OJJAARA
GLAXOSMITHKLINE
dicloridrato de momelotinibe monoidratado
Antineoplásico. Tratamento de mielofibrose risco interm /alto.
Apresentações.
Comprimidos revestidos de 100 mg, 150 mg e 200 mg em embalagens com 30 unidades.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de OJJAARA 100mg contém: momelotinibe 100 mg (equivalente a 121,94 mg de dicloridrato de momelotinibe monoidratado), excipientes* q.s.p. 1 comprimido revestido
Cada comprimido revestido de OJJAARA 150mg contém: momelotinibe 150 mg (equivalente a 182,91 mg de dicloridrato de momelotinibe monoidratado), excipientes* q.s.p. 1 comprimido revestido
Cada comprimido revestido de OJJAARA 200mg contém: momelotinibe 200 mg (equivalente a 243,88 mg de dicloridrato de momelotinibe monoidratado), excipientes* q.s.p. 1 comprimido revestido
*Excipientes: celulose microcristalina, galato de propila, lactose monoidratada, amido glicolato de sódio, dióxido de silício, estearato de magnésio, nitrogênio, Opadry II marrom (álcool polivinílico, macrogol, dióxido de titânio, talco, óxido de ferro amarelo e óxido de ferro vermelho).
Informações técnicas.
1. INDICAÇÕES
OJJAARA é indicado para o tratamento de mielofibrose de risco intermediário e alto, incluindo mielofibrose primária, mielofibrose póspolicitemia vera ou mielofibrose pós-trombocitemia essencial em adultos com anemia.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
A eficácia de momelotinibe no tratamento de pacientes com mielofibrose de risco intermediário-1, intermediário-2 ou de alto risco, incluindo mielofibrose primária, mielofibrose pós-policitemia vera (pós-PV) ou mielofibrose pós-trombocitemia essencial (pós-ET), conforme definido pelo Dynamic International Prognostic Scoring System (DIPSS) ou International Prognostic Scoring System (IPSS) para mielofibrose, foi estabelecida em dois estudos randomizados, controlados ativamente e de fase 3, MOMENTUM e SIMPLIFY 1. Todos os pacientes receberam uma dose inicial de momelotinibe 200 mg uma vez ao dia, independentemente de sua contagem de plaquetas basal (no estudo MOMENTUM, a contagem mínima de plaquetas foi de 25 × 109/L; no estudo SIMPLIFY-1, a contagem mínima de plaquetas foi de 50 × 109/L).
MOMENTUM MOMENTUM foi um estudo duplo-cego, randomizado 2:1 e controlado por comparador ativo em 195 pacientes sintomáticos e anêmicos com mielofibrose que receberam anteriormente um inibidor de JAK. A mediana etária foi de 71 anos (intervalo de 38 a 86 anos); 79% tinham 65 anos ou mais e 63% eram do sexo masculino. Sessenta e quatro por cento (64%) dos pacientes apresentavam mielofibrose primária, 19% apresentavam mielofibrose pós-PV e 17% apresentavam mielofibrose pós-ET. Cinco por cento (5%) dos pacientes apresentavam risco intermediário-1, 57% apresentavam risco intermediário-2 e 35% apresentavam alto risco. Os pacientes eram sintomáticos com uma pontuação total de sintomas (TSS) do Formulário de Avaliação de Sintomas de Mielofibrose (MFSAF) v4.0 de ≥ 10 na triagem (média de MFSAF TSS de 27 na visita basal) e anêmicos com hemoglobina (Hgb) < 10 g/dL. O registro diário do MFSAF coletou os principais sintomas de mielofibrose: suores noturnos, desconforto abdominal, dor sob a costela esquerda, fadiga/cansaço, saciedade prematura, prurido e dor óssea. Dentro de 8 semanas antes da inclusão, 79% receberam transfusões eritrocitárias. No início do estudo, 13% e 15% dos pacientes eram independentes de transfusão nos grupos de momelotinibe e danazol, respectivamente. A Hgb mediana basal foi de 8 g/dL e a contagem mediana de plaquetas foi de 96 × 109/L. O comprimento mediano palpável basal do baço era 11,0 cm abaixo do rebordo costal esquerdo; o volume mediano do baço [medido por imagem por ressonância magnética (IRM) ou tomografia computadorizada (TC)] foi de 2.105 cm3 (intervalo: 610 a 9.717 cm3). Os pacientes foram tratados com momelotinibe 200 mg uma vez ao dia ou danazol 300 mg duas vezes ao dia por 24 semanas, seguido de tratamento aberto com momelotinibe. O desfecho primário de eficácia foi a porcentagem de pacientes com redução de TSS de 50% ou superior desde a visita basal até a semana 24 (Tabela 1). Na semana 24, uma porcentagem significativamente maior de pacientes tratados com momelotinibe alcançou uma redução de TSS de 50% ou superior em relação à visita basal em comparação ao danazol. Os principais desfechos secundários incluíram alteração contínua de TSS, independência de transfusão (definida como nenhuma transfusão e toda a hemoglobina ≥ 8 g/dL nas 12 semanas anteriores à semana 24), resposta do volume do baço (redução de 25% ou superior e redução de 35% ou superior) e taxa de independência transfusional.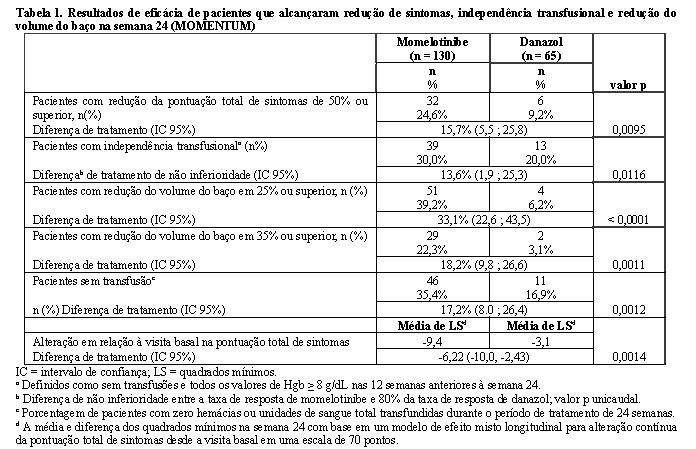
As respostas baseadas nos componentes individuais de MFSAF TSS foram comparadas para momelotinibe e danazol (Figura 1). A pontuação dos sintomas variou de 0 (ausente) a 10 (pior imaginável) para cada componente. Uma porcentagem maior de pacientes tratados com momelotinibe alcançou uma redução de 50% ou superior em relação ao valor basal comparado com danazol para cada sintoma individual.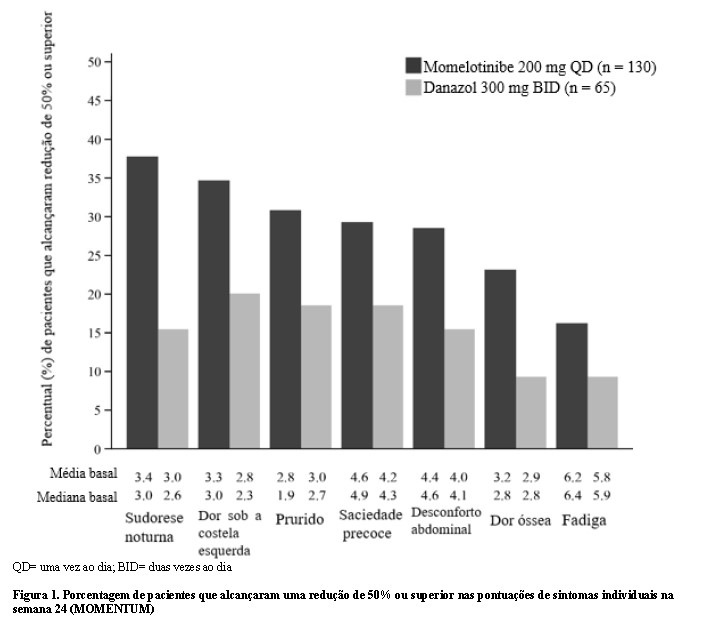
A porcentagem de pacientes com aumentos de Hgb de ≥1 g/dL e aumentos de Hgb de ≥1,5 g/dL foram de 53% e 40% em relação ao valor basal para aqueles tratados com momelotinibe, em comparação com 34% e 23% para aqueles tratados com danazol, respectivamente, até a semana 24.
Análises post-hoc foram conduzidas em pacientes que fizeram a transição para o tratamento aberto com momelotinibe após a semana 24 e foram considerados avaliáveis com base em dados suficientes disponíveis. Estas foram realizadas para a taxa de resposta do TSS do MFSAF (definida como uma redução de ≥ 50% em relação à média basal do TSS do MFSAF durante o período de 28 dias imediatamente antes do final da semana 48), independência de transfusão (TI) até a semana 48 (definida como nenhuma transfusão e todos os valores de Hgb > 8g/dL em qualquer período contínuo de 12 semanas durante o período aberto até a semana 48) e TI na semana 48 (definida como nenhuma transfusão e todos os valores de Hgb ≥8 g/dL nas 12 semanas anteriores à semana 48). Além disso, análises da taxa de resposta esplênica (redução de 35% ou mais) foram realizadas na semana 48.
Na semana 48, para pacientes em momelotinibe na fase duplo-cega que permaneceram em momelotinibe durante a fase aberta, 72% (18/25) dos pacientes que alcançaram uma resposta TSS na semana 24 mantiveram essa resposta. Todos os pacientes (5/5) em danazol na fase duplo-cega mantiveram uma resposta TSS ao fazer a transição para momelotinibe aberto após a semana 24. Vinte e oito por cento (12/43) dos pacientes que estavam no grupo de tratamento com momelotinibe e 40% (10/25) dos pacientes no grupo de tratamento com danazol que não responderam na semana 24 alcançaram uma resposta TSS na semana 48.
Para pacientes em momelotinibe na fase duplo-cega que permaneceram em momelotinibe durante a fase aberta, 94% (34/36) dos pacientes que alcançaram TI na semana 24 mantiveram essa resposta por pelo menos 12 semanas consecutivas nas 24 semanas do período aberto. Para pacientes em danazol durante a fase duplo-cega que transicionaram para momelotinibe em fase aberta, 83% (10/12) dos pacientes que atingiram TI na semana 24 mantiveram TI por pelo menos 12 semanas consecutivas nas 24 semanas do período aberto. Vinte e um por cento (10/48) dos pacientes no grupo de tratamento com momelotinibe e 42% (10/24) dos pacientes no grupo de tratamento com danazol que não responderam na semana 24 alcançaram TI até a semana 48.
Na semana 48, para pacientes em momelotinibe na fase duplo-cega que permaneceram em momelotinibe durante a fase aberta, 88% (30/34) dos pacientes que alcançaram TI na semana 24 mantiveram essa resposta. Para pacientes em danazol durante a fase duplo-cega, 80% (8/10) dos pacientes mantiveram TI ao fazer a transição para momelotinibe aberto após a semana 24. Vinte e quatro por cento (8/33) dos pacientes no grupo de tratamento com momelotinibe e 50% (10/20) dos pacientes no grupo de tratamento com danazol que não responderam na semana 24 alcançaram TI na semana 48.
Na semana 48, para pacientes em momelotinibe na fase duplo-cega que permaneceram em momelotinibe durante a fase aberta, 79% (19/24) dos pacientes que alcançaram uma resposta esplênica na semana 24 mantiveram essa resposta. Para pacientes em danazol na fase duplocega, um paciente (1/1) no grupo de tratamento com danazol manteve uma resposta esplênica ao fazer a transição para momelotinibe aberto após a semana 24. Vinte e três por cento (10/43) dos pacientes que estavam no grupo de tratamento com momelotinibe e 11% (3/28) dos pacientes no grupo de tratamento com danazol que não responderam na semana 24 alcançaram uma resposta esplênica na semana 48.
SIMPLIFY-1
O SIMPLIFY-1 foi um estudo duplo-cego, randomizado e controlado por comparador ativo em 432 pacientes com mielofibrose que não haviam recebido anteriormente um inibidor de JAK. A mediana etária foi de 66 anos (intervalo: 25 a 86 anos), com 57% dos pacientes com mais de 65 anos e 56% do sexo masculino. Cinquenta e seis por cento (56%) dos pacientes apresentavam mielofibrose primária, 23% apresentavam MF pós-PV e 21% apresentavam mielofibrose pós-ET. Vinte e um por cento (21%) dos pacientes apresentavam risco intermediário-1, 33% apresentavam risco intermediário-2 e 46% apresentavam alto risco. A resposta de TSS foi medida pelo registro no Formulário de Avaliação de Sintomas de Neoplasia Mieloproliferativa modificado (MPN-SAF) v2.0 (média de TSS de 19 na visita basal). O registro diário do MPN-SAF coletou os principais sintomas de mielofibrose: sudorese noturna, desconforto abdominal, dor sob a costela esquerda, fadiga/cansaço, saciedade prematura, prurido e dor óssea. Dentro de 8 semanas anteriores à inclusão, 25% dos pacientes receberam transfusões eritrocitárias. A mediana de Hgb basal foi de 10,4 g/dL e a mediana da contagem de plaquetas foi de 243,0 × 109/L na visita basal. O comprimento mediano palpável basal do baço era 12,0 cm abaixo do rebordo costal esquerdo; o volume médio do baço (medido por IRM ou TC) foi de 1.916 cm3 (intervalo: 206 a 9.022 cm3).
Os pacientes foram tratados com momelotinibe 200 mg ou dose ajustada de ruxolitinibe duas vezes ao dia por 24 semanas, seguido de tratamento aberto com momelotinibe sem redução gradual de ruxolitinibe. O desfecho primário de eficácia foi a porcentagem de pacientes com resposta do volume do baço (redução de 35% ou superior) na semana 24; análises também foram realizadas em um subconjunto de pacientes anêmicos (Tabela 2). Uma porcentagem semelhante de pacientes tratados com momelotinibe ou ruxolitinibe alcançou uma resposta de volume do baço em ambas as populações. Outros desfechos incluíram resposta de TSS e requisitos de transfusão eritrocitária.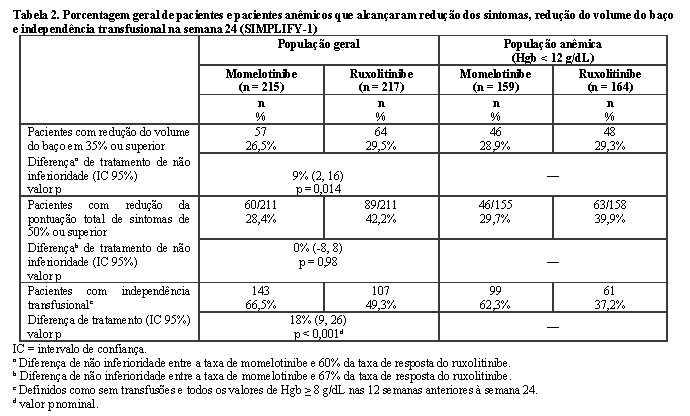
Em pacientes com volume do baço medido na semana 24, a redução mediana no volume do baço foi de 25% em pacientes tratados com momelotinibe em comparação com 24% para ruxolitinibe. A Figura 2 apresenta a alteração percentual em relação à visita basal no volume do baço para cada paciente na semana 24.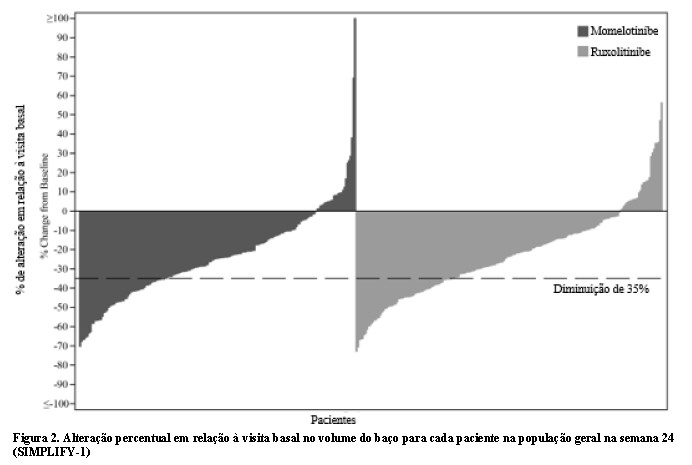
Na população com intenção de tratar, as alterações na carga de sintomas desde a análise basal no TSS médio com base em modelo misto para medidas repetidas foram semelhantes para pacientes tratados com momelotinibe (5,87 pontos) e pacientes tratados com ruxolitinibe (7,11 pontos). A diferença na alteração média de mínimos quadrados entre os grupos momelotinibe e ruxolitinibe na semana 24 foi de 1,24 (IC 95%: -0,40,2,88) na escala de 70 pontos. Na visita basal para a população geral, 68% e 70% dos pacientes tratados com momelotinibe ou ruxolitinibe, respectivamente, eram independentes de transfusão. Na semana 24, a porcentagem de pacientes independentes de transfusão foi de 66,5% com momelotinibe e 49,3% com ruxolitinibe. No geral, a proporção de pacientes com independência de transfusão na semana 24 diminuiu desde a visita basal em 1,9% no grupo momelotinibe e em 20,7% no grupo ruxolitinibe. No subconjunto de pacientes que eram dependentes de transfusão ou necessitavam de transfusão na visita basal, 35,3% dos pacientes no grupo momelotinibe tornaram-se independentes de transfusão na semana 24, em comparação 20,0% dos pacientes no grupo ruxolitinibe. A média basal de Hgb antes de iniciar momelotinibe e ruxolitinibe foi de 10,6 e 10,7 g/dL, respectivamente; na semana 24, a média de Hgb foi de 11,1 e 9,9 g/dL, respectivamente, para pacientes que receberam momelotinibe e ruxolitinibe.
Análises post hoc da taxa de resposta esplênica (definida como redução de 35% ou mais) foram conduzidas em pacientes que passaram para o tratamento aberto com momelotinibe na semana 24 e foram considerados avaliáveis com base em dados suficientes disponíveis. Além disso, análises post hoc de independência de transfusão (definida como a ausência de transfusão de glóbulos vermelhos e nenhum nível de hemoglobina < 8 g/dL em qualquer período contínuo de 12 semanas (ou seja, 84 dias consecutivos) foram conduzidas em pacientes randomizados na fase duplo-cega.
Na semana 48, para pacientes em momelotinibe que permaneceram em momelotinibe durante a fase aberta, 84% (48/57) dos pacientes que alcançaram uma resposta esplênica na semana 24 mantiveram essa resposta. Para pacientes em ruxolitinibe na fase duplo-cega que mudaram para momelotinibe, 50% (32/64) dos pacientes que alcançaram uma resposta esplênica na semana 24 mantiveram essa resposta na semana
48.
Para pacientes randomizados para momelotinibe na fase duplo-cega, incluindo aqueles que permaneceram em momelotinibe durante a fase aberta, 81% (174/215) dos pacientes estavam independentes de transfusão até a semana 48. Para pacientes randomizados para ruxolitinibe, incluindo aqueles que mudaram para momelotinibe, 82% (177/217) dos pacientes estavam independentes de transfusão até a semana 48.
Gerds AT, Verstovsek S, Vannucchi AM, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis previously treated with a JAK inhibitor (MOMENTUM): an updated analysis of an international, double-blind, randomised phase 3 study. Lancet Haemat. 2023;10(9):e735-e746.
Mesa R, Harrison C, Oh ST, et al. Overall survival in the SIMPLIFY-1 and SIMPLIFY-2 phase 3 trials of momelotinib in patients with myelofibrosis. Leukemia. 2022;36(9):2261-2268. Mesa RA, Kiladjian JJ, Catalano JV, et al et al. Simplify-1: A phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naïve patients with myelofibrosis. Journal of Clinical Oncology. 2017;35(34): 3844. Verstovsek S, Gerds AT, Vanucchi AM, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis (MOMENTUM): results from an international, double-blind, randomised, controlled, phase 3 study. Lancet 2023;401: 269-280.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: agentes antineoplásicos, inibidores de proteína quinase.
Código ATC: L01EJ04
Mecanismo de ação
Momelotinibe é um inibidor dos tipos selvagem da Janus quinase 1 e 2 (JAK1, JAK2) e mutante JAK2V617F, que contribuem para a sinalização de uma série de citocinas e fatores de crescimento que são importantes para a hematopoiese e função imunológica. JAK1 e JAK2 recrutam e ativam proteínas STAT (transdutores de sinal e ativador da transcrição) que controlam a transcrição de genes que afetam a inflamação, hematopoiese e regulação imunológica. O momelotinibe e seu principal metabólito circulante humano, M21, adicionalmente inibem o receptor de ativina A tipo 1 (ACVR1), também conhecido como quinase semelhante ao receptor de ativina 2 (ALK2), que subsequentemente regula negativamente a expressão hepática de hepcidina, resultando em maior disponibilidade de ferro e produção de glóbulos vermelhos A mielofibrose é uma neoplasia mieloproliferativa associada com a via de sinalização de JAK desregulada e constitutivamente ativada que contribui para inflamação elevada e hiperativação de ACVR1.
Efeitos farmacodinâmicos
Momelotinibe inibe a fosforilação de STAT3 induzida por citocinas no sangue total de pacientes com mielofibrose. A inibição máxima da fosforilação de STAT3 ocorreu 2 horas após a administração de momelotinibe, com persistência da inibição por pelo menos 6 horas. Momelotinibe também demonstrou redução aguda e prolongada de hepcidina circulante em pacientes com mielofibrose, resultando em aumento da disponibilidade de ferro e eritropoiese.
Efeitos cardiovasculares
Em uma dose 4 vezes superior a dose inicial recomendada de 200 mg, momelotinibe não prolongou o intervalo QT em nenhuma extensão clinicamente relevante.
Farmacocinética
Absorção
Momelotinibe é rapidamente absorvido após administração oral com a concentração plasmática máxima (Cmax) alcançada dentro de 3 horas pós-dose, com aumento das exposições plasmáticas de maneira menos do que proporcional à dose, em doses acima de 300 mg.
Na dosagem máxima recomendada de 200 mg uma vez ao dia no estado estacionário, a média (%CV) de Cmax de momelotinibe é de 479 ng/mL (61%) e AUCtau é 3.288 ng.h/mL (60%) em pacientes com mielofibrose. Após refeições com baixo teor de gordura e alto teor de gordura em voluntários saudáveis, a Cmax de momelotinibe foi 38% e 28% maior, respectivamente, e a AUC foi 16% e 28% maior, respectivamente, em comparação com aqueles em jejum. Essas alterações na exposição não foram clinicamente significativas.
Distribuição
A ligação de momelotinibe às proteínas plasmáticas é de aproximadamente 91% em indivíduos saudáveis.. O volume aparente médio de distribuição de momelotinibe no estado estável foi de 984 L em pacientes com mielofibrose recebendo momelotinibe 200 mg diariamente, sugerindo extensa distribuição tecidual.
Metabolismo
O metabolismo humano do momelotinibe é predominantemente mediado por enzimas CYP com contribuições na seguinte ordem: CYP3A4 (36%), CYP2C8 (19%), CYP2C19 (19%) CYP2C9 (17%) e CYP1A2 (9%). A geração de M21, o metabólito humano ativo que possui aproximadamente 40% da atividade farmacológica do estrutura parental, envolve biotransformação por enzimas CYP seguida de metabolismo por aldeído oxidase. A razão média de M21 para momelotinibe em termos de AUC variou de 1.4 a 2.1.
Eliminação
Após uma dose oral de momelotinibe 200 mg, a meia-vida terminal média (t½) de momelotinibe foi de 4 a 8 horas; a meia-vida de M21 é semelhante. A depuração total aparente (CL/F) de momelotinibe foi de 103 L/h em pacientes com mielofibrose. Momelotinibe é eliminado principalmente pelo metabolismo e depois excretado nas fezes. Após uma dose oral única de momelotinibe marcado com [14C] em indivíduos do sexo masculino saudáveis, 69% da radioatividade foi excretada nas fezes (13% da dose na forma inalterada de momelotinibe) e 28% na urina ( < 1% da dose na forma inalterada de momelotinibe). Aproximadamente 12% da dose administrada foi excretada na urina como M21.
Populações especiais de pacientes
Insuficiência renal
A AUC de momelotinibe diminuiu 13% em indivíduos com insuficiência renal moderada (TFGe: 30-59 mL/min/1,73 m2) e a AUC diminuiu 16% em indivíduos com insuficiência renal grave (TFGe: 15-29 mL/min/1.73 m2) em comparação com indivíduos com função renal normal (TFGe ≥ 90 mL/min/1,73 m2). A AUC do metabólito ativo, M21, aumentou 20% e 41%, respectivamente, em indivíduos com insuficiência renal moderada e grave em comparação com indivíduos com função renal normal. Não há dados em pacientes com doença renal de estágio terminal (ESRD) recebendo diálise.
Insuficiência hepática
A concentração plasmática máxima (Cmax) do momelotinibe aumentou 13% e a AUC aumentou 97% em indivíduos com insuficiência hepática grave (Child-Pugh C). A Cmax do metabólito M21 diminuiu 76% e a AUC diminuiu 48% em indivíduos com insuficiência hepática grave (Child-Pugh C). Uma redução da dose inicial de 200 mg para 150 mg uma vez ao dia é recomendada para levar em consideração o aumento potencial nas exposições plasmáticas de momelotinibe e a diminuição nas exposições plasmáticas de M21 em pacientes com insuficiência hepática grave (consulte Posologia).
Idade, sexo, raça e peso corporal
A idade, sexo, raça ou peso não apresentam um efeito clinicamente significativo na farmacocinética de momelotinibe com base em uma análise farmacocinética da população.
Carcinogênese/mutagênese
O potencial carcinogênico de momelotinibe foi avaliado em um estudo de 6 meses com camundongos transgênicos rasH2 e um estudo de 2 anos de carcinogenicidade em ratos. Não houve evidência de carcinogenicidade em camundongos machos ou fêmeas que receberam doses de momelotinibe de até 100 mg/kg/dia durante 26 semanas. No estudo de carcinogenicidade de 2 anos em ratos Sprague Dawley, momelotinibe oral causou tumores benignos das células de Leydig na dose de 15 mg/kg/dia (aproximadamente 17 vezes a dose máxima recomendada com base na AUC da combinação de momelotinibe e M21). Um aumento no risco para a saúde humana é considerado improvável, uma vez que o aumento nos adenomas de células de Leydig foi considerado relacionado a um achado mecanicista específico da espécie (ou seja, dependência de prolactina de células de Leydig de ratos). Momelotinibe não foi mutagênico em um ensaio de mutação reversa bacteriana ou clastogênico em um ensaio de aberração cromossômica in vitro com linfócitos de sangue periférico humano ou in vivo em um ensaio de micronúcleo de medula óssea de ratos.
Toxicologia reprodutiva
Fertilidade
Em estudos de fertilidade, momelotinibe foi administrado via oral em ratos machos e fêmeas. Nos machos, momelotinibe reduziu a concentração e a motilidade dos espermatozoides e reduziu os pesos dos testículos e das vesículas seminais com doses de 25 mg/kg/dia ou superiores (exposições 13 vezes a dose recomendada de 200 mg com base na combinação de momelotinibe e M21 AUC), levando à redução da fertilidade a 68 mg/kg/dia.. Em fêmeas momelotinibe reduziu a função ovariana (ciclos reprodutivos e ovulação) a 68 mg/kg/dia e diminuiu o número de fêmeas prenhes e aumentou a perda pré e pós-implantação com a maioria das fêmeas prenhes tendo perda total da ninhada na maioria dos animais com 25 mg/kg/dia e superior. Exposições no nível de nenhum efeito adverso observado em ratos machos e fêmeas a 5 mg/kg/dia que é aproximadamente 3 vezes a dose recomendada (com base na combinação de momelotinibe e M21 AUC).
Gestação
Em estudos de reprodução animal, a administração oral de momelotinibe em ratas prenhes durante o período de organogênese causou toxicidade materna na dose de 12 mg/kg/dia e foi associada à morte embrionária, anomalias dos tecidos moles, variações esqueléticas, pesos corporais fetais médios diminuídos; variações esqueléticas foram observadas na exposição de 6 mg/kg/dia 3.5 vezes a exposição na dose recomendada de 200 mg por dia com base na combinação de momelotinibe e M21 (principal metabólito humano) AUC. Não foi observada toxicidade no desenvolvimento com 2 mg/kg/dia em exposições equivalentes à dose recomendada (com base na combinação de momelotinibe e M21 AUC). Em coelhas prenhes, a administração oral de momelotinibe durante o período de organogênese causou toxicidade materna grave e evidência de toxicidade embriofetal (diminuição do peso fetal, ossificação óssea tardia e abortos) em doses de 60 mg/kg/dia, menos do que a exposição equivalente à dose recomendada (com base na combinação de momelotinibe e M21 AUC). Em um estudo de desenvolvimento pré e pós-natal, ratas prenhes receberam momelotinibe oralmente desde a organogênese até o final da lactação. Evidências de toxicidade materna, letalidade embrionária e diminuição do peso do filhote foram observados em 6 e 12 mg/kg/dia. A sobrevivência dos filhotes foi significativamente reduzida com 12 mg/kg/dia desde o nascimento até o dia 4 da lactação e, portanto, foi considerada um efeito direto de momelotinibe via exposição através da lactação. A exposição de mães à momelotinibe a 6 e 12 mg/kg/dia foi aproximadamente 2 vezes a exposição da dose recomendada (com base na combinação de momelotinibe e M21 AUC). A exposição em mães sem desenvolvimento toxicidade foi com uma dose de 2 mg/kg/dia, que foi menor do que a exposição na dose recomendada (com base na combinação de momelotinibe e M21 AUC).
4 CONTRAINDICAÇÕES
OJJAARA (dicloridrato de momelotinibe) é contraindicado em pacientes com hipersensibilidade conhecida a momelotinibe ou a qualquer um de seus excipientes.
5 ADVERTÊNCIAS E PRECAUÇÕES
Infecções
Infecções, incluindo infecções bacterianas e virais graves e às vezes fatais (incluindo COVID-19), ocorreram em pacientes tratados com OJJAARA (consulte Reações adversas). OJJAARA não deve ser iniciado em pacientes com infecções ativas. Os médicos devem monitorar os pacientes recebendo OJJAARA quanto a sinais e sintomas de infecção e iniciar o tratamento apropriado imediatamente.
Reativação da hepatite B
Aumentos da carga viral da hepatite B (título de HBV-DNA), com ou sem elevações associadas na alanina transaminase (ALT) ou aspartato transaminase (AST), foram relatados em pacientes com infecção crônica pelo vírus da hepatite B (HBV) em uso de inibidores de JAK, incluindo OJJAARA. O efeito do OJJAARA na replicação viral em pacientes com infecção crônica pelo HBV é desconhecido. Pacientes com infecção crônica pelo HBV que recebem OJJAARA devem ter sua infecção crônica pelo HBV tratada e monitorada de acordo com as diretrizes clínicas para HBV.
Trombocitopenia e neutropenia
Desencadeamento de trombocitopenia e neutropenia graves (grau ≥ 3) foi observado em pacientes tratados com OJJAARA (consulte Reações adversas) Um hemograma completo, incluindo contagem de plaquetas, deve ser realizado antes de iniciar o tratamento com OJJAARA, periodicamente durante o tratamento e conforme indicação clínica. Pode ser necessária a interrupção ou redução da dose (consulte Posologia).
Monitoramento hepático
Os testes de função hepática devem ser obtidos antes de iniciar o tratamento com OJJAARA, periodicamente durante o tratamento e conforme indicação clínica. Se houver suspeita de aumentos na ALT, AST ou bilirrubina relacionados ao tratamento, pode ser necessária a interrupção ou redução da dose (consulte Posologia).
Eventos cardiovasculares adversos maiores
Eventos cardiovasculares adversos maiores (MACE) foram relatados em pacientes recebendo OJJAARA, no entanto, uma relação causal não foi estabelecida. Antes de iniciar ou continuar a terapia com OJJAARA, os benefícios e riscos para cada paciente devem ser considerados, particularmente em pacientes com 65 anos de idade ou mais, pacientes que são fumantes atuais ou anteriores de longa data, e pacientes com histórico de doença cardiovascular aterosclerótica ou outros fatores de risco cardiovascular.
Trombose
Eventos de trombose venosa profunda e embolia pulmonar foram relatados em pacientes recebendo OJJAARA. No entanto, uma associação causal não foi estabelecida. Antes de iniciar ou continuar a terapia com OJJAARA, os benefícios e riscos para cada paciente devem ser considerados, particularmente em pacientes com fatores de risco cardiovascular. Pacientes com sintomas de trombose devem ser imediatamente avaliados e tratados adequadamente.
Segundas malignidades primárias
Linfoma e outras malignidades foram relatados em pacientes recebendo inibidores da JAK, incluindo OJJAARA. No entanto, uma associação causal não foi estabelecida. Considerar os benefícios e riscos para cada paciente antes de iniciar ou continuar a terapia com OJJAARA, particularmente com malignidade conhecida (exceto câncer de pele não melanoma tratado com sucesso), pacientes que desenvolvem uma malignidade e pacientes que são fumantes atuais ou anteriores.
Gravidez e lactação
Fertilidade
Não existem dados sobre os efeitos do momelotinibe na fertilidade humana masculina ou feminina. Em estudos com animais, momelotinibe comprometeu a fertilidade em ratos (consulte Características Farmacológicas).
Gravidez
Não há dados sobre os efeitos de momelotinibe na gravidez humana. Com base em dados em animais, momelotinibe pode causar toxicidade embriofetal. Em ratos e coelhos, foram observados abortos, morte embrionária e anomalias fetais na presença e ausência de toxicidade materna em exposições equivalentes à dose clínica de 200 mg ao dia (consulte Informações não clínicas). OJJAARA só deve ser usado durante a gravidez se os benefícios esperados para a mãe superarem os possíveis riscos para o feto. Mulheres férteis que não estejam grávidas devem usar métodos contraceptivos altamente eficazes durante o tratamento e por pelo menos 1 semana após a última dose de OJJAARA.
Categoria C: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista. Pacientes que suspeitam da possibilidade de gravidez devem ser advertidas a informar os seus médicos imediatamente.
Lactação
Não há dados sobre a presença de momelotinibe no leite humano. Momelotinibe estava presente em filhotes de ratos após amamentação de mães tratadas com efeitos adversos na prole (consulte Características Farmacológicas). O risco para o lactente não pode ser excluído. As pacientes não devem amamentar durante o tratamento com momelotinibe e por pelo menos 1 semana após o término da última dose de OJJAARA.
Este medicamento é contraindicado durante o aleitamento ou doação de leite, pois é excretado no leite humano e pode causar reações indesejáveis no bebê. Seu médico ou cirurgião-dentista deve apresentar alternativas para o seu tratamento ou para a alimentação do bebê.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não houve estudos para investigar o efeito de OJJAARA no desempenho ao dirigir ou na capacidade de operar máquinas. No entanto, os pacientes que apresentarem tonturas ou visão embaçada após administração de OJJAARA devem ter cuidado ao dirigir ou operar máquinas (consulte Reações adversas).
Aviso: este medicamento contém LACTOSE. Pacientes com problemas hereditários de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glucose-galactose não devem utilizar este medicamento.
Atenção: Contém os corantes óxido de ferro vermelho e óxido de ferro amarelo que podem, eventualmente, causar reações alérgicas.
6. INTERAÇÕES MEDICAMENTOSAS
Indutores fortes de CYP3A4
A coadministração de indutores fortes de CYP3A4 pode levar a uma diminuição da exposição ao momelotinibe e, consequentemente, a um risco de eficácia reduzida. Portanto, o monitoramento adicional é recomendado com o uso concomitante de momelotinibe e indutores fortes de CYP3A4 (incluindo mas não limitado a carbamazepina, fenobarbital, fenitoína e Erva de São João [Hypericum perforatum] (consulte Características Farmacológicas).
Substrato proteico de resistência sensível ao câncer de mama
Como inibidor da proteína de resistência ao câncer de mama (BCRP), OJJAARA tem o potencial de aumentar a concentração plasmática de substratos sensíveis à BCRP, como rosuvastatina e sulfazalazina. As reações adversas em pacientes com esta coadministração devem ser monitoradas.
Inibidores dos transportadores hepáticos de polipeptídeo orgânico de transporte de ânions (OATP) 1B1/1B3
O momelotinibe é um substrato de OATP1B1/B3. O uso concomitante com um inibidor OATP1B1/B3 (como a ciclosporina) resultou eu um aumento moderado na exposição plasmática ao momelotinibe, o que pode aumentar o risco de reações adversas com OJJAARA. Monitore os pacientes que recebem concomitantemente um inibidor OATP1B1/B3 quanto a reações adversas e considere modificações na dose de OJJAARA.
Substratos sensíveis à CYP3A4
A coadministração com múltiplas doses de momelotinibe com midazolan (substrato sensível à CYP3A4) reduziu a Cmax para 8,2% e AUC em 16,2%. Essas mudanças não foram consideradas clinicamente relevantes.
Inibidores da bomba de prótons
Em um estudo com voluntários adultos saudáveis aos quais foi administrado dose única de 200 mg de momelotinibe, coadministrado com múltiplas doses de omeprazol (um inibidor da bomba de prótons) reduziu a Cmax em 35,5% e a AUC em 33,3%. Essas mudanças não são consideradas clinicamente relevantes.
Inibidores potentes da CYP3A4
A coadministração com múltiplas doses de ritonavir (um inibidor potente da CYP3A4) aumentou a Cmax em 23,3% e AUC em 13,5%. Essas mudanças não foram consideradas clinicamente relevantes.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de armazenamento
Manter fora do alcance de crianças Conservar em temperatura ambiente (temperatura entre 15 °C e 30°C). O prazo de validade é de 36 meses a partir da data de fabricação. Conservar no frasco original e proteger da umidade. Não remova o dessecante.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, válido por 30 dias.
Aspectos físicos / características organolépticas
Cada comprimido contém 100 mg, 150 mg ou 200 mg de momelotinibe base livre.
Comprimidos de 100 mg, redondos, marrons, com um "M" sublinhado gravado num dos lados e "100" no outro lado. Comprimidos de 150 mg, triangulares, marrons, com um "M" sublinhado gravado num dos lados e "150" no outro lado. Comprimidos de 200 mg, em forma de cápsula, marrons, com um "M" sublinhado gravado num dos lados e "200" no outro lado.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Posologia
Adultos A dose recomendada de OJJAARA é de 200 mg por via oral uma vez ao dia. OJJAARA pode ser administrado com ou sem alimento.
Dose perdida
Se uma dose de OJJAARA for perdida, a próxima dose agendada deve ser administrada no dia seguinte.
Monitoramento
Um hemograma completo (incluindo contagem de plaquetas) e testes da função hepática devem ser realizados antes de iniciar o tratamento com OJJAARA e, periodicamente durante o tratamento e conforme indicação clínica.
Modificações de dose
Modificações de dose devem ser consideradas para toxicidades hematológicas e não hematológicas (Tabela 3). Descontinue OJJAARA em pacientes incapazes de tolerar 100 mg uma vez ao dia.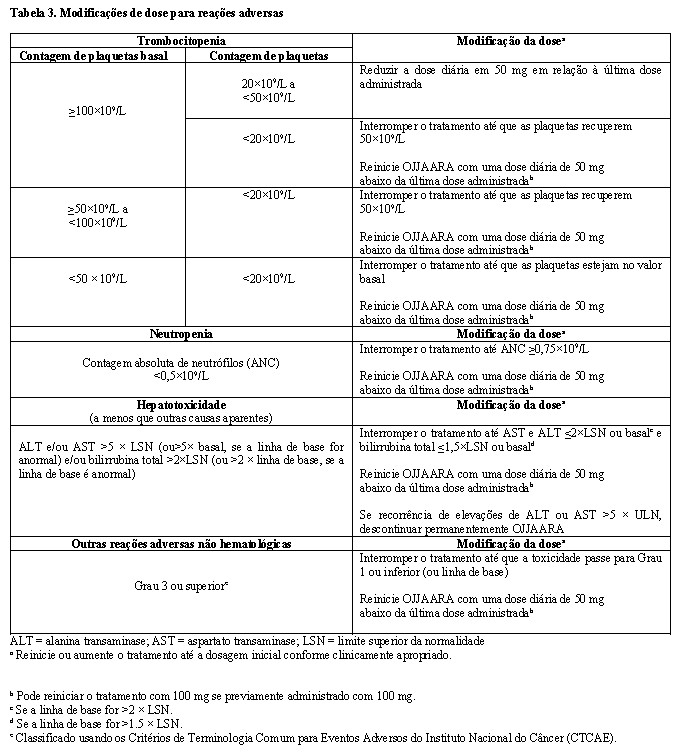
Crianças
A segurança e eficácia de momelotinibe em crianças e adolescentes com menos de 18 anos de idade não foram estabelecidas.
Idosos
Nenhum ajuste de dose é necessário para pacientes com 65 anos ou mais (consulte Farmacocinética - populações especiais de pacientes).
Insuficiência renal
Nenhum ajuste de dose é necessário para pacientes com insuficiência renal (consulte Farmacocinética - populações especiais de pacientes).
Insuficiência hepática
Nenhum ajuste de dose é recomendado para pacientes com insuficiência hepática leve ou moderada. A dose inicial recomendada de OJJAARA é de 150 mg uma vez ao dia em pacientes com insuficiência hepática grave. (Child-Pugh classe C) (consulte Farmacocinética
- populações especiais de pacientes).
Este medicamento não deve ser partido, aberto ou mastigado.
Este medicamento pode causar hepatotoxicidade. Por isso, requer uso cuidadoso, sob vigilância médica estrita e acompanhado por controles periódicos da função hepática durante o tratamento e conforme indicação clínica.
9. REAÇÕES ADVERSAS
Dados de estudos clínicos
A segurança do OJJAARA, com base em três estudos randomizados, multicêntricos e controlados por comparador ativo em adultos com mielofibrose (MOMENTUM, SIMPLIFY-1, SIMPLIFY-2) está apresentada na Tabela 4. Os indivíduos foram inicialmente randomizados para receber OJJAARA 200 mg uma vez ao dia por 24 semanas (n = 448). As reações adversas identificadas para OJJAARA estão listadas por classe de órgãos e sistemas (SOC) do corpo e frequência. As frequências são definidas como: muito comum: (≥ 1/10), comum (≥ 1/100 a < 1/10), incomum (≥ 1/1.000 a < 1/100), rara (≥ 1/10.000 a < 1/1.000)
Durante o tratamento randomizado, os eventos adversos mais relatados para momelotinibe (maior ou igual a 15% dos indivíduos; n = 448) foram diarreia (22,8%), trombocitopenia (19,4%) e náusea (16,7%). Durante o tratamento aberto com momelotinibe, os eventos adversos mais relatados foram diarreia (19,9%) e trombocitopenia (17,5%).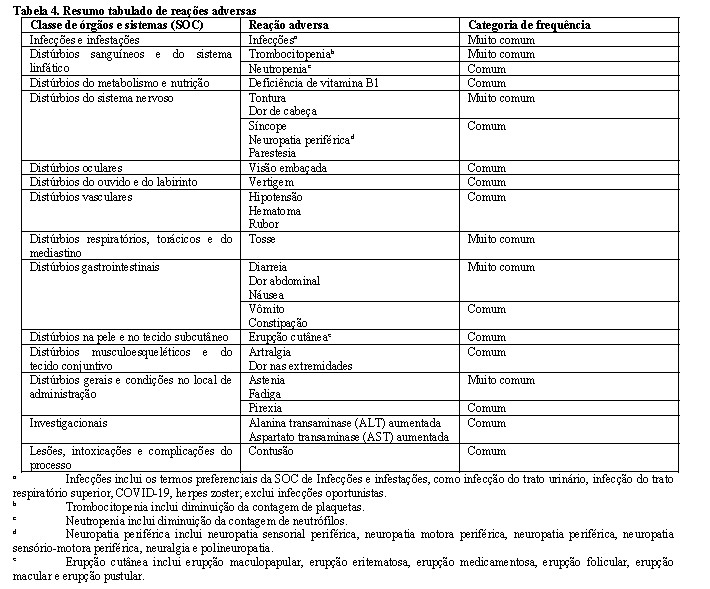
Neuropatia periférica
Nos três estudos clínicos randomizados, 8,7% (39/448) dos pacientes tratados com OJJAARA apresentaram neuropatia periférica. A maioria dos casos de neuropatia periférica foi leve ou moderado, enquanto um dos 39 casos foi grave (≥ Grau 3). A proporção de pacientes que interromperam o tratamento devido a neuropatia periférica foi de 0,7% (3/448).
Elevação das enzimas hepáticas
Nos três estudos clínicos randomizados, elevações novas ou agravadas de ALT e AST (todos os graus) ocorreram em 20% (88/448) e 20% (90/448), respectivamente, dos pacientes tratados com OJJAARA; elevações das transaminases de grau 3 e 4 ocorreram em 1,1% (5/448) e 0,2% (1/448) dos pacientes, respectivamente. Lesão hepática reversível induzida por medicamento foi relatada em pacientes com mielofibrose tratados com OJJAARA em estudos clínicos.
Infecções
Nos três estudos clínicos randomizados, 40% (178/448) dos pacientes tratados com OJJAARA apresentaram infecção. As infecções mais comuns (≥2%) foram infecção do trato urinário (6%), infecção do trato respiratório superior (5%), pneumonia (3,6%), nasofaringite (2,9%), COVID-19 (2,7%), cistite (2,7 %), bronquite (2,5%) e herpes oral (2,5%). A maioria das infecções foi leve ou moderada, enquanto 10% (47/448) dos pacientes apresentaram infecção grave (≥ Grau 3). A proporção de pacientes que interromperam o tratamento devido a uma infecção foi de 2% (9/448). Infecções fatais foram relatadas em 2,2% (10/448) dos pacientes. Nos estudos individuais de MOMENTUM, SIMPLIFY-1 e SIMPLIFY-2, as taxas de infecções para momelotinibe foram de 34%, 36% e 55%, respectivamente, em comparação com 35% para danazol, 43% para ruxolitinibe e 42% para a melhor terapia disponível.
Trombocitopenia
Nos três estudos clínicos randomizados, 21% (94/448) dos pacientes tratados com momelotinibe apresentaram trombocitopenia; 12% (54/448) dos pacientes tratados com momelotinibe apresentaram trombocitopenia grave (≥ grau 3). A proporção de pacientes que descontinuaram o tratamento devido à trombocitopenia foi de 2,5% (11/448). Nos estudos individuais de MOMENTUM, SIMPLIFY-1 e SIMPLIFY-2, as taxas de trombocitopenia para momelotinibe foram de 28%, 19% e 17%, respectivamente, em comparação com 15% para danazol, 29% para ruxolitinibe e 12% para a melhor terapia disponível.
Atenção: este produto é um medicamento no