ONIVYDE
SERVIER
irinotecano
Antineoplásico.
Apresentações.
ONIVYDE® é uma suspensão injetável de liberação prolongada apresentada em embalagem com 1 frasco ampola de 10 mL contendo 43 mg de irinotecano lipossomal peguilado (4,3 mg/mL).
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada mL de ONIVYDE® contém: irinotecano1 4,3 mg. Excipientes2 q.s.p. 1 mL
1 Cada 4,3 mg de irinotecano (como base livre anidra do sal sucralfato de irinotecano em formulação lipossomal peguilada) equivale a 5,0 mg de cloridrato de irinotecano tri-hidratado.
2Excipientes: DSPC (1,2-distearoil-sn-glicero-3-fosfocolina), colesterol, MPEG 2000 DSPE (N-(carbonilmetoxipolietilenoglicol-2000)-1,2-distearoil-sn-glicero-3-fosfoetanolamina), HEPES (ácido 2-[4-(2-hidroxietil)-1-piperazinetanosulfônico), cloreto de sódio, octassulfato de sacarose, água para injetáveis. Traços de dextrose, etanol, hidróxido de sódio e ácido sulfúrico.
Contém 0,144 mmol (3,31 mg) de sódio.
ONIVYDE® é uma formulação lipossomal, não equivalente a outras formulações não lipossomais de irinotecano.
Informações técnicas.
1. INDICAÇÕES
ONYVIDE® é indicado para o tratamento de adenocarcinoma metastático do pâncreas, em combinação com 5-fluoruracila (5-FU) e leucovorina (LV), em pacientes adultos com progressão da doença após terapia à base de gencitabina.
2. RESULTADOS DE EFICÁCIA
A segurança e a eficácia de ONIVYDE® foram investigadas em um estudo clínico multinacional, randomizado, aberto e controlado (NAPOLI-1) que testou dois regimes de tratamento para pacientes com adenocarcinoma pancreático metastático com progressão documentada da doença após tratamento com gencitabina ou esquema quimioterápico contendo gencitabina. O estudo foi desenhado para avaliar a eficácia clínica e a segurança de ONIVYDE® em monoterapia ou ONIVYDE® +5-FU/LV em comparação com um braço controle ativo de 5-FU/LV.
Os pacientes randomizados para ONIVYDE® +5-FU/LV receberam ONIVYDE® (70 mg/m2) através de infusão intravenosa durante 90 minutos, seguido por LV 400 mg/m2 por via intravenosa durante 30 minutos, seguido por 5-FU 2.400 mg/m2 por via intravenosa durante 46 horas, administrados a cada 2 semanas. Os pacientes homozigóticos para o alelo UGT1A1*28 receberam uma dose inicial mais baixa de ONIVYDE® (ver item 8. POSOLOGIA E MODO DE USAR). Os pacientes randomizados para 5-FU/LV receberam leucovorina 200 mg/m2 por via intravenosa durante 30 minutos, seguido por 5-FU 2.000 mg/m2 por via intravenosa durante 24 horas, administrados nos dias 1, 8, 15 e 22 de um ciclo de 6 semanas. Os pacientes randomizados para monoterapia com ONIVYDE® receberam 100 mg/m2 através infusão intravenosa durante 90 minutos a cada 3 semanas.
Os principais critérios de elegibilidade para pacientes com adenocarcinoma metastático do pâncreas no estudo clínico NAPOLI 1 foram Karnofsky Performance Status (KPS) ≥ 70, nível de bilirrubina normal, níveis de transaminase ≤ 2,5 vezes o limite superior normal (LSN) ou ≤ 5 vezes o LSN para pacientes com metástases hepáticas e albumina ≥ 3,0 g/dl.
Um total de 417 pacientes foram randomizados para os seguintes braços: ONIVYDE® +5-FU/LV (N=117), ONIVYDE® em monoterapia (N=151) e 5-FU/LV (N=149). As características clínicas e demográficas basais foram bem equilibradas entre os braços do estudo.
Na população com intenção de tratar (todos os pacientes randomizados), a idade mediana foi de 63 anos (intervalo de 31 a 87 anos), 57% eram do sexo masculino, 61% eram caucasianos e 33% eram asiáticos. O nível basal médio de albumina foi de 3,6 g/dl, e o KPS basal foi de 90-100 em 55% dos pacientes. Com relação às características da doença:68% dos pacientes apresentavam metástases hepáticas e 31% metástases pulmonares; 12% dos pacientes não receberam tratamento em linhas anteriores para doença metastática, 56% dos pacientes receberam 1 linha de tratamento anterior para doença metastática, 32% dos pacientes receberam 2 ou mais linhas prévias de tratamento para doença metastática.
Os pacientes receberam tratamento até à progressão da doença ou toxicidade inaceitável. O desfecho primário foi a sobrevida global (OS). Desfechos adicionais incluíram sobrevida livre de progressão (PFS) e taxa de resposta objetiva (ORR). Os resultados são mostrados na Tabela 1. A sobrevida global é ilustrada na Figura 1.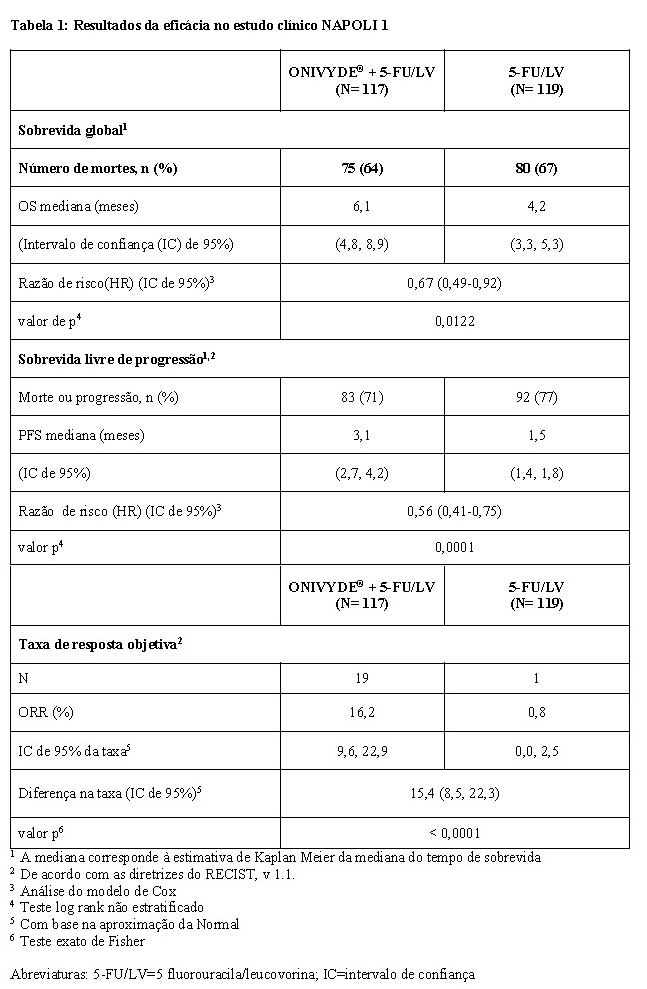
No número limitado de pacientes com exposição prévia ao irinotecano não lipossomal, não foi demonstrado benefício de ONIVYDE®.
População pediátrica
A segurança e a eficácia de ONIVYDE® em crianças e adolescentes com idade inferior a 18 anos não foram estabelecidas. Não há dados disponíveis.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Inibidores da Topoisomerase I (TOP1). Código ATC: L01CE02
Mecanismo de ação
A substância ativa de ONIVYDE® é o irinotecano (inibidor da topoisomerase I) encapsulado em uma vesícula ou lipossoma de bicamada lipídica.
O irinotecano é um derivado da camptotecina. As camptotecinas atuam como inibidores específicos da enzima DNA topoisomerase I. O irinotecano e o seu metabolito ativo SN 38 ligam-se reversivelmente ao complexo topoisomerase I-DNA e induzem lesões na cadeia simples do DNA que bloqueiam a forquilha de replicação do DNA, sendo responsáveis pela citotoxicidade. O irinotecano é metabolizado pela carboxilesterase em SN-38. O SN-38 é aproximadamente 1.000 vezes mais potente que o irinotecano como inibidor da topoisomerase I purificada a partir de linhagens celulares tumorais humanas e de roedores.
Efeitos farmacodinâmicos
Em modelos animais, ONIVYDE® demonstrou aumentar os níveis plasmáticos de irinotecano e prolongar a exposição ao metabólito ativo, SN-38, no local do tumor.
Propriedades Farmacocinéticas
Absorção
A encapsulação de lipossomas de irinotecano estende a circulação e limita a distribuição em relação às formulações não lipossomais do irinotecano.
A farmacocinética plasmática do irinotecano total e SN-38 total foi avaliada em pacientes com câncer que receberam ONIVYDE®, como agente único ou como parte de quimioterapia combinada, em doses entre 50 e 155 mg/m2. Os parâmetros farmacocinéticos do irinotecano total e do SN-38, após a administração de ONIVYDE® 70 mg/m2 são apresentados na Tabela 2.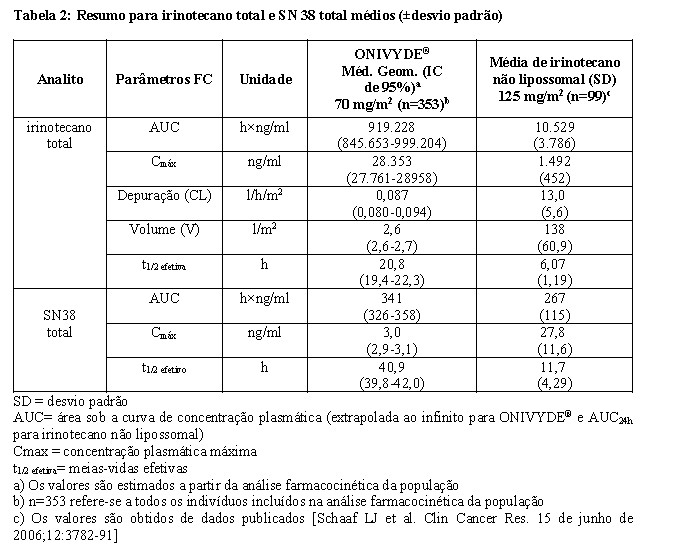
Distribuição
A quantificação direta do irinotecano lipossomal mostra que 95% do irinotecano permanece encapsulado em lipossomas durante a circulação. O irinotecano não lipossomal apresenta um grande volume de distribuição (138 L/m2). O volume de distribuição de ONIVYDE® 70 mg/m2 foi de 2,6 L/m2, o que sugere que ONIVYDE® está amplamente restrito ao fluido vascular. A ligação do ONIVYDE® às proteínas plasmáticas é insignificante ( < 0,44% do irinotecano total do ONIVYDE®). A ligação do irinotecano não lipossomal às proteínas plasmáticas é moderada (30% a 68%) e o SN-38 liga-se fortemente às proteínas plasmáticas humanas (aproximadamente 95%).
Biotransformação
O irinotecano liberado da encapsulação lipossomal segue uma via metabólica semelhante à observada com o irinotecano não lipossomal.
A conversão metabólica do irinotecano para o metabólito ativo SN-38 é mediada por enzimas carboxilesterase. Estudos in vitro indicam que o irinotecano, o SN-38 e um outro metabólito, ácido carboxílico aminopentano (APC), não inibem as isoenzimas do citocromo P-450. Posteriormente, o SN-38 é conjugado, predominantemente pela enzima UDP glucoronil transferase 1A1 (UGT1A1), para formar o metabólito glicuronídeo. A atividade da UGT1A1 é reduzida em indivíduos com polimorfismos genéticos, p. ex., o polimorfismo UGT1A1*28 que resulta numa atividade enzimática diminuída. Na análise farmacocinética populacional em pacientes com ONIVYDE®, utilizando os resultados de um subconjunto com teste genotípico UGT1A1*28, para o qual a análise foi ajustada em função da menor dose administrada a pacientes homozigóticos para o alelo UGT1A1*28, os pacientes homozigóticos (N=14) e não homozigóticos (N=244) para este alelo tiveram concentrações médias de SN-38 total no estado de equilíbrio de 1,06 e 0,95 ng/ml, respetivamente.
Eliminação
A eliminação de ONIVYDE® e do irinotecano não lipossomal não está totalmente elucidada em humanos.
A excreção urinária de irinotecano não lipossomal é de 11% a 20%, < 1% para SN-38 e 3% para o glicuronídeo de SN-38. A excreção biliar e urinária acumulada do irinotecano e respectivos metabólitos (SN-38 e glicuronídeo de SN-38) ao longo de um período de 48 horas após a administração de irinotecano não-lipossomal em dois pacientes variou de aproximadamente 25% (100 mg/m2) a 50% (300 mg/m2).
Insuficiência renal
Nenhum estudo farmacocinético específico foi realizado em pacientes com insuficiência renal. Em uma análise farmacocinética populacional, o comprometimento renal leve a moderado não teve efeito na exposição do SN-38 total após ajuste para a área de superfície corporal (ASC). A análise incluiu 68 pacientes com insuficiência renal moderada (CLcr 30 a 59 ml/min), 147 pacientes com insuficiência renal leve (CLcr 60-89 ml/min) e 135 pacientes com função renal normal (CLcr > 90 ml/min). Não existem dados suficientes em pacientes com comprometimento renal grave (CLcr < 30 ml/min) para avaliar o seu efeito na farmacocinética (ver itens 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES).
Insuficiência hepática
Nenhum estudo farmacocinético específico foi conduzido em pacientes com insuficiência hepática. Em uma análise farmacocinética populacional, os pacientes com concentrações basais de bilirrubina total de 1,2 mg/dl (n=19) apresentaram aumento em 37% (0,98 [IC 95%: 0,94 1,02] e 1,29 [IC95%: 1,11 1,5] ng/ml, respectivamente) nas concentrações médias de SN-38 total no estado de equilíbrio em comparação com pacientes com concentrações basais de bilirrubina < 1 mg/dl (n=329); no entanto, não houve efeito de concentrações elevadas de ALT/AST nas concentrações totais de SN 38. Não há dados disponíveis em pacientes com bilirrubina total superior a 2 vezes o limite superior normal (LSN).
Outras populações especiais
Idade e gênero
A análise farmacocinética populacional em pacientes com idade entre 28 e 87 anos, dos quais 11% tinham ≥75 anos, sugere que a idade não teve efeito clinicamente significativo na exposição ao irinotecano e ao SN-38.
A análise farmacocinética populacional em 196 pacientes do sexo masculino e 157 do sexo feminino sugere que o gênero não teve efeito clinicamente significativo na exposição ao irinotecano e ao SN-38 após o ajuste para a área de superfície corporal (ASC).
Etnia
A análise farmacocinética populacional sugere que os asiáticos têm concentração média de irinotecano total no estado de equilíbrio 56% menor (3,93 [IC 95%: 3,68 4,2] e 1,74 [IC 95%: 1,58 1,93] mg/l, respectivamente) e concentrações média de SN-38 total no estado estacionário 8% maior (0,97 [IC95%: 0,92 1,03] e 1,05 [IC95%: 0,98 1,11] ng/ml, respectivamente) do que os caucasianos.
Relação farmacocinética/farmacodinâmica
Em uma análise agrupada de 353 pacientes, um Cmax plasmático de SN-38 mais elevado foi associado a uma maior probabilidade de apresentar neutropenia, e um Cmax total plasmático mais elevado foi associado a uma probabilidade aumentada de ter diarreia.
No estudo clínico que demonstrou a eficácia de ONIVYDE®, exposições plasmáticas mais altas de irinotecano total e SN-38 em pacientes no braço de tratamento de ONIVYDE® +5-FU/LV foram associadas a OS e PFS mais longos, bem como a ORR mais altas (taxa de resposta objetiva).
Dados de segurança pré-clínica
Em estudos de toxicidade de dose única e repetida em camundongos, ratos e cães, os órgãos-alvo da toxicidade foram o trato gastrointestinal e o sistema hematológico. A gravidade dos efeitos foi dose relacionada e reversível. O nível de efeito adverso não observado (NOAEL) em ratos e cães após 90 minutos de infusão intravenosa de ONIVYDE® administrado uma vez a cada 3 semanas durante 18 semanas foi de 155 mg/m2.
Em estudos farmacológicos de segurança em cães, ONIVYDE® não teve efeito nos parâmetros cardiovasculares, hemodinâmicos, eletrocardiográficos ou respiratórios em doses de até 18 mg/kg ou 360 mg/m2. Não foram observados achados indicativos de toxicidade relacionada ao SNC nos estudos de toxicidade de doses repetidas em ratos.
Potencial genotóxico e carcinogênico
Não foram realizados estudos de genotoxicidade com ONIVYDE®. O irinotecano não lipossomal e o SN 38 foram genotóxicos in vitro no teste de aberração cromossômica em células CHO, bem como no teste de micronúcleos em camundongos. No entanto, em outros estudos com irinotecano, eles demonstraram ser desprovidos de qualquer potencial mutagênico no teste de Ames.
Não foram realizados estudos de carcinogenicidade com ONIVYDE®. Para o irinotecano não lipossomal, em ratos tratados uma vez por semana durante 13 semanas com a dose máxima de 150 mg/m², nenhum tumor relacionado ao tratamento foi relatado 91 semanas após o término do tratamento. Nestas condições, houve uma tendência linear significativa com a dose para a incidência de pólipos do estroma endometrial do corpo uterino combinados e sarcomas do estroma endometrial. Devido ao seu mecanismo de ação, o irinotecano é considerado um potencial carcinógeno.
Toxicidade reprodutiva
Nenhum estudo de toxicidade reprodutiva e de desenvolvimento foi realizado com ONIVYDE.
O irinotecano não lipossomal foi teratogênico em ratos e coelhos em doses inferiores à dose terapêutica humana. Em ratos, os filhotes nascidos de animais tratados e com anormalidades externas apresentaram diminuição da fertilidade. Isso não foi observado em filhotes morfologicamente normais. Em ratas grávidas houve diminuição do peso placentário e na prole, diminuição da viabilidade fetal e aumento de anormalidades comportamentais.
O irinotecano não lipossomal causou atrofia dos órgãos reprodutores masculinos em ratos e cães após doses diárias múltiplas de 20 mg/kg e 0,4 mg/kg, respetivamente. Estes efeitos foram reversíveis após a cessação do tratamento.
4. CONTRAINDICAÇÕES
ONIVYDE® é contraindicado em casos de histórico de hipersensibilidade grave ao irinotecano ou a qualquer um dos excipientes mencionados no item Composição, e durante a amamentação (ver item 5. ADVERTÊNCIAS E PRECAUÇÕES).
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais
ONIVYDE® é uma formulação de irinotecano lipossomal com diferentes propriedades farmacocinéticas em comparação com o irinotecano não lipossomal. A concentração e dosagem são diferentes em comparação com os irinotecanos não lipossomais.
ONIVYDE® não é equivalente a outras formulações não lipossomais de irinotecano e não deve ser trocado.
No número limitado de pacientes com exposição prévia ao irinotecano não lipossomal, não foi demonstrado nenhum benefício de ONIVYDE®.
Mielossupressão/neutropenia
Recomenda-se a monitorização completa da contagem de células sanguíneas durante o tratamento com ONIVYDE®. Os pacientes devem estar cientes do risco de neutropenia e da importância da febre. O tempo mediano até ao nadir para neutropenia de Grau ≥ 3 é de 23 dias (intervalo 8 - 104) após a primeira dose de tratamento com ONIVYDE®. A neutropenia febril (temperatura corporal > 38°C e contagem de neutrófilos ≤ 1.000 células/mm³) deve ser tratada com urgência no hospital com antibióticos intravenosos de amplo espectro. ONIVYDE® deve ser suspenso se ocorrer neutropenia febril ou se a contagem absoluta de neutrófilos cair abaixo de 1.500 células/mm3. Sepse com neutropenia febril e consequente choque séptico com desfecho fatal foi observada em pacientes com adenocarcinoma pancreático metastático tratados com ONIVYDE®.
Em pacientes que apresentaram eventos hematológicos graves, recomenda-se uma redução da dose ou interrupção do tratamento (ver item 8. POSOLOGIA E MODO DE USAR). Pacientes com insuficiência grave da medula óssea não devem ser tratados com ONIVYDE®.
Histórico de irradiação abdominal prévia aumenta o risco de neutropenia grave e neutropenia febril após o tratamento com ONIVYDE®. Monitoramento cuidadoso das contagens sanguíneas é recomendado, e o uso de fatores de crescimento mieloide deve ser considerado para pacientes com histórico de irradiação abdominal. Deve-se ter cautela em pacientes recebendo administração concomitante de ONIVYDE® com irradiação.
Pacientes com glicuronidação deficiente da bilirrubina, como os que possuem síndrome de Gilbert, podem apresentar maior risco de mielossupressão quando recebem terapia com ONIVYDE®.
Em comparação com pacientes caucasianos, os pacientes asiáticos apresentam um risco aumentado de neutropenia grave e neutropenia febril após o tratamento com ONIVYDE® +5-FU/LV (ver itens 9. REAÇÕES ADVERSAS e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Efeitos imunossupressores e vacinas
A administração de vacinas com vírus vivo ou ´vivo-atenuado em pacientes imunocomprometidos por medicamentos quimioterápicos, incluindo ONIVYDE®, pode resultar em infecções graves ou fatais; portanto, a vacinação com uma vacina de vírus vivo ou vivo-atenuado devem ser evitadas. Podem ser administradas vacinas mortas ou inativadas; no entanto, a resposta a essas vacinas podem ser reduzidas.
Interações com indutores fortes do CYP3A4
ONIVYDE® não deve ser administrado com indutores fortes da enzima CYP3A4, como anticonvulsivantes (fenitoína, fenobarbital ou carbamazepina), rifampicina, rifabutina e erva de São João, a menos que não haja alternativas terapêuticas. A dose inicial apropriada para pacientes que tomam esses anticonvulsivantes ou outros indutores fortes não foi definida. Deve-se considerar a substituição por terapias não indutoras de enzimas pelo menos 2 semanas antes do início da terapia com ONIVYDE® (ver seção 6.INTERAÇÕES MEDICAMENTOSAS).
Interações com inibidores fortes de CYP3A4 ou inibidores fortes de UGT1A1
ONIVYDE® não deve ser administrado com fortes inibidores da enzima CYP3A4 (por exemplo, sumo de toranja, claritromicina, indinavir, itraconazol, lopinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telaprevir, voriconazol). Inibidores potentes do CYP3A4 devem ser descontinuados pelo menos 1 semana antes do início da terapia com ONIVYDE®.
ONIVYDE® não deve ser administrado com fortes inibidores da UGT1A (por exemplo, atazanavir, gemfibrozil, indinavir), a menos que não haja alternativas terapêuticas.
Diarreia
A diarreia pode ocorrer precocemente (início em ≤ 24 horas após o início de ONIVYDE®) ou tardiamente ( > 24 horas) (ver item 9. REAÇÕES ADVERSAS).
Em pacientes com diarreia precoce, a atropina terapêutica e profilática deve ser considerada, a menos que seja contraindicada. Os pacientes devem ser alertados sobre o risco de diarreia tardia que pode ser debilitante e, em raras ocasiões, potencialmente fatal, uma vez que fezes moles ou aquosas persistentes podem resultar em desidratação, desequilíbrio eletrolítico, colite, ulceração gastrointestinal, infecção ou sepse.
Assim que ocorrerem as primeiras fezes líquidas, o paciente deve começar a ingerir grandes volumes de bebidas contendo eletrólitos. Os pacientes devem ter loperamida (ou equivalente) prontamente disponível para iniciar o tratamento para diarreia tardia. A loperamida deve ser iniciada na primeira ocorrência de fezes malformadas ou amolecidas ou no primeiro aparecimento de movimentos intestinais mais frequentes do que o normal. A loperamida deve ser administrada até que o paciente esteja sem diarreia por pelo menos 12 horas.
Se a diarreia persistir enquanto o paciente estiver em uso de loperamida por mais de 24 horas, deve-se considerar a adição de suporte antibiótico oral (por exemplo, fluoroquinolona por 7 dias). A loperamida não deve ser usada por mais de 48 horas consecutivas devido ao risco de íleo paralítico. Se a diarreia persistir por mais de 48 horas, interrompa a loperamida, monitore, reponhaos eletrólitos e continue o suporte antibiótico até a resolução dos sintomas associados.
O tratamento com ONIVYDE® deve ser adiado até que a diarreia reduza para ≤ Grau 1 (2-3 evacuações/dia a mais do que a frequência pré-tratamento). ONIVYDE® não deve ser administrado a pacientes com obstrução intestinal e doença inflamatória intestinal crônica, até que esteja resolvida.
Após diarreia de Grau 3 ou 4, a dose subsequente de ONIVYDE® deve ser reduzida (ver item 8. POSOLOGIA E MODO DE USAR).
Reações colinérgicas
A diarreia de início precoce pode ser acompanhada por sintomas colinérgicos, como rinite, aumento da salivação, rubor, diaforese, bradicardia, miose e hiperperistaltismo. Em caso de sintomas colinérgicos, deve-se administrar atropina.
Infusão aguda e reações relacionadas
Reações à infusão consistindo principalmente de erupção cutânea, urticária, edema periorbital ou prurido foram relatadas em pacientes recebendo tratamento com ONIVYDE®. Novos eventos (todos de grau 1 ou grau 2) ocorreram geralmente precocemente durante o tratamento com ONIVYDE® com apenas 2 em 10 pacientes apresentando eventos após a quinta dose. Podem ocorrer reações de hipersensibilidade, incluindo reação aguda à infusão, reação anafilática/anafilactóide e angioedema. ONIVYDE® deve ser descontinuado em caso de reações graves de hipersensibilidade.
Procedimento de Whipple anterior
Os pacientes com histórico de cirurgia de Whipple têm um risco mais elevado de infecções graves após ONIVYDE® em associação com 5-FU e leucovorina (ver item 9. REAÇÕES ADVERSAS). Os pacientes devem ser monitorados quanto a sinais de infecções.
Distúrbios vasculares
ONIVYDE® foi associado a eventos tromboembólicos, como embolia pulmonar, trombose venosa e tromboembolismo arterial. Um histórico médico completo deve ser obtido para identificar pacientes com múltiplos fatores de risco, além da neoplasia subjacente. Os pacientes devem ser informados sobre os sinais e sintomas de tromboembolismo e aconselhados a entrar em contato com seu médico ou enfermeiro imediatamente caso ocorram tais sinais ou sintomas.
Toxicidade pulmonar
Eventos do tipo Doença Pulmonar Intersticial (DPI) levando a fatalidades ocorreram em pacientes recebendo irinotecano não lipossomal. Nenhum caso de eventos do tipo DPI foi relatado com o uso de ONIVYDE® em estudos clínicos. Os fatores de risco incluem doença pulmonar pré-existente, uso de medicamentos pneumotóxicos, uso de fatores estimulante de colônias ou ter recebido radioterapia anteriormente. Pacientes com fatores de risco devem ser cuidadosamente monitorados quanto a sintomas respiratórios antes e durante o tratamento com ONIVYDE®. Um padrão retículo nodular na radiografia de tórax foi observado em uma pequena porcentagem de pacientes incluídos em um estudo clínico com irinotecano. Dispneia recente ou progressiva, tosse e febre devem levar à interrupção do tratamento com ONIVYDE®, enquanto se aguarda avaliação diagnóstica. ONIVYDE® deve ser descontinuado em pacientes com diagnóstico confirmado de DPI.
Insuficiência hepática
Os pacientes com hiperbilirrubinemia apresentaram concentrações mais elevadas de SN-38 total (ver item 3. CARACTERÍSTICAS FARMACOLÓGICAS) e, por conseguinte, o risco de neutropenia é aumentado. O monitoramento regular de hemogramas completos deve ser realizado em pacientes com bilirrubina total de 1,0 a 2,0 mg/dl. Deve-se ter cautela em pacientes com insuficiência hepática (bilirrubina > 2 vezes o limite superior normal [LSN]; transaminases > 5 vezes o LSN). É necessário ter cautela quando ONIVYDE® é administrado em associação com outros medicamentos hepatotóxicos, especialmente em pacientes com comprometimento hepático pré-existente.
Insuficiência renal
A utilização de ONIVYDE® em pacientes com comprometimento renal importante não foi estabelecida (ver item 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Pacientes com baixo peso (índice de massa corporal < 18,5 kg/m2)
No estudo clínico avaliando ONIVYDE® + 5-FU/LV, 5 de 8 pacientes com baixo peso apresentaram reações adversas de grau 3 ou 4, principalmente mielossupressão, enquanto 7 dos 8 pacientes necessitaram de modificação de dose, como atraso da dose, redução ou descontinuação. Deve-se ter cuidado ao usar ONIVYDE® em pacientes com índice de massa corporal < 18,5 kg/m2.
Excipientes
Este medicamento contém 33,1 mg de sódio por frasco, equivalente a 1,65% da ingestão diária máxima recomendada pela OMS de 2 g de sódio para um adulto.
Mulheres com potencial para engravidar/contracepção em homens e mulheres
Mulheres com potencial para engravidar devem usar métodos contraceptivos eficazes durante o tratamento com ONIVYDE® e 7 meses depois. Homens devem usar preservativos durante e 4 meses após o tratamento com ONIVYDE®.
Gravidez
Não existem dados adequados sobre o uso de ONIVYDE® em mulheres grávidas. ONIVYDE® pode causar danos ao feto quando administrado a mulheres grávidas, uma vez que o principal componente, o irinotecano, demonstrou ser embriotóxico e teratogênico em animais (ver o item 3. CARACTERÍSTICAS FARMACOLÓGICAS - Dados de segurança pré-clínica). Portanto, com base nos resultados de estudos em animais e no mecanismo de ação do irinotecano, ONIVYDE® não deve ser usado durante a gravidez, a menos que seja claramente necessário. Se ONIVYDE® for usado durante a gravidez ou se a paciente engravidar durante o tratamento, a paciente deve ser informada sobre o potencial risco para o feto.
Categoria "C" de risco na gravidez. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Amamentação
É desconhecido se ONIVYDE® ou os seus metabolitos são excretados no leite humano. Devido ao potencial para reações adversas graves de ONIVYDE® em lactentes, ONIVYDE® é contraindicado durante a amamentação (ver item 4. CONTRAINDICAÇÕES). As pacientes não devem amamentar até um mês após a última dose.
Fertilidade
Não existem dados sobre o impacto de ONIVYDE® na fertilidade em humanos. Foi demonstrado que o irinotecano não lipossomal causa atrofia dos órgãos reprodutores masculinos e femininos após múltiplas doses diárias de irinotecano em animais (ver item 3. CARACTERÍSTICAS FARMACOLÓGICAS). Antes de iniciar a administração de ONIVYDE®, considere aconselhar os pacientes sobre a preservação dos gametas.
Efeitos na capacidade de dirigir e operar máquinas
ONIVYDE® tem influência moderada na habilidade de dirigir veículos e operar máquinas. Durante o tratamento, os pacientes devem ter cuidado ao conduzir ou utilizar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
As informações sobre interações medicamentosas com ONIVYDE® são referenciadas na literatura científica publicada para o irinotecano não lipossomal.
Interações que afetam o uso de ONIVYDE®:
- Indutores fortes de CYP3A4
Pacientes recebendo concomitantemente irinotecano não lipossomal e anticonvulsivantes indutores da enzima CYP3A4, fenitoína, fenobarbital ou carbamazepina, reduziram substancialmente a exposição ao irinotecano (redução de AUC em 12% com erva de São João, 57% - 79% com fenitoína, fenobarbital ou carbamazepina) e SN-38 (AUC redução de 42% com erva de São João, 36% 92% com fenitoína fenobarbital ou carbamazepina). Portanto, a coadministração de ONIVYDE® com indutores de CYP3A4 pode reduzir a exposição sistêmica de ONIVYDE®.
- Inibidores fortes de CYP3A4 e inibidores de UGT1A1
Pacientes recebendo concomitantemente irinotecano não lipossomal e cetoconazol, um inibidor de CYP3A4 e UGT1A1, aumentaram a exposição a SN 38 em 109%. Portanto, a coadministração de ONIVYDE® com outros inibidores do CYP3A4 (por exemplo, suco de toranja, claritromicina, indinavir, itraconazol, lopinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telaprevir, voriconazol) pode aumentar a exposição sistêmica de ONIVYDE®. Com base na interação medicamentosa de irinotecano não lipossomal e cetoconazol, a coadministração de ONIVYDE® com outros inibidores de UGT1A1 (por exemplo, atazanavir, genfibrozila, indinavir, regorafenibe) também pode aumentar a exposição sistêmica de ONIVYDE®.
A coadministração de ONIVYDE® +5-FU/LV não altera a farmacocinética de ONIVYDE® com base na análise farmacocinética da população.
- Agentes antineoplásicos (incluindo flucitosina como pró-fármaco para 5-fluorouracila)
Os efeitos adversos do irinotecano, como mielossupressão, podem ser exacerbados por outros agentes antineoplásicos com um perfil de efeito adverso semelhante.
Não é conhecida nenhuma interação de ONIVYDE® com outros medicamentos.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
ONIVYDE® deve ser armazenado sob refrigeração, em temperatura entre +2°C e +8°C. Não congelar o produto. Manter o frasco dentro da embalagem original para proteger da luz.
O prazo de validade do medicamento é de 36 meses a partir da data de fabricação.
Sob o ponto de vista microbiológico, recomenda-se que o produto seja utilizado imediatamente após sua diluição.
Qualquer medicamento não utilizado ou resíduos devem ser descartados de acordo com as exigências locais.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após a diluição, manter em temperatura ambiente (até 25°C) por não mais que 6 horas desde o momento da preparação até o final da infusão OU manter sob refrigeração (entre 2 e 8°C) por não mais que 24 horas desde o momento da diluição até o final da infusão. Proteger da luz e não congelar.
CARACTERÍSTICAS FÍSICAS E ORGANOLÉPTICAS
ONYVIDE é uma suspensão lipossomal isotônica, opaca branca a ligeiramente amarela, com um pH de 7,2 e uma osmolaridade de 295 mOsm/kg.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
ONIVYDE® só deve ser prescrito e administrado a pacientes por profissionais de saúde com experiência no uso de terapias anticâncer.
ONIVYDE® não é equivalente a formulações não lipossomais de irinotecano e não deve ser trocado.
POSOLOGIA
ONIVYDE®, leucovorina e 5-fluorouracila devem ser administrados sequencialmente. A dose e regime recomendados de ONIVYDE® é de 70 mg/m2 por via intravenosa durante 90 minutos, seguido de LV 400 mg/m2 por via intravenosa durante 30 minutos, seguido por 5-FU 2.400 mg/m2 por via intravenosa durante 46 horas, administrados a cada 2 semanas. ONIVYDE® não deve ser administrado como agente único.
Uma dose inicial reduzida de ONIVYDE® de 50 mg/m2 deve ser considerada para pacientes homozigotos para o alelo UGT1A1*28 (ver itens 9. REAÇÕES ADVERSAS e 3. CARACTERÍSTICAS FARMACOLÓGICAS). Um aumento da dose de ONIVYDE® para 70 mg/m2 deve ser considerado em ciclos subsequentes, se tolerado.
- Pré-medicação
Recomenda-se que os pacientes recebam pré-medicação com doses padrão de dexametasona (ou um corticosteroide equivalente) juntamente com um antagonista 5-HT3 (ou outro antiemético) pelo menos 30 minutos antes da infusão de ONIVYDE®.
- Ajustes de dose
Todas as modificações de dose devem ser baseadas na pior toxicidade precedente. A dose de LV não requer ajuste. Para toxicidades de Grau 1 e 2, não há recomendações de modificação de dose. Os ajustes de dose, conforme resumidos na Tabela 3 e na Tabela 4, são recomendados para controlar as toxicidades de Grau 3 ou 4 relacionadas ao ONIVYDE®.
Para pacientes que iniciam tratamento com ONIVYDE® 50 mg/m2 e não aumentam a dose para 70 mg/m2, a primeira redução de dose recomendada é para 43 mg/m2 e a segunda redução de dose é para 35 mg/m2. Os pacientes que necessitem de uma redução adicional da dose devem descontinuar o tratamento.
Pacientes que são homozigotos para UGT1A1*28 e sem toxicidades relacionadas ao medicamento durante o primeiro ciclo de terapia (dose reduzida de 50 mg/m2) podem ter a dose de ONIVYDE® aumentada para uma dose total de 70 mg/m2 em ciclos subsequentes com base na tolerância individual do paciente.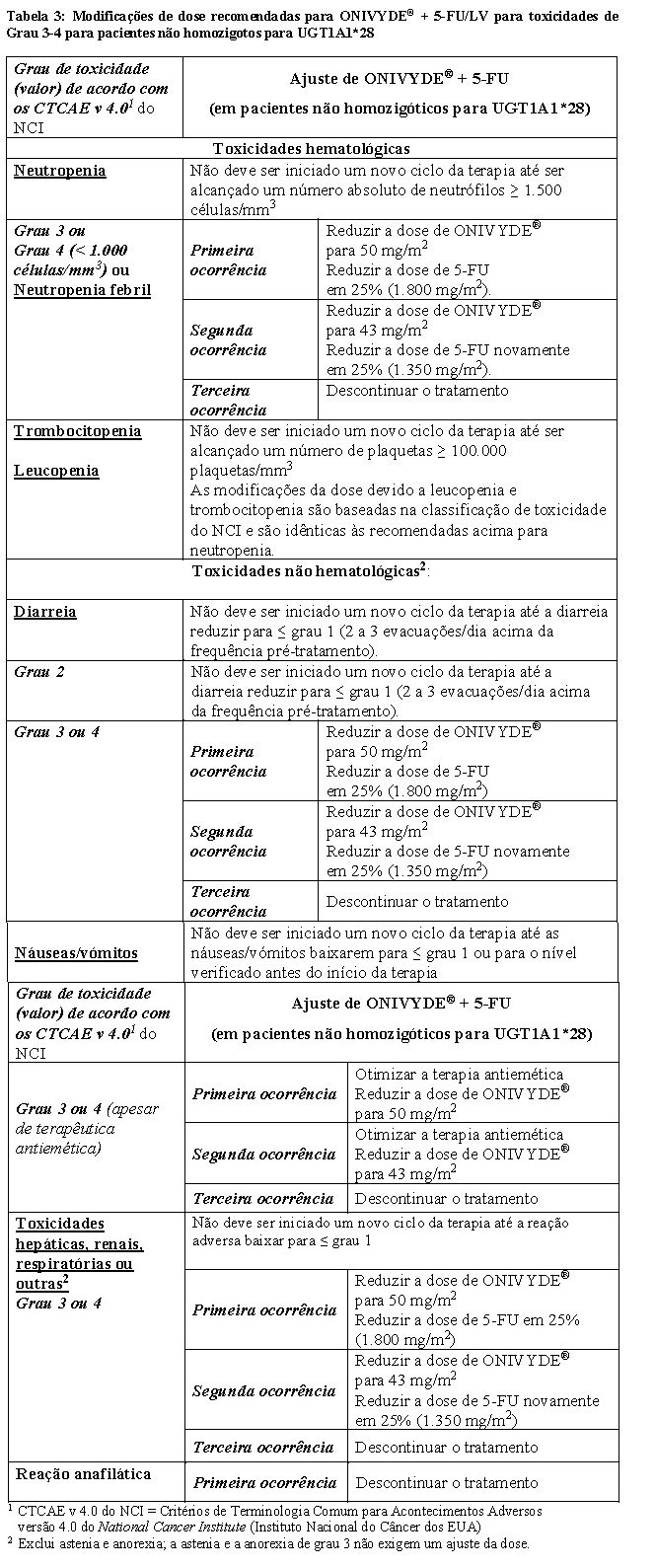

Populações especiais
Insuficiência hepática
Nenhum estudo dedicado à insuficiência hepática foi conduzido com ONIVYDE®. O uso de ONIVYDE® deve ser evitado em pacientes com bilirrubina > 2,0 mg/dL ou aspartato aminotransferase (AST) e alanina aminotransferase (ALT) > 2,5 vezes o limite superior normal (LSN) ou > 5 vezes LSN se houver metástase hepática (ver item 7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO).
Insuficiência renal
Nenhum estudo dedicado à insuficiência renal foi conduzido com ONIVYDE®. Nenhum ajuste de dose é recomendado em pacientes com insuficiência renal leve a moderada (ver itens 7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO e 3. CARACTERÍSTICAS FARMACOLÓGICAS). ONIVYDE® não é recomendado para uso em pacientes com insuficiência renal grave (CLcr < 30 mLmin).
Idoso
Quarenta e um por cento (41%) dos pacientes tratados com ONIVYDE® em todo o programa clínico tinham ≥ 65 anos. Nenhum ajuste de dose é recomendado.
População pediátrica
A segurança e eficácia de ONIVYDE® em crianças e adolescentes com idade ≤ 18 anos ainda não foram estabelecidas. Não há dados disponíveis.
MODO DE USAR
ONIVYDE® é para uso intravenoso. O concentrado deve ser diluído antes da administração e administrado em uma única infusão intravenosa durante 90 minutos.
Precauções a serem adotadas antes de manusear ou administrar o medicamento
ONIVYDE® é um medicamento citotóxico. Recomenda-se o uso de luvas, óculos e roupas protetoras ao manusear ou administrar ONIVYDE®. Se houver contato com a pele, lavar imediatamente e completamente com água e sabão. Se houver contato com mucosas, lavar completamente com água. Grávidas não devem manusear ONIVYDE®.
Instruções de diluição
1. ONIVYDE® é fornecido como uma suspensão lipossomal estéril em uma concentração de 4,3 mg/mL e deve ser diluído antes da administração usando uma agulha de calibre não superior a 21 gauge;
2. Dilua com solução injetável de glicose a 5% ou solução injetável de cloreto de sódio 9 mg/ml (0,9%) para preparar uma dispersão da dose apropriada de ONIVYDE® em um volume final de 500 mL;
3. Misture a dispersão diluída por inversão suave;
4. A dispersão diluída é límpida a ligeiramente branca a ligeiramente opalescente e isenta de partículas visíveis;
5. Para condições de conservação após diluição do medicamento, ver item 7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO.
Instruções de administração:
1. Técnicas assépticas devem ser seguidas durante a preparação da infusão.
2. ONIVYDE® destina-se a uma única administração. Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com os requerimentos locais.
3. ONIVYDE® deve ser administrado antes da leucovorina (LV) que é seguida pela fluorouracila (5-FU).
4. ONIVYDE® não deve ser administrado como injeção em bolus ou sem diluição.
5. Deve-se ter cuidado para evitar extravasamento e o local de infusão deve ser monitorado quanto a sinais de inflamação.
6. Caso ocorra extravasamento, recomenda-se a lavagem do local com solução injetável de cloreto de sódio 9 mg/ml (0,9%) e/ou água estéril e aplicação de gelo.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
O perfil de segurança é baseado no estudo clínico NAPOLI-1. As seguintes reações adversas, consideradas possível ou provavelmente relacionadas com a administração de ONIVYDE®, foram reportadas em 264 pacientes com adenocarcinoma metastático do pâncreas, 147 dos quais receberam ONIVYDE® em monoterapia (100 mg/m2) e 117 receberam ONIVYDE® (70 mg/m2) em combinação com 5-FU/LV.
As reações adversas mais comuns (incidência ≥ 20%) com uso ONIVYDE® + 5-FU/LV foram: diarreia, náuseas, vômitos, diminuição do apetite, neutropenia, fadiga, astenia, anemia, estomatite e pirexia. As reações adversas graves mais comuns (≥ 2%) com uso de ONIVYDE® foram diarreia, vômito, neutropenia febril, náusea, pirexia, sepse, desidratação, choque séptico, pneumonia, insuficiência renal aguda e trombocitopenia.
As taxas de reações adversas que levaram à descontinuação permanente do tratamento foram de 11% para o braço de ONIVYDE® +5-FU/LV e de 12% para o braço de monoterapia.
As reações adversas relatadas com maior frequência que levaram à descontinuação foram infecção e diarreia para o braço de ONIVYDE® +5-FU/LV, e vômitos e diarreia para o braço de monoterapia.
Lista tabelada de reações adversas
As reações adversas descritas nesta seção são derivadas dos dados do estudo e da experiência pós-comercialização de ONIVYDE®.
As reações adversas que podem ocorrer durante o tratamento com ONIVYDE® estão resumidas abaixo e são apresentadas por classe de sistema de órgãos e categoria de frequência (Tabela 5). Dentro de cada classe de sistema de órgãos e categoria de frequência, as reações adversas são apresentadas em ordem decrescente de gravidade. Dentro de cada classe de sistemas de órgãos e categoria de frequência, as reações adversas são apresentadas por ordem decrescente de gravidade. As categorias de frequência utilizadas para reações adversas são: muito comum (≥1/10); comum (≥1/100, < 1/10); incomum (≥1/1.000, < 1/100); raros (≥1/10.000, < 1/1.000)* e frequência desconhecida (a frequência não pode ser calculada a partir dos dados disponíveis).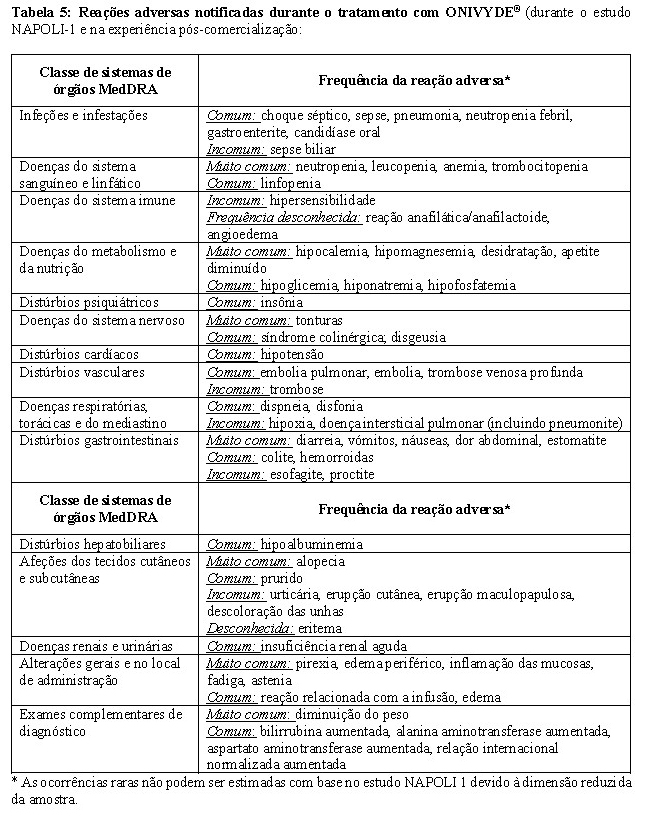
Descrição de reações adversas selecionadas
As seguintes reações