PLUVICTO
NOVARTIS
vipivotida tetraxetana
Antineoplásico.
Apresentações.
Pluvicto® 1.000 MBq/mL (27 mCi/mL) - Solução injetável para uso intravenoso.
Pluvicto® (vipivotida tetraxetana (177 Lu)) apresenta-se em forma de solução injetável/infusão estéril disponível em frasco-ampola.
O frasco de vidro está disponível dentro de um recipiente de chumbo para proteção.
Este medicamento é um produto radiofarmacêutico somente para terapia.
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada mL da solução contém 1 GBq (1.000 MBq) (27 mCi) de vipivotida tetraxetana (177Lu) na data e hora da calibração.
Pluvicto® (vipivotida tetraxetana (177 Lu)) é um agente terapêutico radioligante. Vipivotida tetraxetana (177 Lu) é um PSMA ligante, associado a um quelante DOTA, radiomarcado com lutécio-177.
A quantidade total de radioatividade por frasco de dose única é de 7,4 GBq (7.400 MBq) (200 mCi) ± 10% na data e hora da administração. Dada a atividade volumétrica fixa de 1 GBq/mL (1.000 MBq / mL) (27 mCi/mL) na data e hora da calibração, o volume da solução no frasco é ajustado entre 7,5 mL e 12,5 mL, a fim de fornecer a quantidade necessária de radioatividade na data e hora da administração.
Excipientes: ácido acético, acetato de sódio, ácido gentísico, ascorbato de sódio, ácido pentético e água para injetáveis.
Pluvicto® contém sódio
Cada mL da solução contém até 0,312 mmol (7,1 mg) de sódio. Cada frasco contém até 88,75 mg de sódio (principal componente do sal de cozinha). Isso equivale a 4,4% da ingestão diária máxima recomendada pela OMS de 2 g de sódio para um adulto.
Características físicas
O lutécio-177 decai para um háfnio-177 estável com uma meia-vida física de 6,647 dias emitindo radiação beta-menos com uma energia máxima de 0,498 MeV (79%) e radiação fotônica (c) de 0,208 MeV (11%) e 0,113 MeV (6,4%).
As principais radiações do lutécio-177 estão detalhadas na Tabela 1.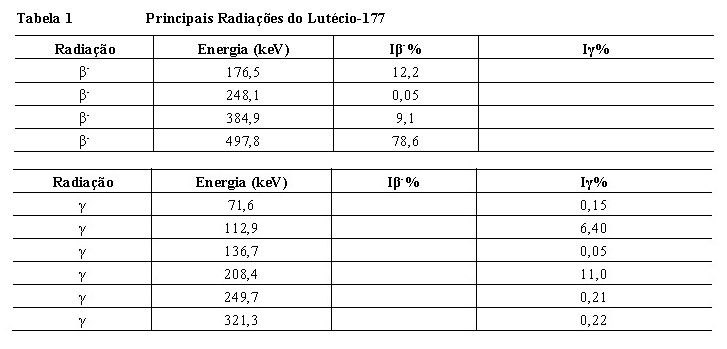
Radiação externa
A Tabela 2 resume as propriedades de decaimento radioativo do lutécio-177.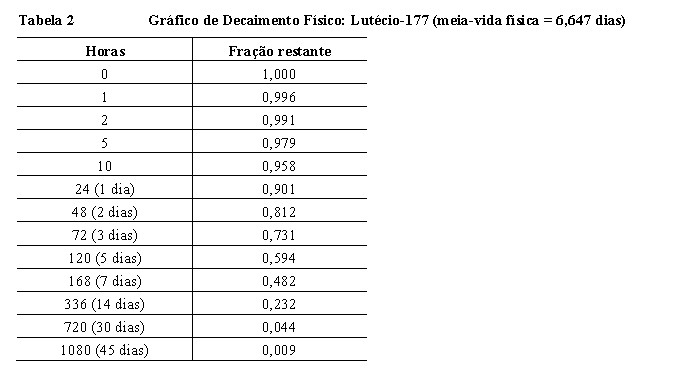
Informações técnicas.
1. INDICAÇÕES
Pluvicto® é indicado para o tratamento de pacientes adultos com câncer de próstata metastático resistente à castração (mCRPC), positivo para antígeno de membrana específico da próstata (PSMA) que foram tratados com inibição da via do receptor de andrógeno (AR) e quimioterapia baseada em taxano.
2. RESULTADOS DE EFICÁCIA
A eficácia de Pluvicto® em pacientes com mCRPC progressivo, PSMA positivo, foi estabelecida no estudo VISION, um estudo de Fase III aberto, multicêntrico e randomizado. Oitocentos e trinta e um (N = 831) pacientes foram randomizados (2:1) para receber Pluvicto® 7,4 GBq (7.400 MBq) (200 mCi) a cada 6 semanas para um total de 6 doses em adição ao melhor padrão de tratamento (BSOC) (N = 551) ou BSOC sozinho (N = 280).
Os pacientes elegíveis deveriam ter mCRPC, PSMA positivo, com status de desempenho (SD) do Eastern Cooperative Oncology Group Performance Status (ECOG PS) de 0 a 2, pelo menos uma lesão metastática presente na tomografia computadorizada (TC), ressonância magnética (MRI) ou cintilografia de imagem óssea, e função renal, hepática e hematológica adequada. Todos os pacientes receberam um análogo de GnRH (hormônio liberador de gonadotrofinas) ou tiveram orquiectomia bilateral prévia. Os pacientes elegíveis também deveriam ter recebido pelo menos um inibidor da via AR, como acetato de abiraterona ou enzalutamida, e 1 ou 2 regimes anteriores de quimioterapia à base de taxano (com um regime definido como uma exposição mínima de 2 ciclos de um taxano). Pacientes com metástases instáveis sintomáticas do sistema nervoso central ou compressão da medula espinhal sintomática ou clinicamente / radiologicamente iminente não eram elegíveis para o estudo. Os pacientes foram submetidos a uma tomografia por emissão de pósitrons (PET) com gálio (68Ga) gozetotida para avaliar a expressão de PSMA em lesões definidas por critérios de leitura central. Os pacientes elegíveis deveriam apresentar mCRPC PSMA positivo definido como tendo pelo menos uma lesão tumoral com captação de gozetotida (68 Ga) maior que a do fígado normal. Os pacientes foram excluídos se quaisquer lesões que excedessem os critérios de tamanho no eixo curto [órgãos ≥ 1 cm, gânglios linfáticos ≥ 2,5 cm, ossos (componentes de tecido mole) ≥ 1 cm] tivessem captação inferior ou igual à captação do fígado normal.
O BSOC administrado a critério do médico incluiu: medidas de suporte, incluindo medicamentos para dor, hidratação, transfusões de sangue, etc.; cetoconazol; terapia de radiação (incluindo forma semeada ou qualquer radioterapia de feixe externo [incluindo radioterapia estereotáxica corporal e feixe externo paliativo]) para alvos de câncer de próstata localizados; agentes direcionados aos ossos, incluindo ácido zoledrônico, denosumabe e quaisquer bisfosfonatos; agentes redutores de andrógeno incluindo qualquer corticosteroide e 5-alfa redutases; inibidores da via AR.
Os pacientes continuaram o tratamento por até 4 - 6 doses, ou até progressão da doença ou toxicidade inaceitável. Pacientes com doença estável ou resposta parcial após 4 doses de Pluvicto® mais BSOC receberam até 2 doses adicionais a critério do investigador.
Os desfechos de eficácia primários alternativos foram sobrevida global (SG) e sobrevida livre de progressão radiográfica (SLPr) pela revisão central independente cega (BICR) de acordo com os critérios do PCWG3. Os desfechos de eficácia secundários adicionais foram a taxa de resposta geral (TRG) por BICR através dos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST) v1.1 e o tempo para o primeiro evento esquelético sintomático (EES) definido como a primeira nova fratura óssea patológica sintomática, compressão da medula espinhal, intervenção cirúrgica ortopédica relacionada ao tumor, necessidade de radioterapia para aliviar a dor óssea ou morte por qualquer causa, o que ocorresse primeiro.
As características demográficas e de linha de base da doença foram equilibradas entre os braços de tratamento. A mediana da idade foi de 71 anos (variação: 40 a 94 anos); 86,8% branco; 6,6% negro ou afro-americano; 2,4% asiático; 92,4% tinham ECOG PS 0-1; 7,6% tinham ECOG PS 2. A randomização foi estratificada pela lactase desidrogenase basal (LDH), presença de metástases hepáticas, pontuação ECOG PS e inclusão de um inibidor da via AR como parte do BSOC no momento da randomização. Na randomização, todos os pacientes (100,0%) receberam pelo menos um regime anterior de quimioterapia à base de taxano e 41,2% dos pacientes receberam dois. Na randomização, 51,3% dos pacientes receberam um inibidor da via AR anterior, 41,0% dos pacientes receberam 2, e 7,7% dos pacientes receberam 3 ou mais. Durante o período de tratamento randomizado, 52,6% dos pacientes no braço Pluvicto® mais BSOC e 67,8% dos pacientes no braço BSOC sozinho receberam pelo menos um inibidor da via AR.
Os resultados de eficácia do VISION são apresentados na Tabela 3 e nas Figuras 1 e 2. As análises finais de Sobrevida Global (SG) e Sobrevida Livre de Progressão radiográfica (SLPr) foram orientadas por eventos e conduzidas após a ocorrência de 530 mortes e 347 eventos, respectivamente. O tratamento com Pluvicto® mais BSOC demonstrou uma melhora estatisticamente significativa em SG e SLPr pelo BICR em comparação ao tratamento com BSOC sozinho. Os resultados de eficácia primária são suportados por uma diferença estatisticamente significativa entre os braços de tratamento no tempo para o primeiro EES (p < 0,001) e ORR (p < 0,001). Houve uma redução de risco estimada de 38% de morte, uma redução de risco estimada de 60% de progressão de doença radiográfica ou morte, e uma redução de risco estimada de EES ou morte de 50% com base nas razões de risco em favor do tratamento de Pluvicto® mais BSOC.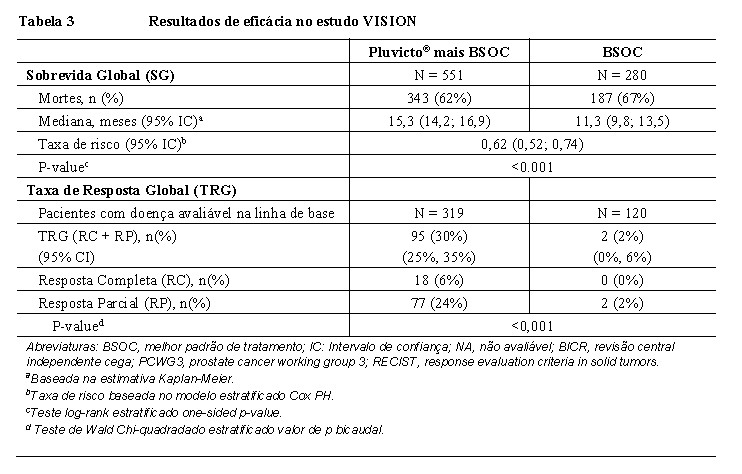
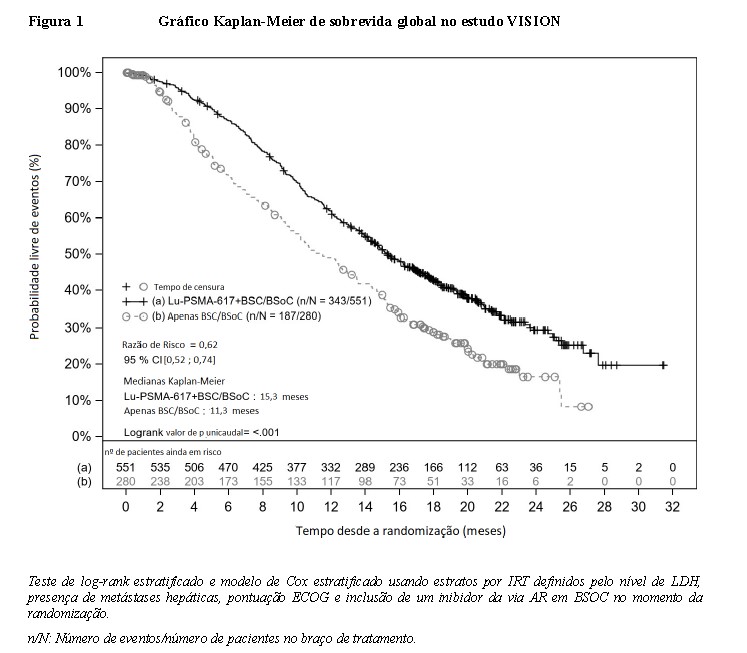
A interpretação da magnitude do efeito na SLPr foi limitada devido à ocorrência elevada de censura pela retirada precoce de pacientes do braço controle.
Referências bibliográficas
1. [Clinical Overview] AAA617 ([177Lu]Lu-PSMA-617]) - 2.5 Clinical Overview in prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer. AAA. 29-Jun-2021.
2. [Clinical Study Report] PSMA-617-01 - VISION: An international, prospective, open-label, multicenter, randomized phase 3 study of 177Lu-PSMA-617 in the treatment of patients with progressive PSMA-positive metastatic castration-resistant prostate cancer (mCRPC). AAA. 28-Jun-2021.
3. [Clinical Study Report - Appendix 16.1.1] PSMA-617-01 - VISION: An international, prospective, open-label, multicenter, randomized phase 3 study of 177Lu-PSMA-617 in the treatment of patients with progressive PSMA-positive metastatic castration-resistant prostate cancer (mCRPC). AAA. 28-Jun-2021.
4. [Summary of Clinical Safety] AAA617 ([177Lu]Lu-PSMA-617]) - 2.7.4 Summary of Clinical Safety in prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer. AAA. 28-Jun-2021. AAA. 2021.
5. [Summary of Clinical Efficacy] AAA617 ([177Lu]Lu-PSMA-617]) - 2.7.3 Summary of Clinical Efficacy in prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer. AAA. 25-Jun-2021.
6. [Clinical Study Report - Appendix 16.1.3] PSMA-617-01 - VISION: An international, prospective, open-label, multicenter, randomized phase 3 study of 177Lu-PSMA-617 in the treatment of patients with progressive PSMA-positive metastatic castration-resistant prostate cancer (mCRPC). AAA. 28-Jun-2021.
7. [Summary of Clinical Safety - Appendix 1] AAA617 ([177Lu]Lu-PSMA-617]) - 2.7.4 Summary of Clinical Safety in prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer. AAA. 28-Jun-2021. AAA. 2021.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico
Grupo farmacoterapêutico: Outros radiofármacos terapêuticos, código ATC: V10XX05
Mecanismo de ação
A porção ativa de Pluvicto® é o radionuclídeo lutécio-177 que está ligado a uma fração de direcionamento que se conecta com alta afinidade ao PSMA, uma proteína transmembrana que é altamente expressa no câncer de próstata, incluindo mCRPC. Após a ligação de Pluvicto® às células cancerígenas que expressam PSMA, a emissão beta menos do lutécio-177 fornece radiação terapêutica à célula alvo, bem como às células vizinhas, e induz danos no DNA que podem levar à morte celular.
Propriedades farmacodinâmicas
Não há dados sobre as relações de exposição-eficácia de vipivotida tetraxetana (177Lu) e o tempo para a resposta farmacodinâmica.
Existem dados limitados sobre as relações de segurança de exposição a vipivotida tetraxetana (177Lu) e o tempo para a resposta farmacodinâmica.
A vipivotida tetraxetana não marcada não possui atividade farmacodinâmica.
Eletrofisiologia cardíaca
A capacidade do Pluvicto® de prolongar o intervalo QTc na dose recomendada foi avaliada em 30 pacientes no sub-estudo Fase III VISION. Na dose recomendada, Pluvicto® não levou a um maior aumento médio ( > 20 ms) no intervalo QTc.
Propriedades farmacocinéticas (PK)
A farmacocinética de vipivotida tetraxetana (177Lu) foi investigada em 30 pacientes no sub-estudo do estudo de fase III VISION.
Absorção
Pluvicto® é administrado por via intravenosa e está imediata e completamente biodisponível.
A média geométrica de exposição sanguínea (área sob a curva [ASCinf]) para vipivotida tetraxetana (177Lu) na dose recomendada é de 52,3 ng.h/mL (coeficiente de variação geométrico médio [CV] 31,4%). A média geométrica da concentração sanguínea máxima (Cmax) para vipivotida tetraxetana (177Lu) é de 6,58 ng/mL (CV 43,5%).
Distribuição
A média geométrica do volume de distribuição (Vz) para vipivotida tetraxetana (177Lu) é de 123 L (CV 78,1%).
Vipivotida tetraxetana não marcada e vipivotida tetraxetana marcada com lutécio não radioativo (175Lu) estão, cada uma, 60% a 70% ligadas às proteínas plasmáticas humanas.
Absorção nos órgãos
Dentro de 2,5 horas após a administração, vipivotida tetraxetana (177Lu) distribui-se no trato gastrointestinal, fígado, pulmões, rins, parede do coração, medula óssea e glândulas salivares.
Eliminação
A média geométrica do clearance (CL) para vipivotida tetraxetana (177Lu) é 2,04 L/h (CV 31,5%).
Meia-vida
Pluvicto® apresenta eliminação bi-exponencial com média geométrica de meia vida de eliminação terminal (T½) de 41,6 horas (CV 68,8%).
Metabolismo
Vipivotida tetraxetana (177Lu) não sofre metabolismo hepático ou renal.
Eliminação
Vipivotida tetraxetana (177Lu) é eliminado principalmente por via renal.
Efeitos da idade, peso e insuficiência renal
Não foram identificados efeitos clinicamente significativos nos parâmetros farmacocinéticos de vipivotida tetraxetana (177Lu) para as seguintes covariáveis avaliadas em 30 pacientes no sub-estudo Fase III VISION: idade (mediana: 67 anos; intervalo: 52 a 80 anos), peso corporal (mediana: 88,8 kg; intervalo: 63,8 a 143,0 kg), insuficiência renal leve a moderada (CLcr basal 30 a 89 mL/min por Cockcroft-Gault). A exposição (ASC) da vipivotida tetraxetana (177Lu) aumentou com a diminuição da depuração da creatinina (CLcr). O efeito na linha de base CLcr < 54 mL/min na farmacocinética do vipivotida tetraxetana (177Lu) não foi estudado.
Dados de segurança não clínica
Farmacologia de segurança e toxicologia animal
Nenhum efeito toxicológico foi observado em estudos de farmacologia de segurança ou de toxicidade de dose única em ratos e mini suínos nos quais foi administrada uma formulação não radioativa contendo vipivotida tetraxetana não marcada e vipivotida tetraxetana marcada com lutécio não radioativo (175Lu), ou em estudos de toxicidade de dose repetida em ratos que receberam vipivotida tetraxetana não marcada.
Carcinogenicidade e mutagenicidade
Estudos de mutagenicidade e carcinogenicidade de longo prazo não foram realizados com vipivotida tetraxetana (177Lu), no entanto, a radiação é cancerígena e mutagênica.
Toxidade reprodutiva
Para informações sobre toxicidade reprodutiva, ver item "Gravidez, lactação e mulheres e homens com potencial reprodutivo" da seção "5. ADVERTÊNCIAS E PRECAUÇÕES".
Avaliação de potenciais interações medicamentosas in vitro
Enzimas CYP450
Vipivotida tetraxetana não é um substrato das enzimas do citocromo P450 (CYP450).
Ele não induz o citocromo P450 (CYP) 1A2, 2B6 ou 3A4, e não inibe o citocromo P450 (CYP) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 ou 3A4/5 in vitro.
Transportadores
Vipivotida tetraxetana não é um substrato de BCRP, P-gp, MATE1, MATE2-K, OAT1, OAT3 ou OCT2, e não é um inibidor de BCRP, P-gp, BSEP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 ou OCT2 in vitro.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a algum dos excipientes listados no início da bula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Risco de exposição à radiação
Pluvicto® contribui para a exposição cumulativa geral de longo prazo do paciente à radiação. A exposição cumulativa de longo prazo à radiação está associada a um risco aumentado de câncer.
A exposição à radiação de pacientes, pessoal médico e contatos domiciliares deve ser minimizada durante e após o tratamento com Pluvicto®, consistente com as boas práticas institucionais de radioproteção, procedimentos de gerenciamento de pacientes e instruções ao paciente para o acompanhamento da proteção a radiação em casa.
Os pacientes devem ser encorajados a aumentar a ingestão de líquidos e orientados a urinar com a maior frequência possível para reduzir a radiação da bexiga, especialmente após atividades intensas, por exemplo, terapia com radionuclídeos.
Antes do paciente ser liberado, o médico de medicina nuclear ou prestador de cuidados de saúde deve explicar as precauções necessárias de radioproteção que o paciente deve seguir para minimizar a exposição à radiação a outros.
Após a administração de Pluvicto®, os pacientes devem ser orientados a:
• limitar o contato próximo (pelo menos 1 metro) com contatos domésticos por 2 dias, ou com crianças e gestantes por 7 dias.
• abster-se de atividade sexual por 7 dias.
• dormir em um quarto separado dos contatos domésticos por 3 dias, de crianças por 7 dias, ou de gestantes por 15 dias.
Mielossupressão
Pluvicto® pode causar mielossupressão grave e potencialmente fatal, incluindo anemia, trombocitopenia, leucopenia e neutropenia. No estudo VISION, a mielossupressão ocorreu com mais frequência em pacientes que receberam Pluvicto® mais o melhor padrão de tratamento (BSOC) em comparação com pacientes que receberam apenas BSOC.
Testes laboratoriais de hematologia, incluindo hemoglobina, contagem de glóbulos brancos, contagem absoluta de neutrófilos e contagem de plaquetas, devem ser realizados antes e durante o tratamento com Pluvicto®. Pluvicto® deve ser suspenso, a dose reduzida ou descontinuada permanentemente e os pacientes devem ser clinicamente manejados, conforme apropriado com base na gravidade da mielossupressão (ver seção "8 POSOLOGIA E MODO DE USAR").
Toxicidade renal
Pluvicto® pode causar toxicidade renal grave. No estudo VISION, a toxicidade renal ocorreu com mais frequência em pacientes que receberam Pluvicto® mais BSOC em comparação com pacientes que receberam apenas BSOC (ver seção "9 REAÇÕES ADVERSAS").
Os pacientes devem ser orientados a manter-se bem hidratados e a urinar frequentemente antes e após a administração de Pluvicto®. Monitore frequentemente a função renal e as reações adversas em pacientes com insuficiência renal leve (CLcr 60 a 89 mL/min de base por Cockcroft-Gault) a moderada (CLcr 30 a 59 mL/min). Testes laboratoriais de função renal, incluindo creatinina sérica e CLcr calculado, devem ser realizados antes e durante o tratamento com Pluvicto®.
Pluvicto® deve ser suspenso, a dose reduzida ou descontinuada permanentemente com base na gravidade da toxicidade renal (ver seção "8 POSOLOGIA E MODO DE USAR").
Monitoramento do tratamento
Testes laboratoriais devem ser realizados antes e durante o tratamento com Pluvicto®.
• Hematologia (hemoglobina, contagem de leucócitos, contagem absoluta de neutrófilos, contagem de plaquetas);
• Função renal (creatinina sérica, depuração de creatinina calculada [CLcr]);
• Função hepática (alanina aminotransferase, aspartato aminotransferase, fosfatase alcalina, albumina sérica sanguínea, bilirrubina total no sangue);
Gravidez, lactação e mulheres e homens com potencial reprodutivo
Gravidez
Resumo do risco
A segurança e eficácia de Pluvicto® não foram estabelecidas em mulheres, uma vez que Pluvicto® não é indicado para uso em mulheres. Não foram conduzidos estudos em animais usando vipivotida tetraxetana (177Lu) para avaliar seu efeito na reprodução feminina e no desenvolvimento embrio-fetal; no entanto, todos os radiofármacos, incluindo o Pluvicto®, têm o potencial de causar dano fetal. Com base no seu mecanismo de ação, Pluvicto® pode causar danos fetais quando administrado a mulheres grávidas (ver seção "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Lactação
Resumo de Risco
A segurança e eficácia de Pluvicto® não foram estabelecidas em mulheres, pois Pluvicto® não é indicado para uso em mulheres. Não há dados sobre a presença de vipivotida tetraxetana (177Lu) no leite humano, seus efeitos na criança amamentada ou na produção de leite.
Contracepção
Sexo masculino
Com base no seu mecanismo de ação, os pacientes do sexo masculino devem ser aconselhados a não conceberem e a utilizarem preservativos nas relações sexuais durante o tratamento com Pluvicto® e durante 14 semanas após a última dose (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Infertilidade
Não foram realizados estudos para determinar os efeitos de vipivotida tetraxetana (177Lu) na fertilidade. A dose cumulativa recomendada de 44,4 GBq (44.400 MBq) de Pluvicto® resulta em uma dose de radiação absorvida nos testículos dentro do intervalo em que Pluvicto® pode causar infertilidade.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos clínicos de interação medicamentosa.
Incompatibilidades
Este medicamento não deve ser misturado com outros medicamentos, exceto os mencionados na seção 8 -POSOLOGIA E MODO DE USAR.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
O armazenamento de Pluvicto® deve estar de acordo com as regulações nacionais sobre materiais radioativos.
Conservar em temperatura abaixo de 30°C. Não congelar. Conservar na embalagem original para proteger da radiação ionizante (blindagem de chumbo).
O prazo de validade é de 120 horas (5 dias) a partir da data e hora da calibração.
Não use Pluvicto® após a data e hora de validade do produto que está descrito na embalagem.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
Pluvicto® é fornecido em frasco de vidro tipo I transparente e incolor, fechado com rolha de borracha bromobutílica e selo de alumínio.
Cada frasco contém um volume de solução que pode variar de 7,5 mL a 12,5 mL, correspondendo a uma radioatividade de 7,4 GBq (7.400 MBq) (200 mCi) ± 10% na data e hora da administração.
O frasco é fechado dentro de um recipiente de chumbo com função de blindagem protetora.
Pluvicto® é uma solução límpida transparente ou levemente amarelada de pH 4,5 a 7,0.
Precauções especiais de descarte
Qualquer medicamento não utilizado ou resíduos devem ser descartados de acordo com os regulamentos nacionais.
O lutécio-177 pode ser preparado usando duas fontes diferentes de isótopos estáveis (ou lutécio-176 ou itérbio-176) que requerem gerenciamento de resíduos diferente. O lutécio-177 é preparado usando itérbio-176 ("sem adição de carreador"), a menos que comunicado de outra forma no certificado de liberação do lote do produto.
Antes de usar, observe o aspecto do medicamento.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
8. POSOLOGIA E MODO DE USAR
Instruções de segurança importantes
Pluvicto® é um radiofármaco e deve ser manuseado com as medidas de segurança adequadas para minimizar a exposição à radiação. Luvas impermeáveis e proteção eficaz contra radiação devem ser usadas ao manusear Pluvicto®.
Os radiofármacos, incluindo o Pluvicto®, devem ser usados por ou sob o controle de profissionais de saúde qualificados por treinamento específico e experiência no uso e manuseio seguro de radiofármacos, e devidamente certificado para tal atividade pelo órgão competente.
Identificação do paciente
Os pacientes com mCRPC previamente tratados devem ser identificados para tratamento com Pluvicto® com o uso de um agente de imagem com PSMA regularizado, com base na expressão de PSMA nos tumores. Critérios de seleção adicionais foram usados no estudo VISION.
Regime de dose
A dose recomendada de Pluvicto® é de 7,4 GBq (7.400 MBq) (200 mCi) por via intravenosa a cada 6 semanas (± 1 semana), para um total de 6 doses.
Modificações de dose para reações adversas
As modificações de dose recomendadas de Pluvicto® para pacientes que apresentem reações adversas são fornecidas na Tabela 4. O tratamento de reações adversas graves ou intoleráveis pode exigir a interrupção temporária da dose (prolongando o intervalo entre as doses de a cada 6 semanas para até a cada 10 semanas), redução da dose ou descontinuação permanente do tratamento com Pluvicto®. Se um atraso no tratamento devido a uma reação adversa persistir por > 4 semanas, o tratamento com Pluvicto® deve ser descontinuado. A dose de Pluvicto® pode ser reduzida uma vez em 20% para 5,9 GBq (160 mCi).; a dose não deve ser re-escalada. Se o paciente apresentar outras reações adversas ao medicamento que exijam uma redução adicional da dose, o tratamento com Pluvicto® deve ser descontinuado.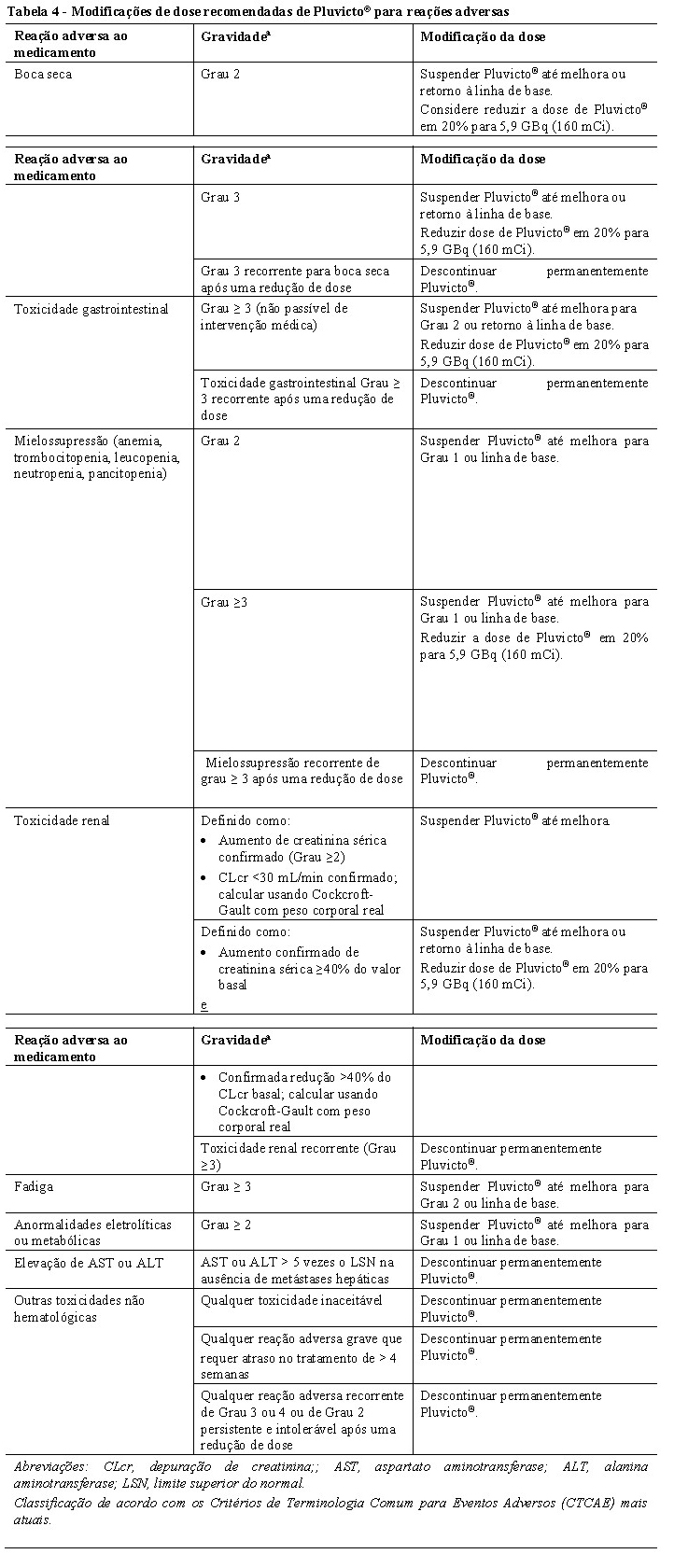
Populações especiais
Insuficiência renal
Nenhum ajuste de dose é recomendado para pacientes com insuficiência renal leve (CLcr de linha basal 60 a 89 mL/min por Cockcroft-Gault) a moderada (CLcr 30 a 59 mL/min). O perfil farmacocinético e a segurança de Pluvicto® não foram estudados em pacientes com insuficiência renal grave (CLcr 15 a 29 mL/min) ou doença renal em estágio terminal.
Insuficiência hepática
Nenhum ajuste de dose é recomendado para pacientes com insuficiência hepática.
Pacientes pediátricos (menores de 18 anos)
A segurança e a eficácia do Pluvicto® em pacientes pediátricos não foram estabelecidas.
Pacientes geriátricos (65 anos ou mais)
Não é recomendado ajuste de dose em pacientes com 65 anos ou mais.
Modo de Usar
Instruções de preparo
• A técnica asséptica e a blindagem por radiação devem ser usadas ao manusear ou administrar Pluvicto®, usando pinças conforme necessário para minimizar a exposição à radiação.
• O frasco deve ser inspecionado visualmente, sob uma tela blindada, para material particulado e descoloração antes da administração. O frasco deve ser descartado se partículas ou descoloração estiverem presentes.
• Pluvicto® é uma solução pronta para uso de uso único. A solução de Pluvicto® não deve ser injetada diretamente em qualquer outra solução intravenosa.
• A quantidade de radioatividade administrada ao paciente deve ser confirmada com um calibrador de dose devidamente calibrado antes e depois da administração de Pluvicto®.
• Qualquer medicamento não usado ou material residual deve ser descartado de acordo com as normas nacionais.
Instruções de administração
A dose recomendada de Pluvicto® pode ser administrada por via intravenosa como uma injeção usando uma seringa descartável equipada com um protetor de seringa (com ou sem uma bomba de seringa), como uma infusão usando o método de gravidade (com ou sem uma bomba de infusão), ou como uma infusão usando o frasco (com uma bomba de infusão peristáltica).
Uma dose reduzida de Pluvicto® deve ser administrada utilizando o método de seringa (com ou sem uma bomba de seringa) ou o método de frasco (com uma bomba de infusão peristáltica). O uso do método de gravidade para administrar uma dose reduzida de Pluvicto® não é recomendado, pois pode resultar na administração do volume incorreto de Pluvicto® se a dose não for ajustada antes da administração.
Antes da administração, lave o cateter intravenoso utilizado exclusivamente para a administração de Pluvicto® com ≥ 10 mL de solução de cloreto de sódio estéril a 0,9% para garantir a desobstrução e minimizar o risco de extravasamento. Os casos de extravasamento devem ser gerenciados de acordo com as diretrizes institucionais.
Métodos de administração intravenosa
Instruções para o método de seringa (com ou sem bomba de seringa)
• Após desinfetar a tampa do frasco, retire um volume adequado de solução de Pluvicto® para fornecer a radioatividade desejada usando uma seringa descartável equipada com um protetor de seringa e uma agulha estéril descartável.
• Administre Pluvicto® ao paciente por injeção intravenosa lenta dentro de aproximadamente 1 a 10 minutos (com uma bomba de seringa ou manualmente sem uma bomba de seringa) através de um cateter intravenoso que é pré-preenchido com solução de cloreto de sódio estéril a 0,9% e que é usado exclusivamente para a administração de Pluvicto® ao paciente.
• Uma vez que a radioatividade desejada de Pluvicto® tenha sido administrada, realize uma lavagem intravenosa de ≥10 mL de solução estéril de cloreto de sódio a 0,9% através do cateter intravenoso para o paciente.
Instruções para o método de gravidade (com ou sem bomba de infusão)
• Insira uma agulha de 2,5 cm de calibre 20 (agulha curta) no frasco de Pluvicto® e conecte-a através de um cateter a uma solução de cloreto de sódio estéril a 0,9% de 500 mL (usada para transportar a solução de Pluvicto® durante a infusão). Certifique-se de que a agulha curta não toque a solução de Pluvicto® no frasco e não conecte a agulha curta diretamente ao paciente. Não permita que a solução de cloreto de sódio flua para o frasco de Pluvicto® antes do início da perfusão de Pluvicto® e não injete a solução de Pluvicto® diretamente na solução de cloreto de sódio.
• Insira uma segunda agulha de 9 cm, calibre 18 (agulha longa) no frasco de Pluvicto®, assegurando-se de que a agulha longa toque e esteja presa ao fundo do frasco de Pluvicto® durante toda a infusão. Conecte a agulha longa ao paciente por meio de um cateter intravenoso pré-preenchido com solução estéril de cloreto de sódio a 0,9% e que é usado exclusivamente para a infusão de Pluvicto® no paciente.
• Utilize uma pinça ou uma bomba de perfusão para regular o fluxo da solução de cloreto de sódio através da agulha curta para o frasco de Pluvicto® (a solução de cloreto de sódio que entra no frasco através da agulha curta transportará a solução de Pluvicto® do frasco para o paciente através do tubo cateter conectado à agulha longa em aproximadamente 30 minutos).
• Durante a infusão, certifique-se de que o nível de solução no frasco de Pluvicto® permanece constante.
• Desconecte o frasco do cateter da agulha longa e interrompa o fluxo do cateter da solução salina assim que o nível de radioatividade estiver estável por pelo menos cinco minutos.
• Siga a infusão com uma lavagem intravenosa de ≥10 mL de solução estéril de cloreto de sódio a 0,9% através do cateter intravenoso até o paciente.
Instruções para o método do frasco (com bomba de infusão peristáltica)
• Insira uma agulha de calibre 20 de 2,5 cm (agulha de ventilação curta) no frasco de Pluvicto®. Certifique-se de que a agulha curta não toque na solução de Pluvicto® no frasco e não conecte a agulha curta diretamente ao paciente ou à bomba de infusão peristáltica.
• Insira uma segunda agulha de 9 cm, calibre 18 (agulha longa) no frasco de Pluvicto®, assegurando-se de que a agulha longa toque e esteja presa ao fundo do frasco de Pluvicto® durante toda a infusão. Conecte a agulha longa e uma solução de cloreto de sódio estéril a 0,9% a uma válvula de 3 vias através de um tubo apropriado.
• Conecte a saída da válvula de 3 vias à tubulação instalada no lado de entrada da bomba de infusão peristáltica seguindo as instruções do fabricante da bomba.
• Preencha a linha abrindo a torneira de 3 vias e bombeando a solução de Pluvicto® pela tubulação até atingir a saída da válvula.
• Preencha o cateter intravenoso que será conectado ao paciente abrindo a válvula de 3 vias para a solução de cloreto de sódio estéril a 0,9% e bombeando a solução de cloreto de sódio estéril a 0,9% até que ela saia da extremidade do tubo do cateter.
• Conecte o cateter intravenoso pré-preenchido ao paciente e ajuste a válvula de 3 vias de modo que a solução de Pluvicto® fique alinhada com a bomba de infusão peristáltica.
• Infundir um volume apropriado de solução de Pluvicto® a aproximadamente 25 mL/h para administrar a radioatividade desejada.
• Quando a radioatividade de Pluvicto® desejada for administrada, pare a bomba de infusão peristáltica e, em seguida, mude a posição da válvula de 3 vias para que a bomba de infusão peristáltica fique alinhada com a solução de cloreto de sódio estéril a 0,9%. Reinicie a bomba de infusão peristáltica e infunda uma irrigação intravenosa de ≥10 mL de solução de cloreto de sódio estéril a 0,9% através do cateter intravenoso para o paciente.
Dosimetria de radiação
Dados de dosimetria de vipivotida tetraxetana (177Lu) foram coletados em 29 pacientes no sub-estudo do estudo Fase III VISION, a fim de calcular a dosimetria de radiação de corpo inteiro e órgãos. A média e o desvio padrão (DP) das doses estimadas de radiação absorvida em diferentes órgãos para pacientes adultos recebendo Pluvicto® são mostrados na Tabela 5. Os órgãos com as maiores doses absorvidas de radiação são as glândulas lacrimais, glândulas salivares, intestino grosso (cólon esquerdo e direito), rins e parede da bexiga urinária.
A penetração máxima do lutécio-177 no tecido é de aproximadamente 2 mm e a penetração média é de 0,67 mm.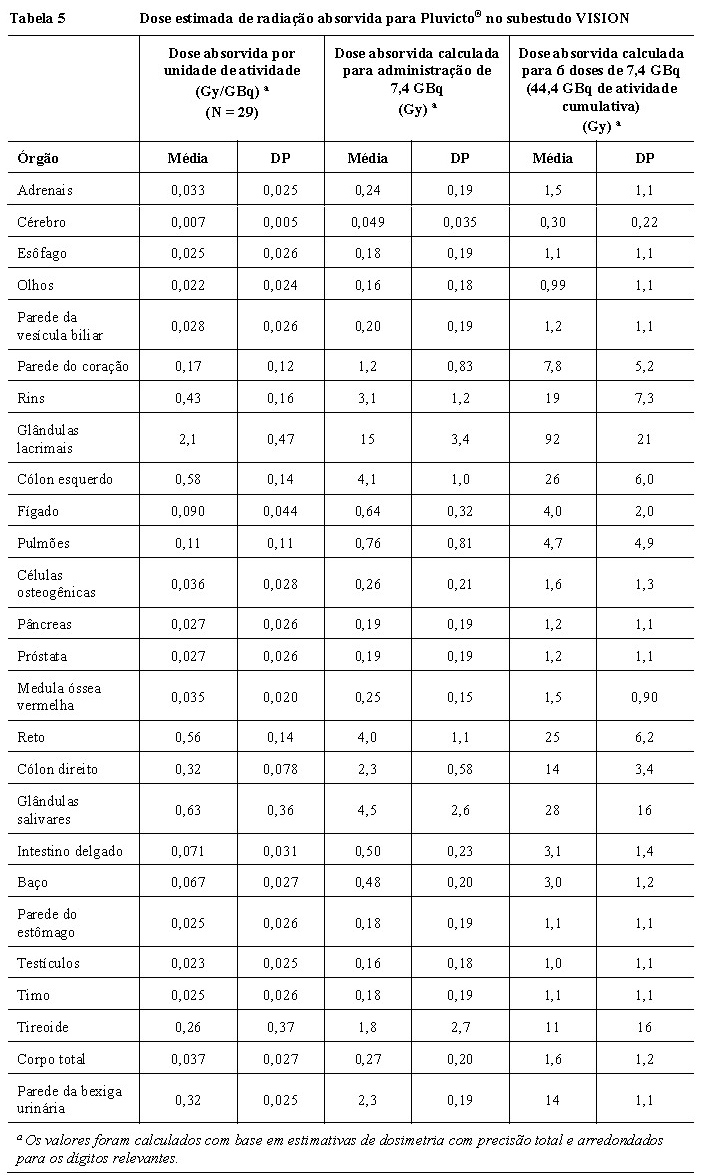
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A segurança de Pluvicto® foi avaliada no estudo de Fase III VISION em pacientes com mCRPC progressivo PSMA-positivo. Dos 831 pacientes randomizados, 734 pacientes receberam pelo menos uma dose de tratamento randomizado. Os pacientes receberam pelo menos uma dose de Pluvicto® 7,4 GBq (7.400 MBq) (200 mCi) administrado a cada 6 a 10 semanas mais BSOC (N = 529) ou BSOC sozinho (N = 205).
Entre os pacientes que receberam Pluvicto® mais BSOC, o número mediano de doses de Pluvicto® recebido foi de 5 (intervalo: 1 a 6), sendo que 67,7% dos pacientes receberam pelo menos 4 doses de Pluvicto® e 46,5% dos pacientes receberam um total de 6 doses de Pluvicto®. A dose cumulativa mediana de Pluvicto® foi de 37,5 GBq (intervalo: 7,0 a 48,3). A duração mediana da exposição ao tratamento randomizado foi de 7,8 meses (intervalo: 0,3 a 24,9) para pacientes que receberam Pluvicto® mais BSOC e 2,1 meses (intervalo: 0,0 a 26,0) para pacientes que receberam apenas BSOC, totalizando um período de seguimento mediano de 14,8 meses no estudo, para o braço Pluvicto® mais BSoC.
A Tabela 6 resume a incidência de reações adversas. As reações adversas mais comuns (≥20%) que ocorrem com maior incidência em pacientes que receberam Pluvicto® mais BSOC em comparação com BSOC sozinho incluem: fadiga (43,1%), boca seca (39,3%), náusea (35,3%), anemia (31,8%), diminuição do apetite (21,2%) e constipação (20,2%). As reações adversas de Grau 3 a 4 mais comuns (≥5%) que ocorreram com maior incidência em pacientes que receberam Pluvicto® mais BSOC em comparação com BSOC sozinho incluem: anemia (12,9%), trombocitopenia (7,9%), linfopenia (7,8%) e fadiga (5,9%).
Reações adversas graves, ocorreram em 36% dos pacientes que receberam Pluvicto® mais BSoC. As reações adversas graves em > 1% dos pacientes que receberam Pluvicto® mais BSoC incluíram hemorragia (4%), dor musculoesquelética (3,8%), sepse (3,2%), anemia (2,8%), infecção do trato urinário (2,6%), lesão renal aguda (1,7%), pneumonia (1,7%), pancitopenia (1,3%), pirexia (1,3%), compressão da medula espinhal (1,1%) e embolia pulmonar (1,1%).
Reações adversas fatais, ocorreram em 2,8% dos pacientes que receberam Pluvicto® mais BSoC, incluindo sepse (0,9%), pancitopenia (0,6%), insuficiência hepática (0,4%), hemorragia intracraniana (0,2%), hematoma subdural (0,2%), acidente vascular cerebral isquêmico (0,2%), COVID-19 (0,2%) e pneumonia por aspiração (0,2%).
Pluvicto® foi permanentemente descontinuado devido a reações adversas, em 12% dos pacientes. As reações adversas que levaram à descontinuação permanente de Pluvicto® em ≥ 1% dos pacientes que receberam Pluvicto® mais BSoC foram anemia (2,8%), trombocitopenia (2,8%) e leucopenia (incluindo neutropenia) (1,7%).
As reações adversas, que levaram à interrupção da dose de Pluvicto® ocorreram em 16% dos pacientes. As reações adversas mais frequentes (≥ 3%) que levaram à interrupção da dose de Pluvicto® em pacientes que receberam Pluvicto® mais BSoC foram anemia (5%) e trombocitopenia (3,6%). As reações adversas que levaram à redução da dose de Pluvicto® ocorreram em 6% dos pacientes. As reações adversas mais frequentes (≥ 1%) que levaram a uma redução da dose de Pluvicto® em pacientes que receberam Pluvicto® mais BSoC foram trombocitopenia (1,9%) e anemia (1,3%).
As reações adversas (Tabela 6) estão listadas por classe de sistema de órgãos MedDRA. Dentro de cada classe de sistema de órgãos, as reações adversas são classificadas por frequência, com as reações mais frequentes em primeiro lugar. Além disso, a categoria de frequência correspondente para cada reação adversa é baseada na seguinte convenção (CIOMS III): muito comum (≥ 1/10); comum (≥ 1/100 a < 1/10); incomum (≥ 1/1.000 a < 1/100); raros (≥ 1/10.000 a < 1/1.000); muito raros ( < 1/10.000).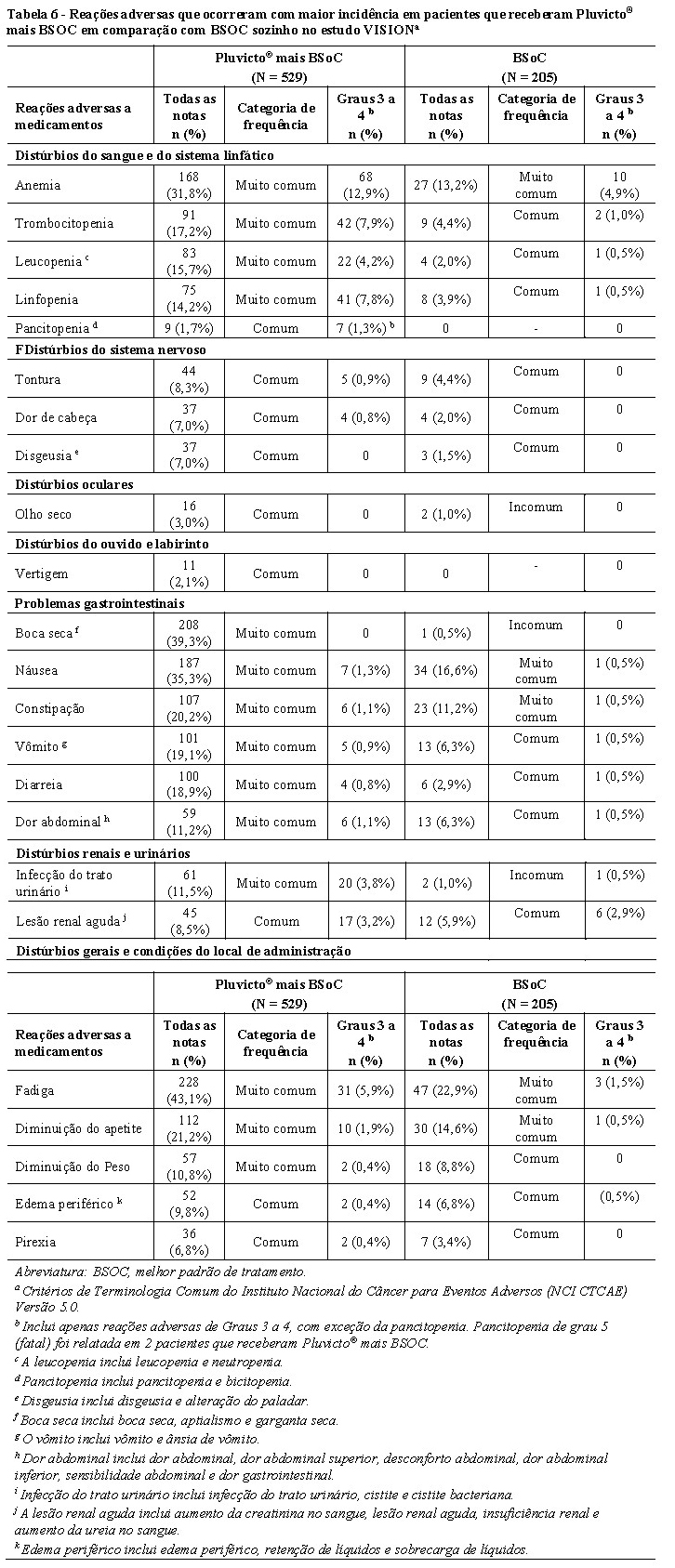
Descrição de algumas reações adversas ao medicamento
Mielossupressão
No estudo VISION, a mielossupressão ocorreu com mais frequência em pacientes que receberam Pluvicto® mais BSOC em comparação com pacientes que receberam apenas BSOC (todos os Graus/Grau ≥3): anemia (31,8% /12,9%) versus (13,2%/4,9%); trombocitopenia (17,2%/7,9%) versus (4,4%/1,0%); leucopenia (12,5%/2,5%) versus (2,0%/0,5%); linfopenia (14,2%/7,8%) versus (3,9%/0,5%); neutropenia (8,5%/3,4%) versus (1,5%/0,5%); pancitopenia (1,5%/1,1%) versus (0%/0%) incluindo dois eventos fatais de pancitopenia em pacientes que receberam Pluvicto® mais BSOC; e bicitopenia (0,2%/0,2%) versus (0%/0%).
As reações adversas de mielossupressão que levaram à descontinuação permanente em ≥ 0,5% dos pacientes que receberam Pluvicto® mais BSOC incluíram: anemia (2,8%), trombocitopenia