratamento destes pacientes com pomalidomida, incluindo a monitorização periódica de sinais ou sintomas de eventos cardíacos.
Reações Alérgicas e Reações Cutâneas Graves
Angioedema, anafilaxia e reações dermatológicas severas, incluindo síndrome de Stevens-Johnson (SJS), necrólise epidérmica tóxica (NET) e reação medicamentosa com eosinofilia e sintomas sistêmicos (DRESS) foram reportados. DRESS pode se apresentar com uma reação cutânea (como uma erupção ou dermatite esfoliativa), eosinofilia, febre e/ou linfadenopatia com complicações sistêmicas como hepatite, nefrite, pneumonite, miocardite e/ou pericardite. Esses eventos podem ser fatais.
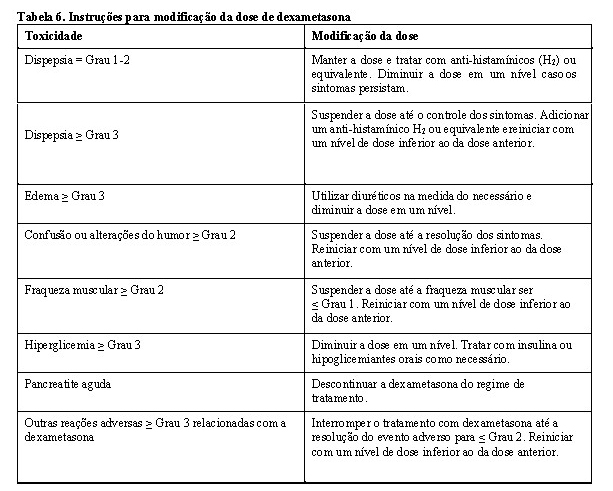
Pacientes com história prévia de reações alérgicas graves associadas à talidomida ou à lenalidomida foram excluídos dos estudos clínicos, pois podem apresentar risco mais elevado de hipersensibilidade e não devem receber pomalidomida. A interrupção ou descontinuação de pomalidomida deve ser considerada para a erupção cutânea Grau 2-3. A pomalidomida deve ser descontinuada em caso de angioedema, anafilaxia, erupção Grau 4, erupção esfoliativa ou bolhosa, ou se houver suspeita de SJS, NET ou DRESS, e não deve ser reiniciada mesmo após a descontinuação destas reações.
Tontura e Confusão
Tontura e confusão foram reportadas. Oriente os pacientes a serem cautelosos em situações em que tontura ou confusão podem ser prejudiciais.
Doença pulmonar intersticial (DPI)
Foi observada DPI e casos relacionados, incluindo casos de pneumonite, com a pomalidomida. Deve efetuar-se uma avaliação cuidadosa dos pacientes com início agudo ou agravamento inexplicável dos sintomas pulmonares de modo a excluir a DPI. A pomalidomida deve ser interrompida mediante a investigação destes sintomas e se houver confirmação de DPI, deve iniciar-se um tratamento apropriado. A pomalidomida só deverá ser reiniciada após uma avaliação rigorosa dos benefícios e dos riscos.
Segunda Neoplasia Primária
Segunda Neoplasia Primária (SNP), tais como câncer de pele não-melanoma, foram reportadas em pacientes que receberam pomalidomida. A significância clínica dessas observações é incerta. Os médicos devem avaliar cuidadosamente os pacientes antes e durante o tratamento usando a triagem padrão do câncer para a ocorrência de SNP e instituir o tratamento conforme indicado.
Distúrbios Hepáticos
Níveis acentuadamente elevados de alanina aminotransferase e bilirrubina foram observados em pacientes tratados com pomalidomida (vide item "9 - REAÇÕES ADVERSAS"). Também houve casos de hepatite que resultaram na descontinuação de Pomalyst®. O monitoramento regular da função hepática é recomendado durante os primeiros 6 meses de tratamento com pomalidomida, e conforme indicado clinicamente a partir de então.
Infecção
Reativação de hepatite B foi raramente reportada em pacientes que receberam pomalidomida em combinação com dexametasona que foram previamente infectados com o vírus da Hepatite B (HBV). Alguns destes casos progrediram para insuficiência hepática aguda, resultando na descontinuação de Pomalyst®. Recomenda-se cautela quando pomalidomida em combinação com dexametasona for utilizada em pacientes previamente infectados com HBV. Esses pacientes devem ser monitorados atentamente quanto aos sinais e sintomas de infecção ativa por HBV durante toda a terapia.
Leucoencefalopatia multifocal progressiva (LMP)
Foram notificados casos de leucoencefalopatia multifocal progressiva, incluindo casos fatais, com pomalidomida. A LMP foi notificada a partir de vários meses até vários anos após o início do tratamento com pomalidomida. Os casos foram normalmente relatados em doentes que tomavam concomitantemente dexametasona ou com tratamento anterior com outras quimioterapias imunossupressoras. Os médicos devem monitorar os pacientes em intervalos regulares e considerar a possibilidade de LMP no diagnóstico diferencial em pacientes que apresentem novos ou agravamento de sinais ou simtomas neurológicos, cognitivos ou comportamentais. Os pacientes também devem ser aconselhados a informar os companheiros ou cuidadores sobre o seu tratamento, dado que estes podem aperceber-se de sintomas de que o paciente não tenha consciência.
A avaliação para LMP deve basear-se num exame neurológico, na imagem de ressonância magnética do cérebro e na análise do líquido cefalorraquidiano, para detecção de DNA do vírus JC (JCV) através da técnica de reação em cadeia da polimerase (PCR) ou da biópsia cerebral com pesquisa de JCV. Um resultado de PCR negativo para a presença de JCV não exclui a possibilidade de LMP. Poderá ser necessário o acompanhamento e avaliação adicional, caso não seja possível estabelecer um diagnóstico alternativo.
Se houver suspeita de LMP, o tratamento com pomalidomida deve ser suspenso até ter sido excluída a
existência de LMP. Se a LMP se confirmar, a pomalidomida deve ser descontinuada de forma permanente.
Não Compartilhe/Devolva Cápsulas Não Utilizadas
Os pacientes devem ser instruídos a nunca compartilhar Pomalyst® com outra pessoa e a devolver todas cápsulas não utilizadas no local onde o Pomalyst® foi retirado ao final do tratamento.
Doação de Sangue ou Esperma
Todos os indivíduos (homens e mulheres férteis ou não) devem concordar em abster-se de doar sangue ou esperma enquanto estiverem em tratamento com Pomalyst® (incluindo interrupções da dose) e por pelo menos 30 dias após a última dose de Pomalyst®.
Síndrome de Lise Tumoral
Síndrome de lise tumoral (SLT) pode ocorrer em pacientes tratados com Pomalyst®. Pacientes em risco de SLT são aqueles que apresentam carga tumoral elevada antes do tratamento. Estes pacientes devem ser monitorados atentamente e precauções adequadas devem ser adotadas.
Para informações sobre os medicamentos utilizados em combinação com pomalidomida, consultar a bula do respectivo produto.
5.2 Mieloma Múltiplo Recidivado/R