NUVYOR
EUROFARMA
lenalidomida
Imunomodulador.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
Cápsula dura 5 mg: embalagens com 21 cápsulas.
Cápsula dura 10 mg: embalagens com 21 e 28 cápsulas.
Cápsula dura 15 mg: embalagens com 21 e 28 cápsulas.
Cápsula dura 25 mg: embalagens com 14 e 21 cápsulas.
USO ORAL
USO ADULTO ACIMA DE 18 ANOS
Composição.
Cada cápsula dura de 5 mg contém: lenalidomida 5 mg, excipientes* q.s.p. 1 cápsula dura
*Excipientes: lactose, celulose microcristalina, croscarmelose sódica, estearato de magnésio, dióxido de titânio, óxido de ferro amarelo, óxido de ferro vermelho, óxido de ferro preto, azul brilhante, amarelo crepúsculo e gelatina.
Cada cápsula dura de 10 mg contém: lenalidomida10 mg, excipientes** q.s.p. 1 cápsula dura
**Excipientes: lactose, celulose microcristalina, croscarmelose sódica, estearato de magnésio, dióxido de titânio, vermelho allura 129, azul brilhante, amarelo crepúsculo, amarelo de tartrazina e gelatina.
Cada cápsula dura de 15 mg contém: lenalidomida 15 mg, excipientes*** q.s.p. 1 cápsula dura
***Excipientes: lactose, celulose microcristalina, croscarmelose sódica, estearato de magnésio, dióxido de titânio, óxido de ferro amarelo, óxido de ferro vermelho, óxido de ferro preto, azul brilhante, vermelho allura 129, amarelo de tartrazina e gelatina.
Cada cápsula dura de 25 mg contém: lenalidomida 25 mg, excipientes**** q.s.p. 1 cápsula dura
****Excipientes: lactose, celulose microcristalina, croscarmelose sódica, estearato de magnésio, dióxido de titânio e gelatina.
Proibido para mulheres grávidas. Este medicamento pode causar o nascimento de crianças sem braços e sem pernas. Este medicamento é somente seu. Não passe para ninguém. Este medicamento não provoca aborto e não evita filhos.
Informações técnicas.
1. INDICAÇÕES
1.1 Mieloma múltiplo
A lenalidomida em terapia combinada (vide item 8. "POSOLOGIA E MODO DE USAR" para mais detalhes sobre as combinações e doses), é indicado para o tratamento de pacientes com mieloma múltiplo que não receberam tratamento prévio e não são elegíveis a transplante.
A lenalidomida, em combinação com bortezomibe e dexametasona, é indicado para o tratamento de pacientes com mieloma múltiplo que não receberam tratamento prévio.
A lenalidomida em monoterapia é indicado para o tratamento de manutenção de pacientes com mieloma múltiplo recém-diagnosticado que foram submetidos a transplante autólogo de células-tronco.
A lenalidomida, em combinação com dexametasona, é indicado para o tratamento de pacientes com mieloma múltiplo refratário/recidivado que receberam ao menos um esquema prévio de tratamento.
1.2 Linfoma folicular ou linfoma de zona marginal
A lenalidomida em combinação com rituximabe (anticorpo anti-CD20) é indicado para o tratamento de pacientes com linfoma folicular ou linfoma de zona marginal previamente tratados.
1.3 Linfoma de células do manto
A lenalidomida é indicado para o tratamento de pacientes com linfoma de células do manto refratário/recidivado.
2. RESULTADOS DE EFICÁCIA
2.1 Mieloma múltiplo recém-diagnosticado não elegível a transplante
2.1.1 Estudo MM-015
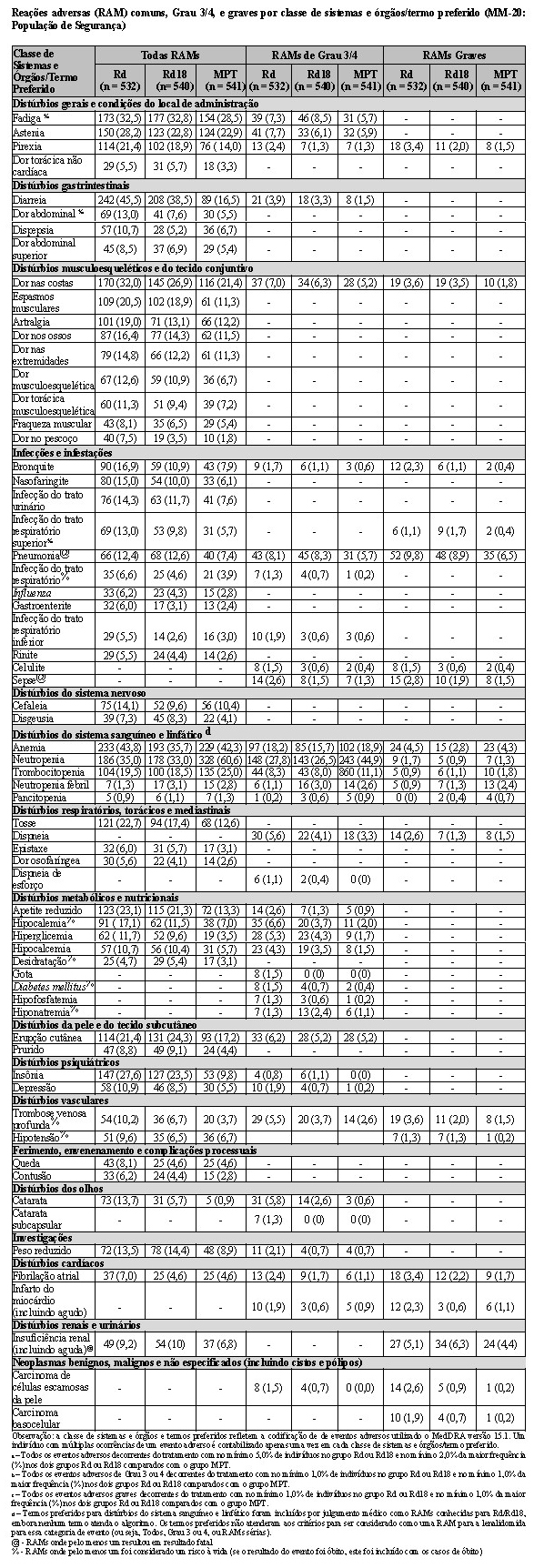
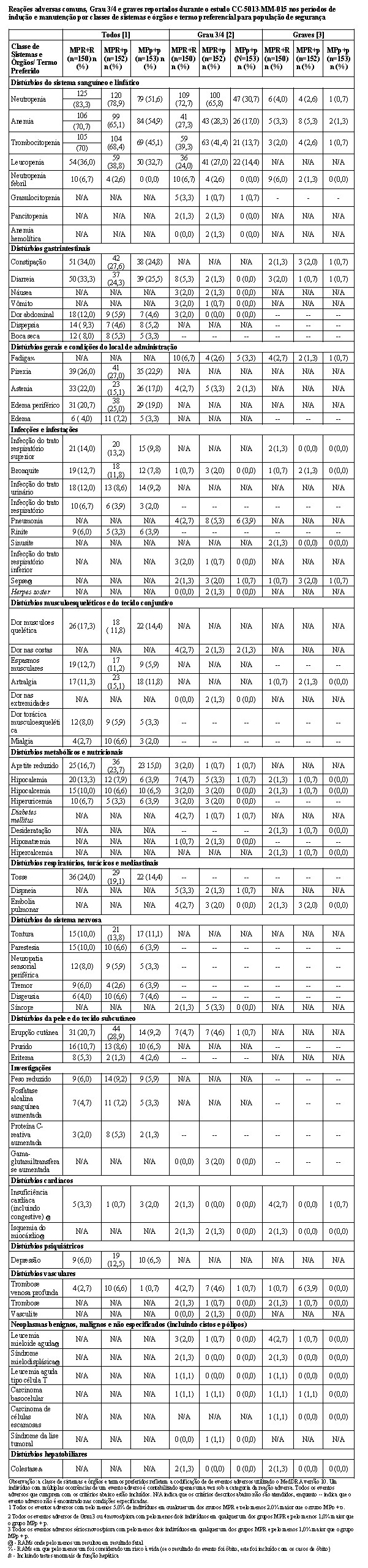
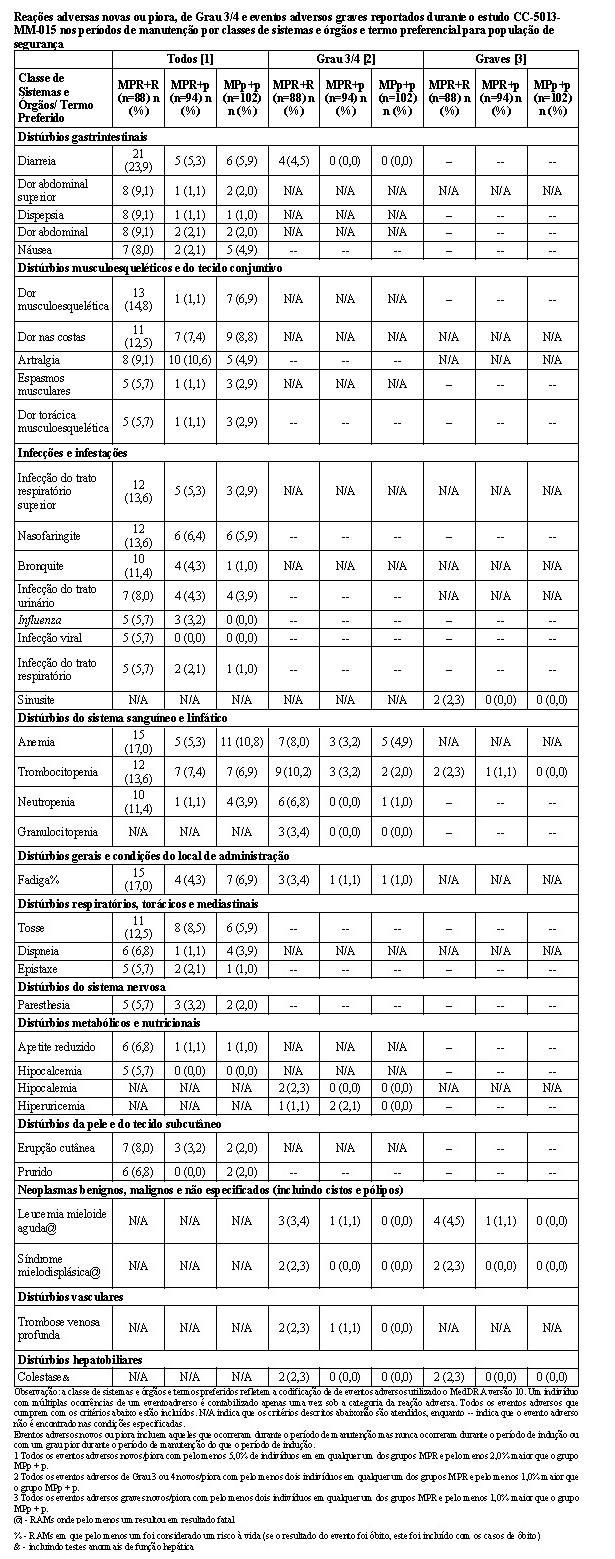
MM-015 foi conduzido para avaliar a segurança e a eficácia da terapia de combinação para melfalano, prednisona e lenalidomida (MPR), seguida pela monoterapia de manutenção com lenalidomida. Este foi um estudo de Fase 3, multicêntrico, randomizado, duplo-cego, controlado por placebo, de 3 grupos paralelos, em pacientes com mieloma múltiplo sintomático, recém-diagnosticado que tinham no mínimo 65 anos de idade. Os pacientes foram randomizados em uma proporção 1:1:1 para um dos 3 grupos de tratamento: Grupo MPR+R - terapia de combinação de indução oral com MPR seguida por tratamento de manutenção com lenalidomida; Grupo MPR+p - terapia de combinação de indução oral com MPR seguida por tratamento de manutenção com placebo; ou Grupo MPp+p - terapia de combinação de indução oral com MPp (MP + placebo) seguida por tratamento de manutenção com placebo.
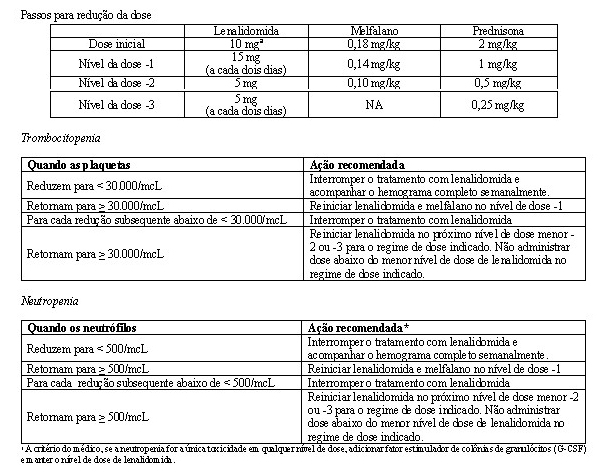
Este estudo investigou a utilização da terapia de combinação de MPR (0,18 mg/kg de melfalano via oral nos Dias 1-4 dos ciclos repetidos de 28 dias; 2 mg/kg de prednisona via oral nos Dias 1-4 dos ciclos repetidos de 28 dias; e 10 mg/dia de lenalidomida via oral nos Dias 1-21 dos ciclos repetidos de 28 dias) para terapia de indução, por até 9 ciclos. Os pacientes que concluíram 9 ciclos ou que foram incapazes de concluir 9 ciclos em decorrência da intolerância procederam para a monoterapia de manutenção, iniciando com 10 mg de lenalidomida via oral nos Dias 1-21 dos ciclos repetidos de 28 dias, administrados até a progressão da doença.
O estudo incluiu pacientes com contagem absoluta de neutrófilos (ANC - absolut neutrophil count) ≥ 1500 células/mcL, contagens de plaquetas ≥ 75.000/mcL, hemoglobina ≥ 8 g/dL, creatinina sérica ≤ 2,5 mg/dL, e TGO/AST ou TGP/ALT séricas < 3,0 x limite superior da normalidade (LSN).
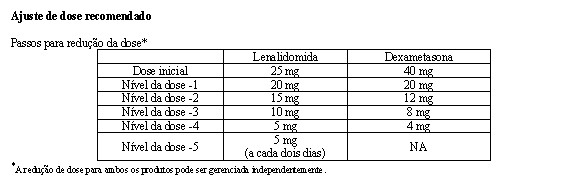
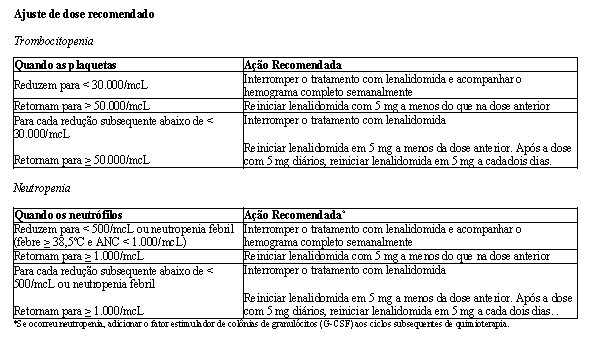
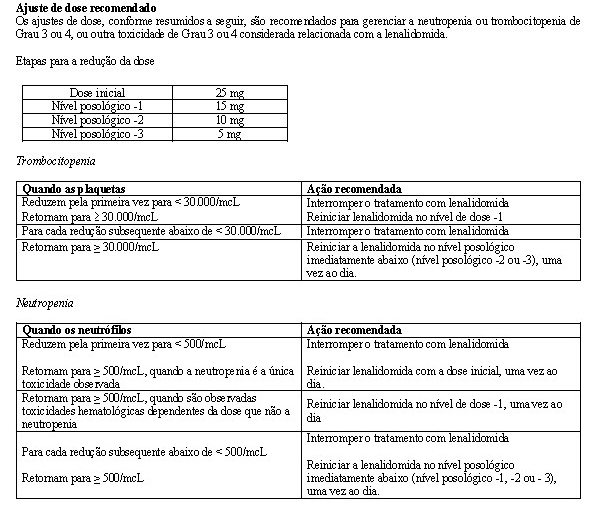
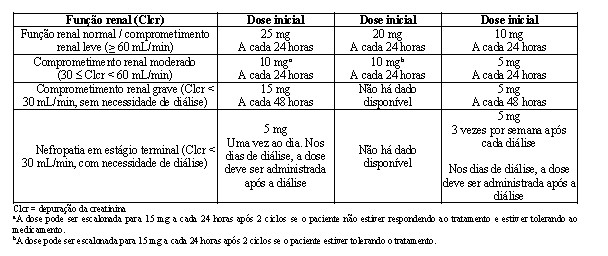
Ajustes de dose foram permitidos com base em achados clínicos e laboratoriais. As reduções sequenciais de dose foram permitidas para lenalidomida com uma dose inicial de 10 mg diariamente e com redução para 7,5 mg, 5 mg e 2,5 mg diariamente, e foram baseadas em uma avaliação das toxicidades. Se as toxicidades retornavam, a dose não podia ser reduzida para menos do que 2,5 mg diariamente, e lenalidomida deveria ser interrompida.
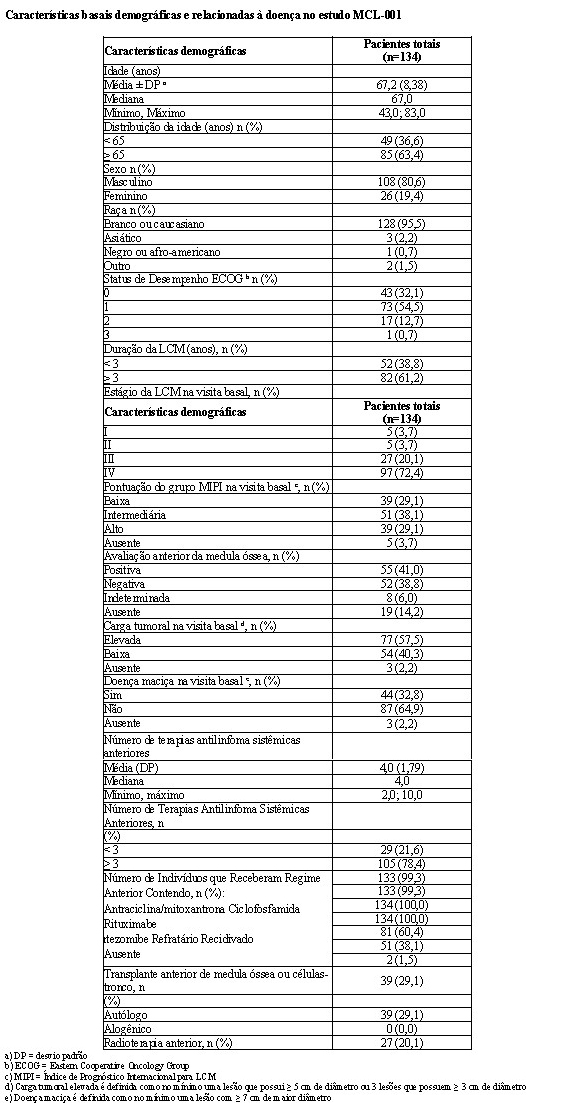
A tabela a seguir resume as características demográficas e relacionadas às características basais da doença. Em geral, os 3 grupos de tratamento (MPR+R, MPR+p e MPp+p) foram bem equilibrados em relação às características demográficas e relacionadas à doença.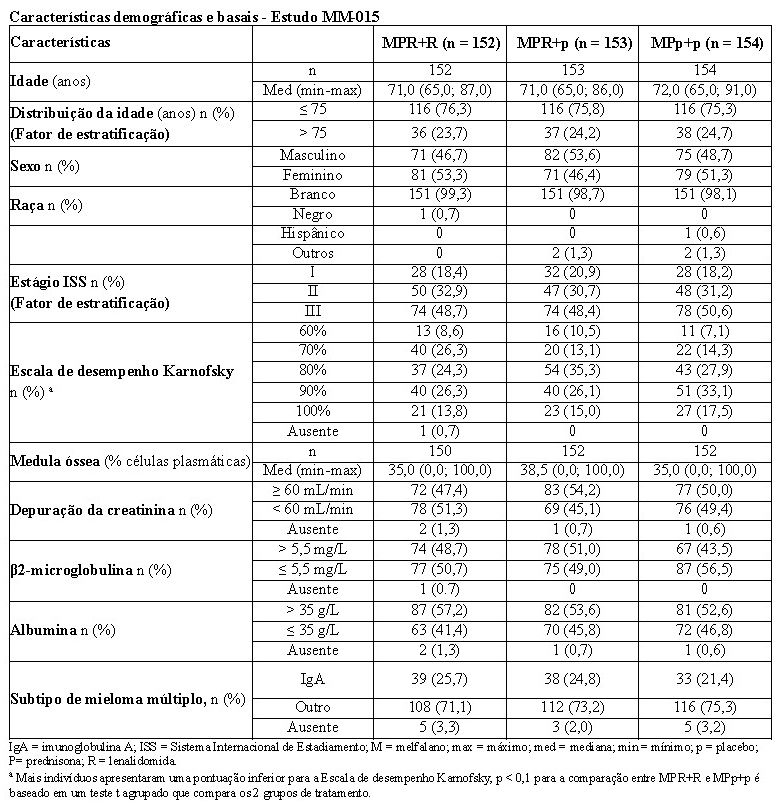
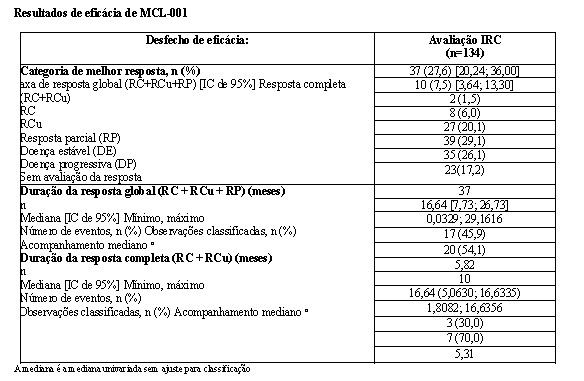
A análise primária do desfecho primário sobrevida livre de progressão (SLP) foi conduzida na data de corte de 11 de maio de 2010 com base na avaliação do Comitê Central de Avaliação. Os resultados de eficácia estão resumidos na tabela a seguir. A sobrevida livre de progressão por revisão independente cega foi significativamente maior com MPR+R do que com MPp+p, com uma HR de 0,388 (IC de 95% = 0,274, 0,550; p < 0,001) indicando 61% de redução no risco de doença progressiva ou morte para MPR+R em comparação com MPp+p.
Para a avaliação do investigador com > 98% de eventos de SLP especificados no protocolo (abril de 2013), a SLP foi significativamente maior com o tratamento MPR+R versus MPp+p: HR 0,371 [IC de 0,271; 0,503] (p < 0,001) indicando 63% de redução no risco de progressão da doença ou morte.
A taxa de resposta global (comparação de resposta completa + resposta parcial) foi maior em MPR+R (78,9%) do que em MPp+p (54,5%) (p < 0,001). Um percentual maior de pacientes obteve no mínimo uma resposta completa em MPR+R do que em MPp+p (19,7% versus 5,8%, respectivamente).
A duração mediana da resposta foi 26,5 meses para MPR+R e 12,0 meses para MPp+p. Mais do que a metade dos responsivos em MPR+R (55%) apresentou respostas que duraram no mínimo 2 anos em comparação com somente 15,5% daqueles que receberam MPp+p.
A sobrevida livre de progressão na terapia de próxima linha (SLP2) calculada como o tempo desde a randomização até o início da terapia de terceira linha ou morte para todos os pacientes randomizados também foi significativamente melhorada no tratamento MPR+R versus MPp+p [HR = 0,701 (IC de 0,536-0,916)], o que significa uma mediana de 39,7 meses para MPR+R versus 28,8 meses para MP+p+p.
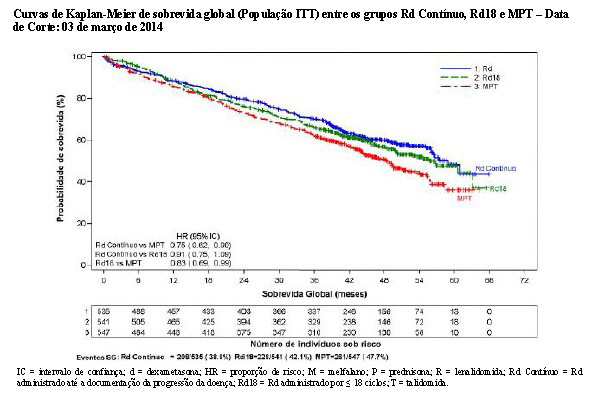
Não houve diferenças significativas entre os grupos de tratamento na sobrevida global.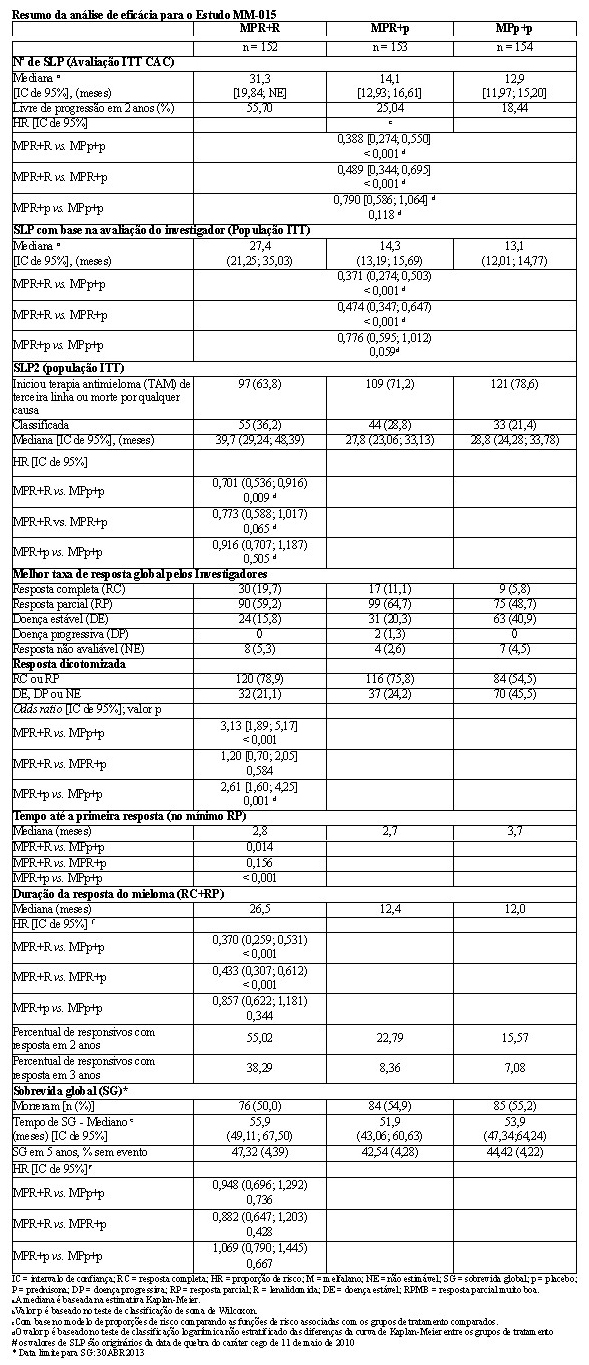
Estimativa Kaplan-Meier do tempo de sobrevida livre de progressão para MPR+R, MPR+p e MPp+p – Estudo MM-015 - Avaliação CAC na quebra do caráter cego do estudo (População ITT)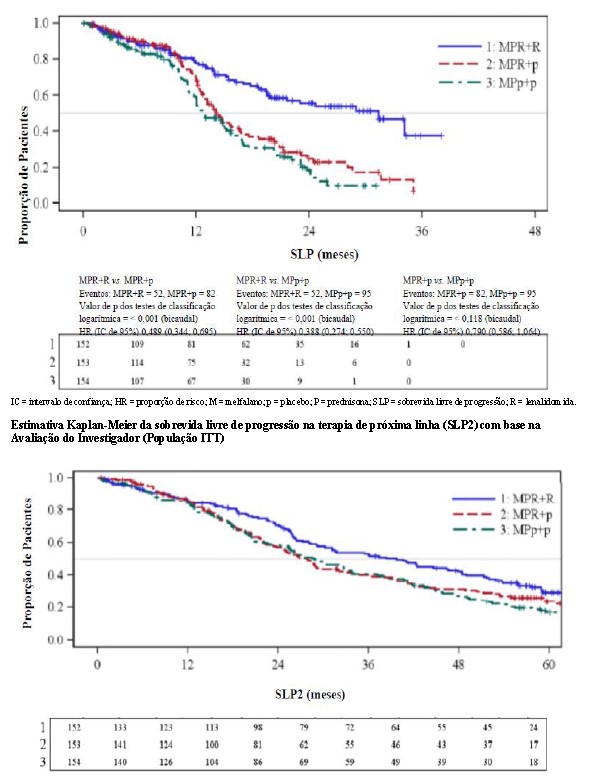
2.1.2 Estudo MM-020
O estudo clínico MM-020 foi um estudo de Fase 3, multicêntrico, randomizado, aberto, de 3 grupos, para comparar a eficácia e a segurança de lenalidomida e da dexametasona (Rd) administradas por 2 períodos com durações diferentes [ou seja, até a progressão de doença (grupo Rd) ou por até dezoito ciclos de 28 dias (72 semanas, grupo Rd18)] com aquelas de melfalano, prednisona e talidomida (MPT) por um máximo de doze ciclos de 42 dias (72 semanas). Os principais critérios de elegibilidade incluíram pacientes com mieloma múltiplo recém-diagnosticado sintomático que apresentaram a proteína do mieloma (proteína M) mensurável através de análises eletroforéticas de proteínas [no soro (SPEP) e/ou na urina (UPEP)] e que tinham 65 anos de idade ou mais, ou que não eram elegíveis a transplante de células-tronco (TCT). Os pacientes que apresentaram mieloma múltiplo não secretor nas análises SPEP e UPEP não foram elegíveis para este estudo. Para os fins deste estudo, um paciente que tinha < 65 anos de idade não era um candidato ao TCT se o paciente se recusasse e se submeter à terapia TCT ou se o paciente não tinha acesso ao TCT em decorrência dos custos ou de quaisquer outras razões. Os pacientes com status de desempenho fraco (status ECOG de 3 ou 4) ou com condições médicas coexistentes sérias, conforme considerado pelo médico investigador, foram excluídos deste estudo.
Os pacientes elegíveis foram randomizados em uma proporção 1:1:1 para 1 de 3 grupos de tratamento. Os pacientes foram estratificados na randomização por idade (≤ 75 versus > 75 anos), estágio (Estágio ISS I e II versus Estágio III), e país. A resposta, incluindo progressão da doença, foi avaliada de acordo com os critérios do Grupo Internacional de Trabalho de Mieloma (IMWG), com base em valores laboratoriais centrais de medidas da proteína M.
Os pacientes nos grupos Rd e Rd18 receberam 25 mg de lenalidomida uma vez ao dia nos Dias 1 a 21 dos ciclos de 28 dias. Dexametasona foi administrada em 40 mg uma vez ao dia nos Dias 1, 8, 15 e 22 de cada ciclo de 28 dias. A dose inicial e os regimes para Rd e Rd18 foram ajustados de acordo com a idade e a função renal. Todos os pacientes receberam anticoagulação profilática (heparina de baixo peso molecular, varfarina, heparina, aspirina de dose baixa) durante o estudo.
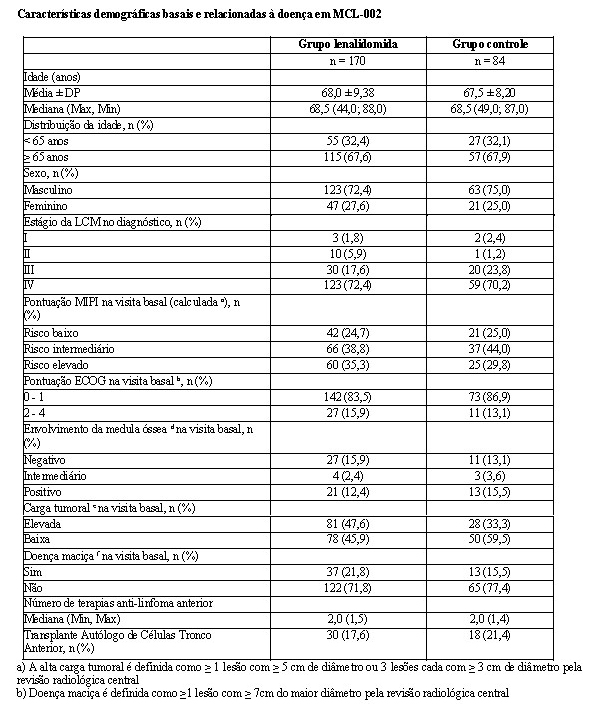
As características demográficas e basais da doença para a população com intenção de tratamento (ITT) estão resumidas nas tabelas a seguir. Em geral, não foram observadas diferenças clinicamente significativas nas características demográficas e relacionadas à doença, e os grupos de tratamento foram equilibrados quanto às características demográficas e relacionadas à doença.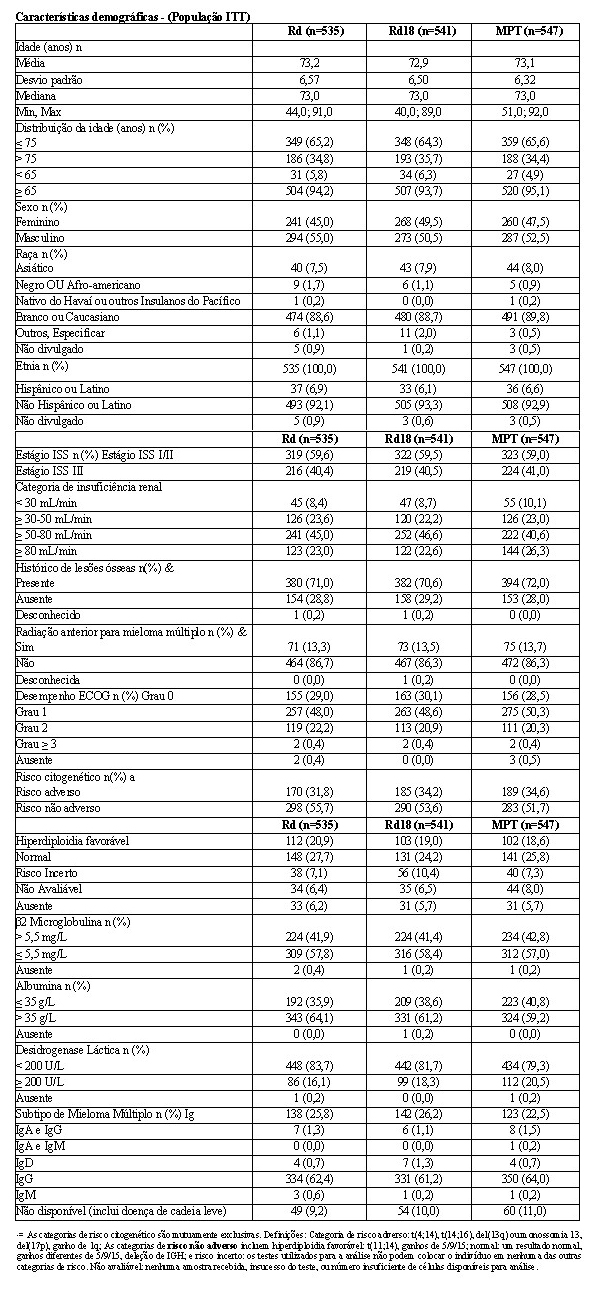
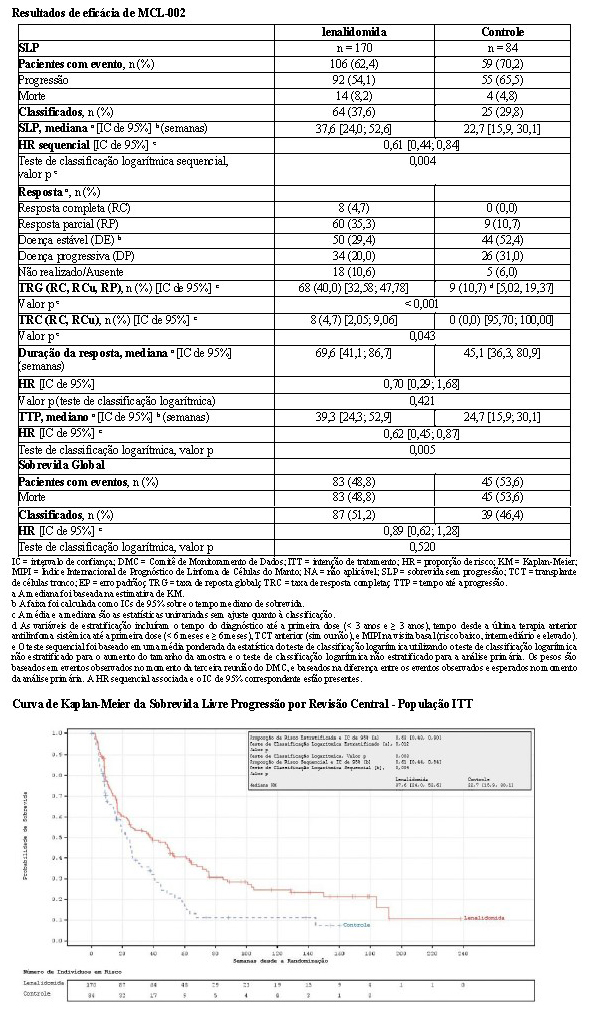
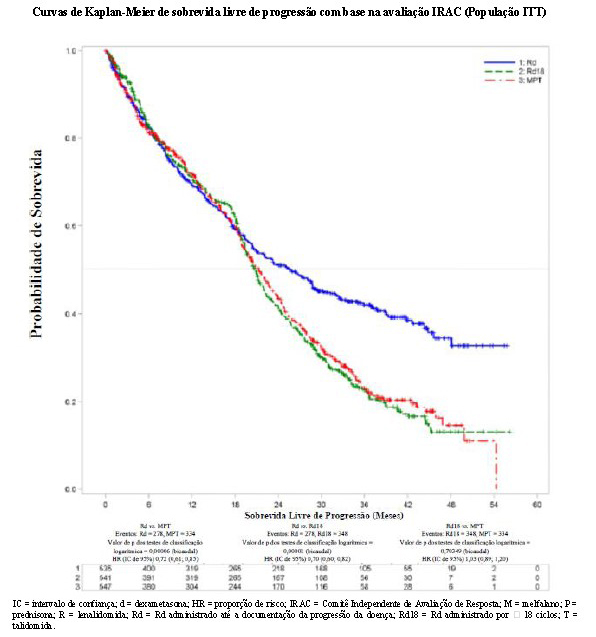
O desfecho de eficácia primário (SLP) foi definido como o tempo da randomização até a primeira documentação de progressão da doença (com base nos critérios IMWG) ou morte decorrente de qualquer causa durante o estudo até o fim da fase de acompanhamento da SLP. A análise primária da SLP foi baseada na avaliação independente IRAC (Comitê Independente de Avaliação de Resposta) para Rd versus MPT. Para a análise de eficácia de todos os desfechos, a comparação primária foi realizada entre os grupos Rd e MPT.
Os resultados de eficácia estão resumidos na tabela a seguir. A SLP foi significativamente maior com Rd do que com MPT: HR 0,72 (IC de 95%: 0,61-0,85 p = 0,00006) indicando 28% de redução no risco de progressão da doença ou morte. Um percentual inferior de pacientes no grupo Rd em comparação com o grupo MPT apresentou eventos da SLP (52% versus 61%, respectivamente). A mesma proporção (10%) de eventos de morte durante o estudo contribuiu para a SLP em ambos os grupos de tratamento. A melhora do tempo mediano de SLP no grupo Rd em comparação com o grupo MPT foi 4,3 meses. A taxa de resposta do mieloma foi significativamente maior com Rd em comparação com MPT (75,1% versus 62,3%; p < 0,00001) com uma resposta completa em 15,1% de pacientes no grupo Rd versus 9,3% dos pacientes no grupo MPT. O tempo mediano até a primeira resposta foi 1,8 meses no grupo Rd versus 2,8 meses no grupo MPT.
Para a análise de SG, o tempo mediano de acompanhamento para todos os pacientes que sobreviveram é 37,0 meses, com 574 eventos de morte, em 64% de ocorrência (574/896) dos eventos finais de SG. A HR observada foi 0,78 para Rd versus MPT (IC de 95% = 0,64; 0,96; nominal p = 0,01685) indicando 22% de redução no risco de morte.
Os parâmetros de qualidade de vida melhoraram após o início do tratamento e no geral foram mantidos enquanto os pacientes estavam livres de progressão, porém pioraram com a progressão da doença.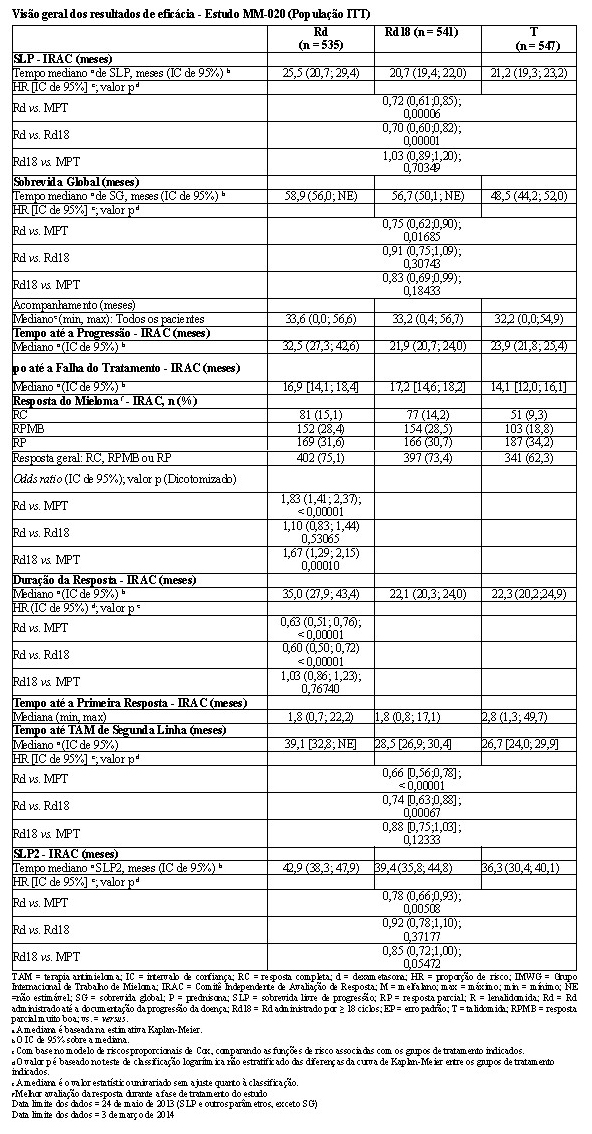
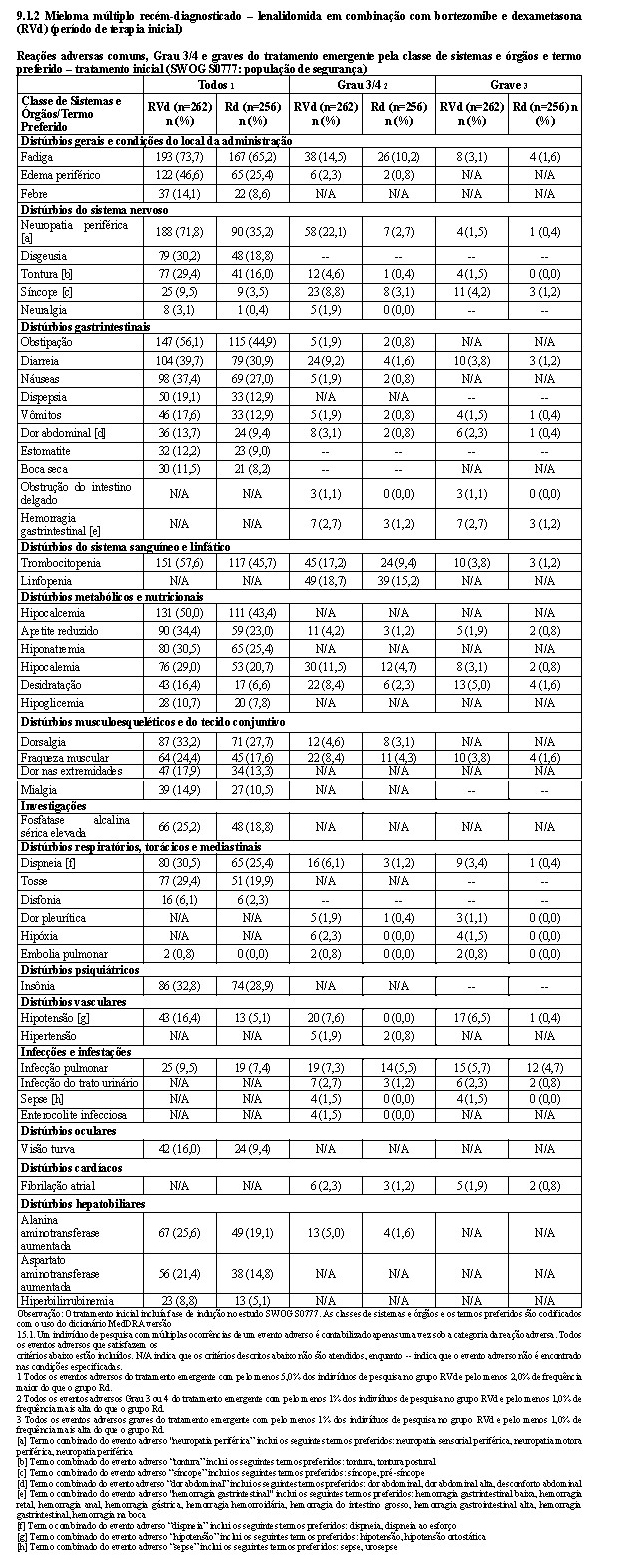
2.2 Adição de bortezomibe a lenalidomida em combinação com dexametasona (RVd) (período inicial de tratamento)
2.2.1 Estudo SWOG S0777
Um estudo clínico randomizado, multicêntrico, aberto, com 2 braços (Estudo SWOG S0777, NCT no. 00644228) de 523 pacientes comparou a eficácia e a segurança de lenalidomida, bortezomibe e dexametasona (RVd) à de lenalidomida e dexametasona (Rd) em pacientes com mieloma múltiplo recém-diagnosticado, sem considerar a elegibilidade para transplante. Pacientes no grupo RVd receberam até oito ciclos de 21 dias com lenalidomida 25 mg/dia, via oral, nos Dias 1-14, bortezomibe intravenoso, 1,3 mg/m2, nos Dias 1, 4, 8 e 11, e dexametasona 20 mg/dia, via oral, nos Dias 1, 2, 4, 5, 8, 9, 11 e 12. Pacientes no grupo Rd receberam até seis ciclos de 28 dias com lenalidomida 25 mg/dia, via oral, nos Dias 1-21 e dexametasona 40 mg/dia, via oral, nos Dias 1, 8, 15 e 22. Após o tratamento inicial (24 semanas para cada braço de tratamento), Rd foi continuado até a progressão da doença para todos os pacientes em ciclos de 28 dias com lenalidomida 25 mg, via oral, nos Dias 1-21 e dexametasona 40 mg, nos Dias 1, 8, 15 e 22. As doses foram reduzidas, o tratamento foi temporariamente interrompido ou suspenso, conforme necessário, para o controle da toxicidade.
As características basais demográficas e relacionadas à doença dos pacientes foram semelhantes nos dois grupos de tratamento e refletiram uma população mais ampla de pacientes com mieloma múltiplo recém-diagnosticado (vide a tabela a seguir). Os pacientes com mieloma múltiplo recém-diagnosticado incluídos neste estudo incluíam os pacientes elegíveis e os não elegíveis para transplante autólogo de células-tronco imediato.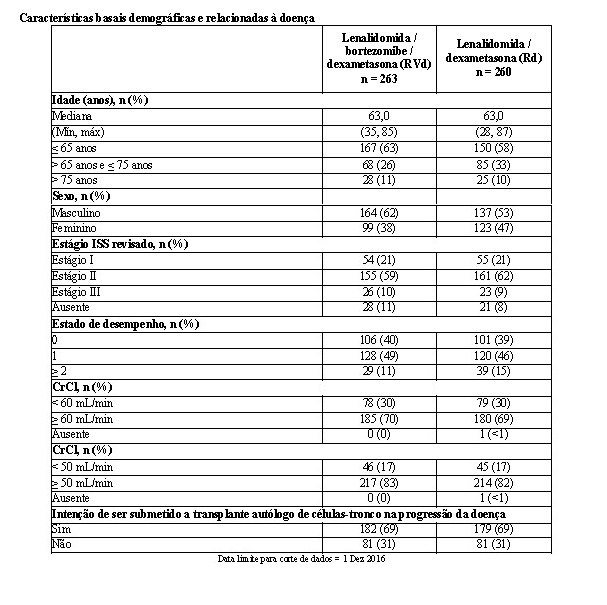
O desfecho primário do estudo foi definido pela SLP como o tempo da data de randomização à data de progressão da doença (incluindo deterioração sintomática), conforme determinado pelos critérios de resposta IMWG ou óbito (qualquer causa). A SLP foi avaliada retrospectivamente pelo IRAC, usando a população com intenção de tratamento. A sobrevida global (tempo da randomização ao óbito por qualquer causa) foi o desfecho de eficácia secundário.
Os resultados de eficácia estão resumidos na tabela a seguir. A SLP foi significativamente maior com RVd comparada a Rd (sem considerar as regras de classificação utilizadas). Uma porcentagem menor de indivíduos no grupo RVd comparada com o grupo Rd teve eventos de SLP (novamente, sem considerar as regras de classificação utilizadas). A melhora no tempo mediano de SLP no grupo RVd comparada com o grupo Rd foi de 12,6 meses.
A sobrevida global foi maior com RVd comparada a Rd: mediana (meses) 89,1 (IC de 95%; NE 76,1) e 67,2 (IC de 95%: 58,4, 90,8), respectivamente.
A taxa de resposta global (≥ RP) foi maior com RVd (tratamento inicial mais Rd) comparada com Rd (81% versus 73%) com uma resposta completa em 19% do grupo RVd versus 9% no grupo Rd.
A taxa de resposta global (≥ RPMB) foi maior com RVd (tratamento inicial mais Rd) comparada Rd (68% versus 50%).
O tempo mediano até, pelo menos, uma resposta parcial foi de 5,3 semanas no grupo RVd versus 5,1 semanas no grupo Rd. O tempo mediano até, pelo menos, uma resposta parcial muito boa foi de 8,2 semanas no grupo RVd versus 12,2 semanas no grupo Rd.
O benefício para RVd versus Rd foi observado sem considerar a elegibilidade para transplante de células-tronco.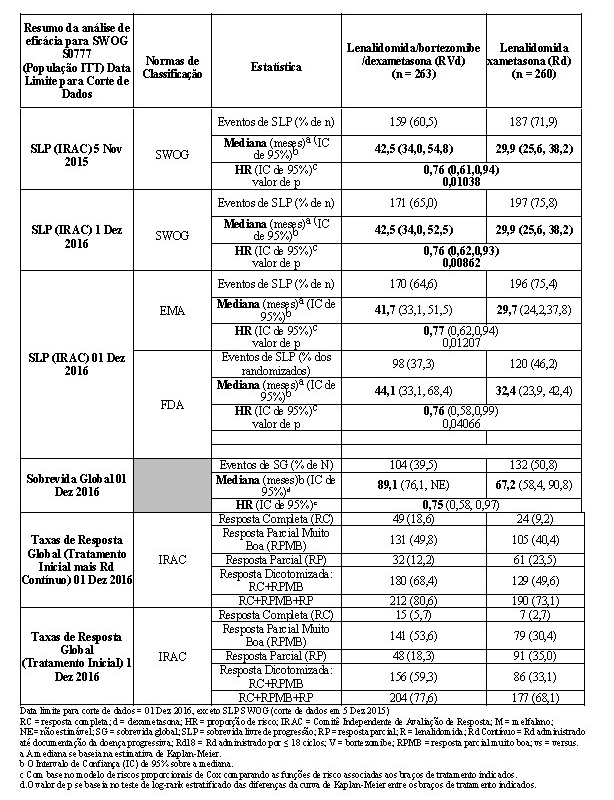
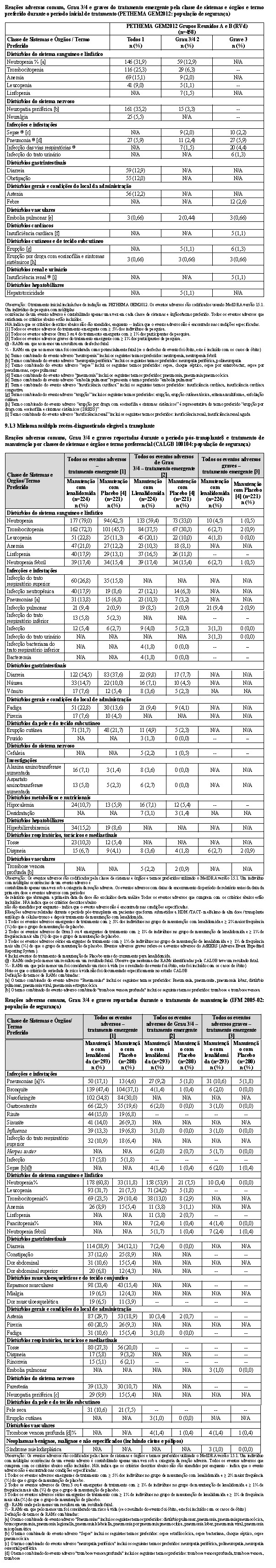
2.2.2 Estudos PETHEMA GEM2012 e IFM 2009
A eficácia e segurança de lenalidomida em combinação com bortezomibe e dexametasona (RVd) foram avaliadas em dois estudos multicêntricos Fase 3: PETHEMA GEM2012 e IFM 2009.
O estudo PETHEMA GEM2012 foi um estudo Fase 3, randomizado, controlado, aberto, multicêntrico que comparou 2 esquemas de condicionamento pré-transplante (bussulfano-melfalano e MEL200) em pacientes que receberam RVd (lenalidomida, bortezomibe e dexametasona) como terapia inicial. RVd foi administrado como seis ciclos de 4 semanas (24 semanas). Os pacientes receberam lenalidomida 25 mg/dia, via oral, nos Dias 1-21, bortezomibe 1,3 mg/m2, nos Dias 1, 4, 8 e 11 e dexametasona 40 mg/dia, via oral, nos Dias 1-4, 9-12 de ciclos repetidos de 28 dias. A seguir ao tratamento inicial, os pacientes receberam bussulfano-melfalano ou esquema de condicionamento MEL200 (randomização 1:1) e TACT. Os pacientes também receberam dois ciclos adicionais de 4 semanas de RVd em seguida a TACT. No total, 458 pacientes foram incluídos no estudo.
O estudo IFM 2009 foi um estudo Fase 3, randomizado, controlado, aberto, multicêntrico que comparou RVd com e sem transplante autólogo de células-tronco (TACT) como tratamento inicial para pacientes que apresentam mieloma múltiplo não tratado previamente, que são elegíveis para transplante. Os pacientes receberam lenalidomida 25 mg/dia, via oral, nos Dias 1-14, bortezomibe intravenoso 1,3 mg/m2, nos Dias 1, 4, 8 e 11 e dexametasona 20 mg/dia, via oral, nos Dias 1, 2, 4, 5, 8, 9,11 e 12 de ciclos repetidos de 21 dias. RVd foi administrado como oito ciclos de 3 semanas (24 semanas) sem TACT imediato (Braço A) ou três ciclos de 3 semanas (9 semanas) antes do TACT (Braço B). Os pacientes no Braço B também receberam e dois ciclos adicionais de 3 semanas de RVd em seguida a TACT. No total, 700 pacientes foram incluídos no estudo.
Um resumo das taxas de resposta do mieloma para os braços de tratamento utilizando até 24 semanas do tratamento inicial com RVd (isto é, seis ciclos de 28 dias ou oito ciclos de 21 dias) para os estudos PETHEMA GEM2012 e IFM 2009, usando um corte de dados nas datas 31 de março de 2017 e 01 de dezembro de 2016, respectivamente, é apresentado na tabela a seguir.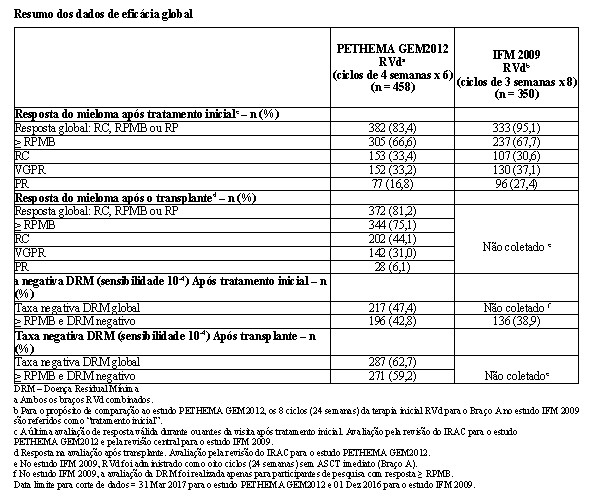
No estudo PETHEMA GEM2012, no final do tratamento inicial com RVd, 66,6% (305/458) dos pacientes apresentavam ≥ RPMB, dos quais 38,7% (118/305) eram DRM negativa (sensibilidade 10-6). Após transplante, 75,1% (344/458) dos pacientes apresentavam ≥ RPMB, dos quais 54,4% (187/344) eram DRM negativa (sensibilidade 10-6).
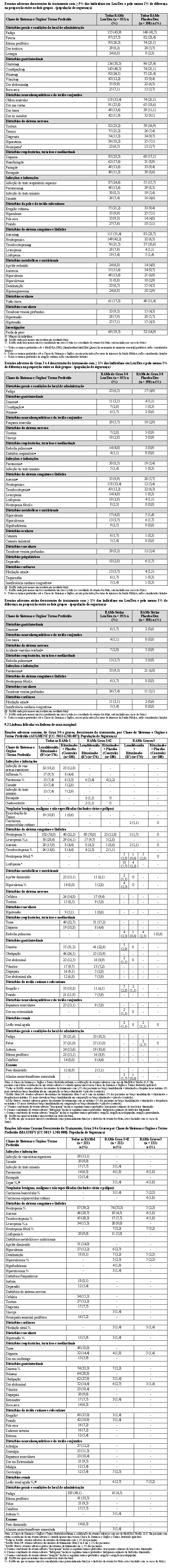
2.2.3 Análise integrada para terapia inicial do mieloma múltiplo recém-diagnosticado elegível a transplante
Uma análise integrada foi conduzida para comparar RVd (lenalidomida, bortezomibe e dexametasona) versus VTD (bortezomibe, talidomida e dexametasona) como terapia inicial antes de uma terapia de alta dose/auto transplante de células-tronco hematopoéticas em pacientes elegíveis a transplante com mieloma múltiplo recém-diagnosticado, baseada em dados individuais do paciente, usando análise estatística baseada no método de escores de propensão. Esta metodologia pode minimizar os efeitos dos fatores de confusão basais observados e melhorar a comparabilidade das populações de pacientes entre coortes de tratamento, conduzindo à conclusões confiáveis para comparações transversais do estudo.
Dois estudos, PETHEMA GEM2012 (RVd) e PETHEMA GEM2005 (VTD), forneceram dados para a comparação principal para as análises integradas. Dados de suporte também são fornecidos de 2 estudos adicionais, IFM 2009 (RVd) e IFM 2013-04 (VTD). Os resultados para os estudos PETHEMA são apresentados na tabela a seguir.
O desfecho primário para a análise integrada foi a taxa de resposta após tratamento inicial de ≥ RPMB (conforme avaliado pelo IRAC e baseado nos critérios IMWG) para RVd versus VTD nos dois estudos PETHEMA.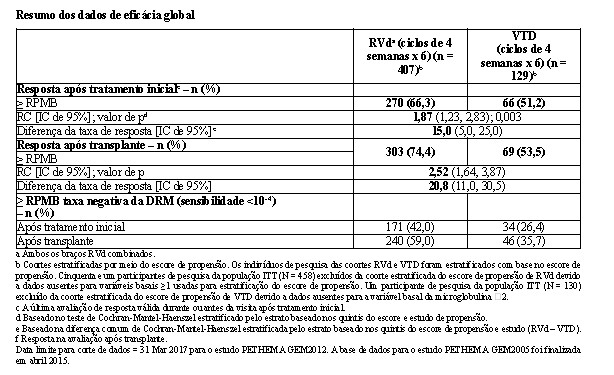
Os resultados dos estudos comparativos IFM (IFM 2009 [RVd] e IFM 2013-04 [VTD]), baseados na revisão central, são compatíveis com a comparação correspondente entre 2 estudos PETHEMA e apoiam os benefícios de RVd versus VTD como o tratamento inicial na população com mieloma múltiplo recém-diagnosticado elegível a transplante. Os resultados são mostrados na tabela a seguir.
Também, a taxa de resposta com RVd aumentou com o tempo. Entre os 300 pacientes na coorte RVd (Braço A IFM 2009) que iniciaram o Ciclo 8, a porcentagem de pacientes que alcançou a resposta ≥ RPMB aumentou de 54,3% com 3 ciclos de tratamento inicial para 60,7% com 4 ciclos, e para 72,7% com 8 ciclos.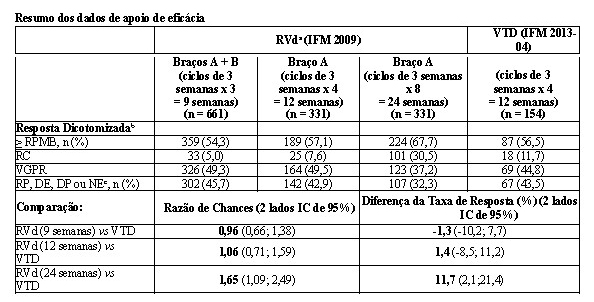
2.3 Tratamento de manutenção do mieloma múltiplo recém-diagnosticado após transplante autólogo de células-tronco
Dois estudos foram realizados como suporte para a eficácia e segurança do tratamento de manutenção com lenalidomida no tratamento de pacientes com mieloma múltiplo recém-diagnosticado após transplante autólogo de células-tronco.
2.3.1 Estudo CALGB100104
CALGB100104 foi um estudo de Fase 3 multicêntrico, randomizado, duplo cego e controlado com placebo para avaliar a eficácia e segurança do tratamento de manutenção com lenalidomida ou placebo após transplante autólogo de células-tronco (TACT) para pacientes que apresentam mieloma múltiplo recém-diagnosticado. O objetivo primário foi determinar se o tratamento de manutenção com lenalidomida prolongaria o tempo até a progressão (TTP); a definição de TTP no protocolo era equivalente a uma definição geral de sobrevida livre de progressão (SLP).
Os pacientes com idades entre 18 e 70 anos com mieloma múltiplo ativo requerendo tratamento e doença estável ou receptividade há pelo menos 2 meses de qualquer tratamento de indução, e sem progressão prévia após o tratamento inicial, eram elegíveis. O tratamento de indução deve ter ocorrido dentro de 12 meses.
A dose de lenalidomida foi 10 mg uma vez ao dia (aumentada para 15 mg uma vez ao dia após 3 meses, para os pacientes que toleraram o tratamento de manutenção). Um aumento de dose para 15 mg ocorreu em 135 pacientes (58%). O tratamento foi continuado até a progressão da doença ou a retirada do paciente por outro motivo. O tratamento foi interrompido, pausado ou a dose foi reduzida conforme necessário para controlar a toxicidade.
O desfecho primário do estudo foi a SLP (definida desde a randomização até a data de progressão ou óbito, o que ocorrer primeiro) conforme analisado pelos investigadores e avaliado de acordo com os critérios do Grupo Internacional de Trabalho de Mieloma - IMWG (Durie, 2006).
As características demográficas e da doença basal para a população com intenção de tratamento são resumidas nas tabelas a seguir. As características demográficas e da doença refletiram uma população típica de pacientes elegíveis a transplante que apresentam mieloma múltiplo recém-diagnosticado.
Houve um desequilíbrio (p < 0,1) entre os grupos de tratamento na distribuição das categorias do estágio do ISS favorecendo o grupo do placebo e a proporção de indivíduos de pesquisa com resposta completa (RC) / resposta parcial muito boa (RPMB) pós-TACT foi significativamente menor (p < 0,1) para os participantes de pesquisa randomizados para o braço de manutenção de lenalidomida, em comparação com os participantes de pesquisa randomizados para o braço de placebo. Nos demais aspectos, não foram observadas diferenças clinicamente significativas nas características demográficas, da doença no diagnóstico ou da terapia prévia.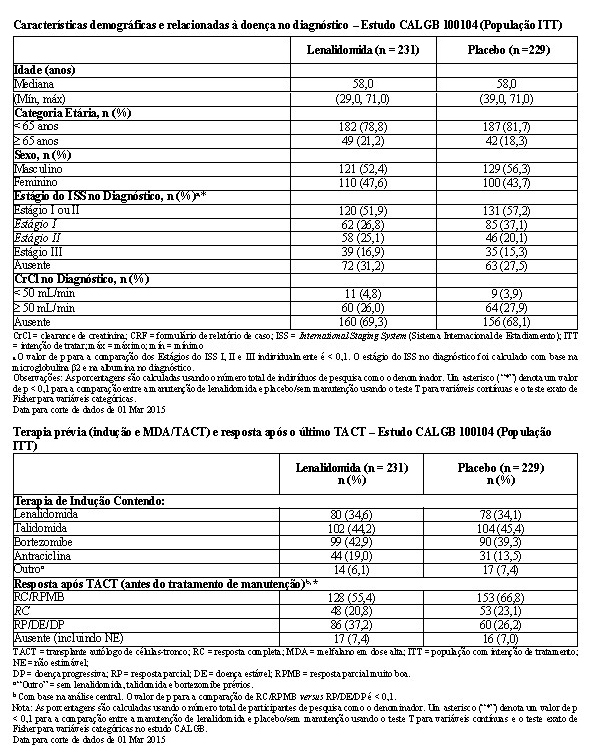
O acompanhamento mediano dos pacientes sobreviventes na data para corte de dados de 01 Mar 2015 foi 72,4 meses. Usando as normas de classificação da EMA, houve uma redução de 42% no risco de progressão da doença ou óbito, favorecendo lenalidomida (HR = 0,58; IC de 95%, 0,46 a 0,73). O tempo de SLP foi 58,4 meses no grupo de lenalidomida versus uma estimativa de 28,9 meses no braço de placebo.
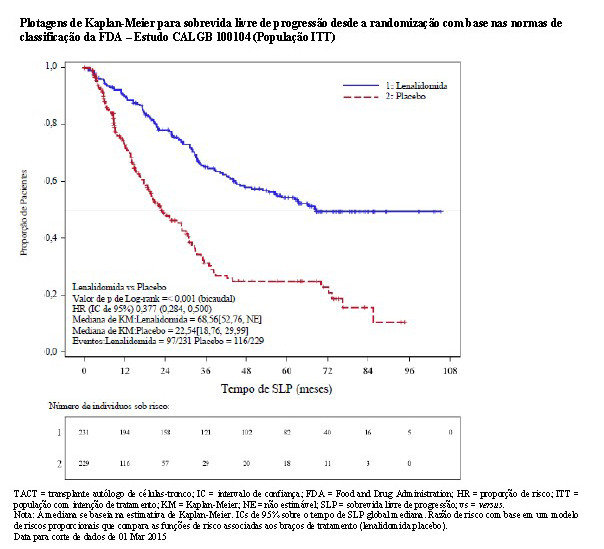
Usando as normas de classificação do FDA, houve uma redução de 62% no risco de progressão da doença ou óbito, favorecendo lenalidomida (HR = 0,38; IC de 95%, 0,28 a 0,50). Diferente da análise com uso das normas de classificação do EMA, as análises com as do FDA também censuraram 76 pacientes no braço de placebo no momento do cruzamento para lenalidomida (antes da PD). O tempo de SLP foi 68,6 meses no grupo de lenalidomida versus uma estimativa de 22,5 meses no braço de placebo. O estudo foi sem mascaramento devido ao cruzamento de um limite pré-especificado para SLP
em uma análise parcial planejada. Os pacientes que receberam placebo foram autorizados a fazer o cruzamento para a terapia com lenalidomida antes da doença progressiva (76 pacientes o fizeram).
A SLP na próxima linha de terapia (SLP2) calculada como o tempo desde a randomização até o início da 3ª linha de tratamento ou óbito para todos os pacientes randomizados, também foi significativamente melhorada no braço de lenalidomida versus placebo (HR = 0,64 [IC de 0,50-0,82]) resultando em uma mediana de 78,3 meses para lenalidomida versus 52,8 meses para placebo.
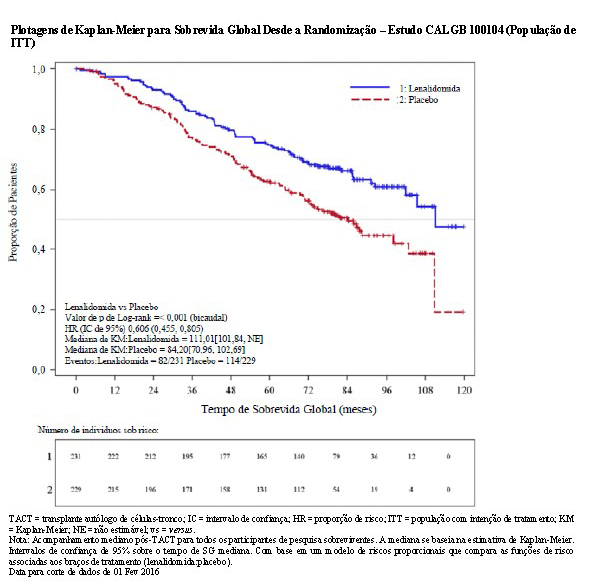
Para a análise da sobrevida global (SG), a HR observada foi 0,57 (IC de 95%, 0,42 a 0,76) para lenalidomida versus placebo, indicando uma redução de 43% no risco de óbito. O tempo de sobrevida global mediana não foi alcançado no grupo de lenalidomida versus uma estimativa de Kaplan-Meier (KM) de 73,0 meses no braço de placebo.
O benefício da manutenção de lenalidomida na SLP e na SG foi observado em todos os subgrupos examinados. Esses resultados, bem como os resultados de eficácia atualizados desde a data para corte de dados de 1 de fevereiro de 2016, estão resumidos na tabela a seguir.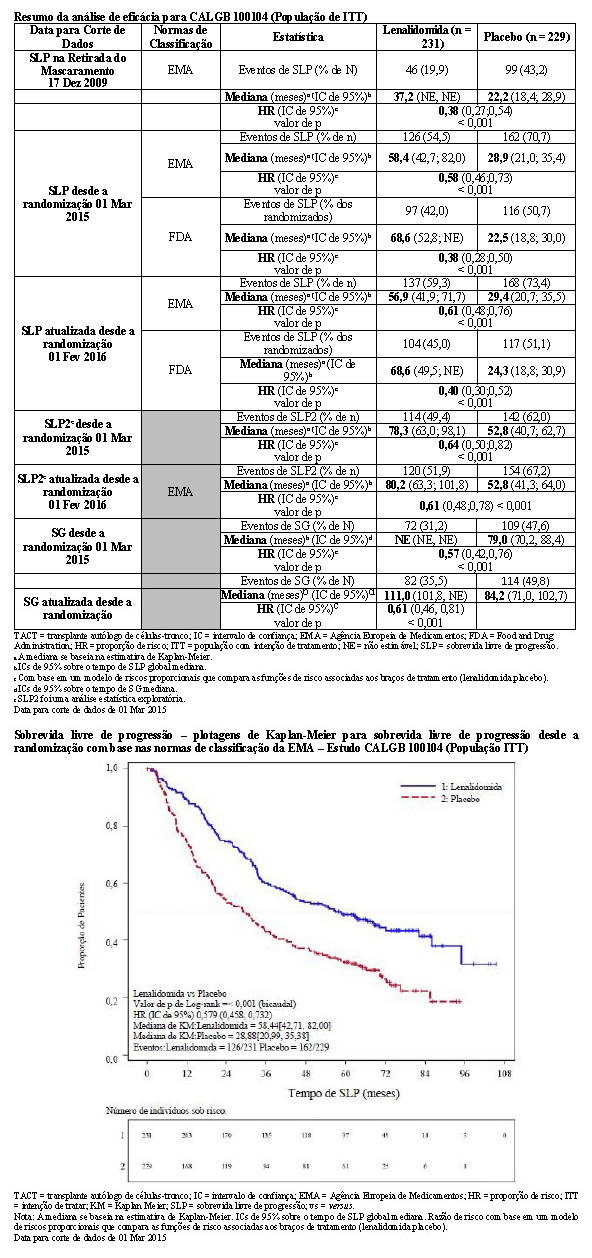
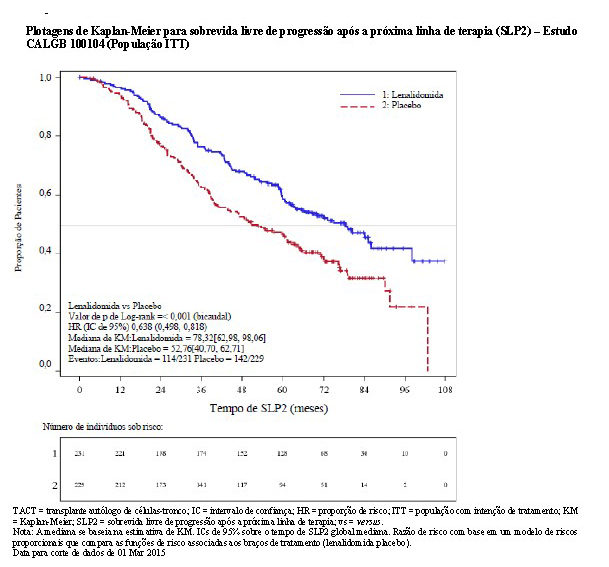
2.1.3 Estudo IFM 2005-02
O Estudo IFM 2005-02 foi um estudo de Fase 3, multicêntrico, randomizado, duplo cego, controlado com placebo para investigar a eficácia e a segurança do tratamento de manutenção com lenalidomida depois que os pacientes foram submetidos ao TACT. O objetivo primário do estudo foi avaliar a eficácia do tratamento de manutenção com lenalidomida após o TACT na extensão da SLP pós-transplante.
Os pacientes tinham idade < 65 anos no diagnóstico e foram submetidos ao tratamento com quimioterapia de alta dose apoiada pelo TACT e alcançaram pelo menos uma resposta de doença estável no momento da recuperação hematológica.
Dentro de 6 meses após o TACT, os pacientes foram randomizados para receber o tratamento de manutenção com lenalidomida ou placebo. Após 2 ciclos de consolidação de lenalidomida (25 mg/dia, Dias 1-21 de um ciclo de 28 dias), a dose de manutenção de lenalidomida foi 10 mg uma vez ao dia (aumentada para 15 mg uma vez ao dia após 3 meses para pacientes que toleraram a terapia). Um aumento de dose para 15 mg ocorreu em 185 pacientes (60%). O tratamento foi continuado até a progressão da doença ou a retirada do paciente por outro motivo. O tratamento foi interrompido, pausado ou a dose foi reduzida conforme necessário para controlar a toxicidade.
O desfecho primário foi a SLP (definida desde a randomização até a data de progressão ou óbito, o que ocorreu primeiro) conforme analisado pelos investigadores e avaliado de acordo com os critérios de IMWG (Durie, 2006). O estudo foi sem mascaramento devido ao cruzamento de um limite pré-especificado para SLP em uma análise parcial planejada. Os pacientes que receberam placebo não fizeram o cruzamento para a terapia com lenalidomida antes da doença progressiva. O grupo de lenalidomida foi descontinuado, como medida proativa de segurança, após observar um desequilíbrio nas segundas malignidades primárias.
As características demográficas e da doença basal para a população ITT são resumidas nas tabelas a seguir. Essas refletiram uma população típica de pacientes elegíveis para transplante que apresentam mieloma múltiplo recém diagnosticado. Foram observadas algumas diferenças nas características demográficas e relacionadas à doença. No diagnóstico, houve um desequilíbrio entre os braços de tratamento na distribuição das categorias de estágio do ISS, citogenética de risco adverso, definida como translocação envolvendo os cromossomos 4 e 14 (t[4;14]) ou deleção no cromossomo 17p (del 17p) e participantes de pesquisa apresentando uma depuração de creatinina reduzida ( < 50 mL/min). Esses desequilíbrios podem ter influenciado positivamente o desfecho de longo prazo do grupo placebo. Nos demais aspectos, os grupos de lenalidomida e placebo foram geralmente comparáveis em relação às características demográficas ou da doença no diagnóstico e na terapia prévia.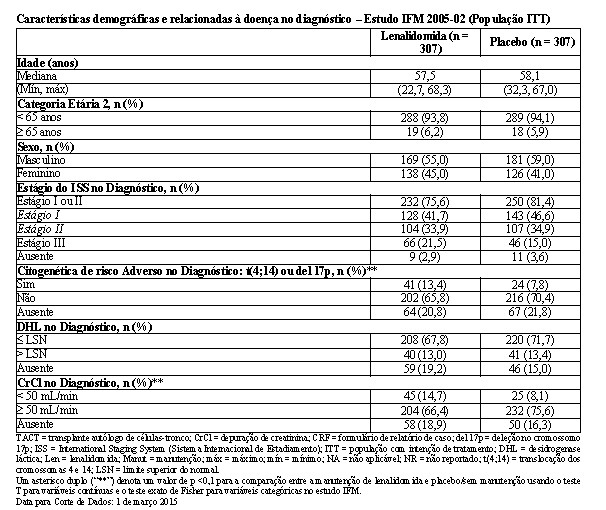
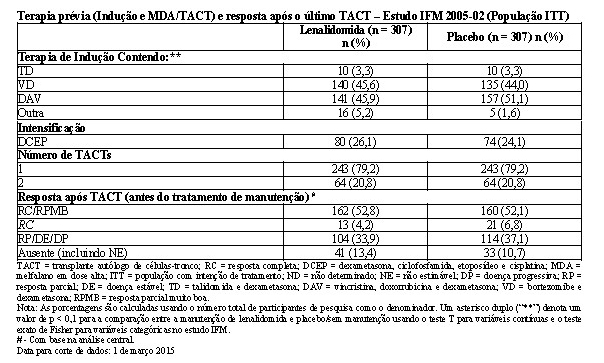
O acompanhamento mediano dos indivíduos de pesquisa sobreviventes na presente data limite para corte de dados de 01 Mar 2015 foi 86,0 meses. Usando as normas de classificação da EMA, houve uma redução de 45% no risco de progressão da doença ou óbito, favorecendo lenalidomida (HR = 0,55; IC de 95%, 0,46 a 0,66). O tempo de sobrevida livre de progressão foi 44,4 meses no grupo de lenalidomida versus 23,8 meses no braço de placebo.
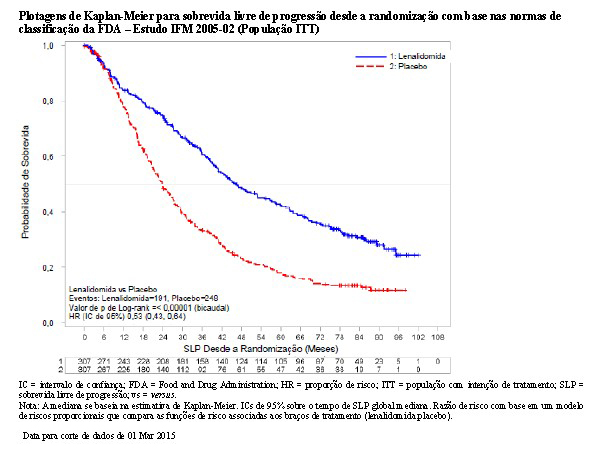
Usando as normas de classificação da FDA, houve uma redução de 47% no risco de progressão da doença ou óbito, favorecendo lenalidomida (HR = 0,53; IC de 95%, 0,43 a 0,64). O tempo de sobrevida livre de progressão foi 46,3 meses no grupo de lenalidomida versus uma estimativa de 23,8 meses no grupo de placebo.
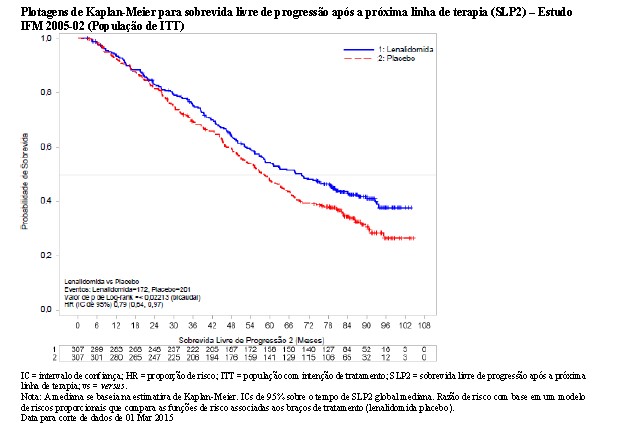
O benefício da manutenção de lenalidomida na SLP foi observado em todos os subgrupos examinados. A SLP na próxima linha de terapia (SLP2) calculada como o tempo desde a randomização até progressão na 2ª linha de terapia ou óbito para todos os pacientes randomizados, também foi significativamente melhorada no grupo de lenalidomida versus placebo (HR = 0,79 [C1 de 0,64-0,99]) traduzindo-se em uma mediana de 70,0 meses para lenalidomida versus 58,4 meses para placebo.
Para a análise da SG, a HR observada foi 0,91 (IC de 95%, 0,72 a 1,15) para lenalidomida versus placebo; mais pacientes no grupo de placebo (46,9%) foram a óbito, quando comparados ao grupo de lenalidomida (41,7%). O tempo de sobrevida global mediana não foi alcançado no grupo de lenalidomida versus uma estimativa de KM de 90,9 meses no grupo de placebo. Esses resultados, bem como os resultados de eficácia atualizados desde a data limite para corte de dados de 1 de fevereiro de 2016, estão resumidos na tabela a seguir.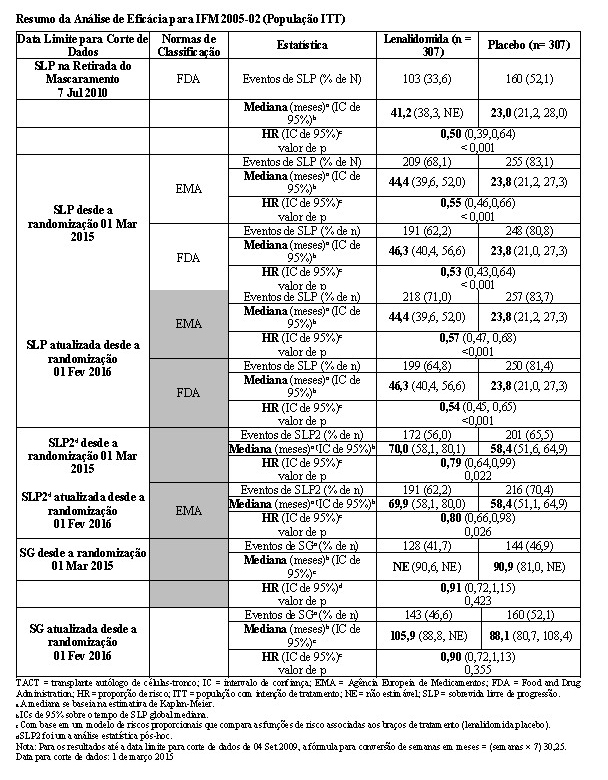
O tratamento foi interrompido para os demais 119 participantes de pesquisa que receberam manutenção de lenalidomida (duração mínima do tratamento de 27 meses) devido a um desequilíbrio observado das segundas malignidades primárias.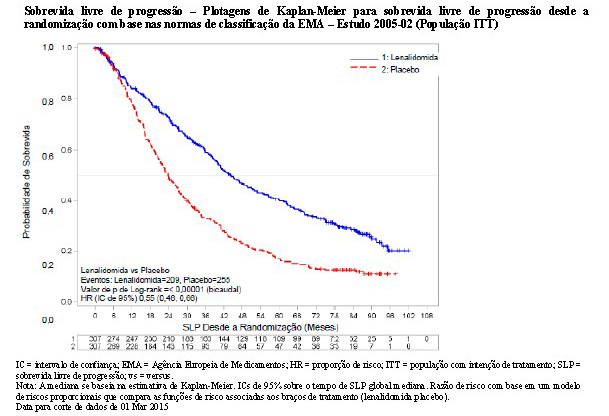
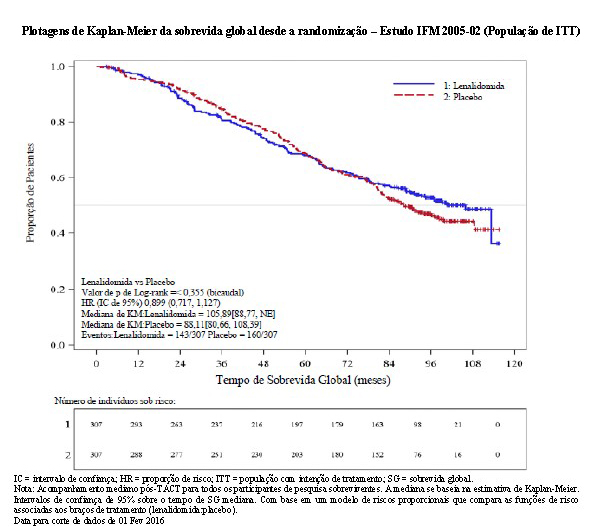
2.3.3 Metanálise de eficácia dos estudos controlados randomizados de sobrevida global no mieloma múltiplo recém-diagnosticado em pacientes após transplante autólogo de células-tronco
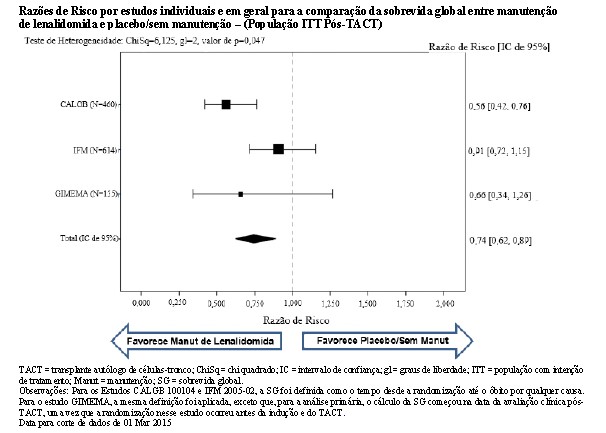
Foi realizada uma metanálise de estudos controlados randomizados que estudaram a eficácia do tratamento de manutenção de lenalidomida, após melfalano de dose alta (MDA) e o transplante autólogo de células-tronco (TACT). O objetivo da metanálise foi comparar a eficácia, conforme medida pela sobrevida global (SG), para os pacientes que apresentam mieloma múltiplo recém-diagnosticado tratados com tratamento de manutenção de lenalidomida versus placebo/sem manutenção pós-MDA/TACT. Com base em critérios predefinidos, três estudos foram elegíveis para inclusão: IFM-2005-02, CALGB 100104 e GIMEMA. Os três estudos foram semelhantes em relação às populações de pacientes, abordagens de tratamento e desenhos gerais do estudo, incluindo o desfecho primário da sobrevida livre de progressão (SLP). Nenhum dos 3 estudos foi potencializado para SG.
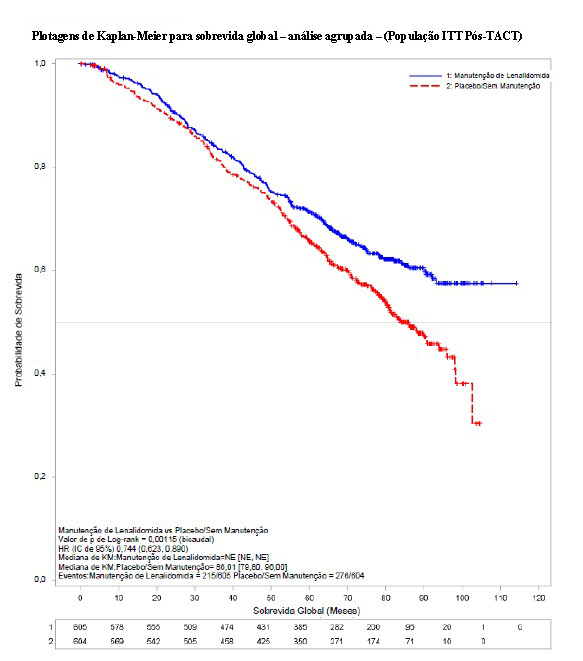
A metanálise foi realizada a partir dos dados demográficos e de sobrevida no nível do paciente em todos os 3 estudos clínicos. Um total de 1209 pacientes foram incluídos na população ITT pós-TACT: 460 pacientes do CALGB 100104, 614 pacientes do IFM 2005-02 e 135 pacientes do GIMEMA.
As características demográficas e da doença basal para a população de ITT pós-TACT são resumidas nas tabelas a seguir. As características demográficas e da doença refletiram uma população típica de pacientes elegíveis para transplante que apresentam mieloma múltiplo recém-diagnosticado. Algumas características relacionadas ao desfecho de risco desfavorável pareceram favorecer o Agrupamento de Placebo/Sem Manutenção para os participantes de pesquisa com dados não ausentes (todos p < 0,1): estágio do ISS, citogenética de risco adverso e função renal (CrCl). Nos demais aspectos, não foram observadas diferenças clinicamente significativas nas características demográficas, da doença ou da terapia prévia.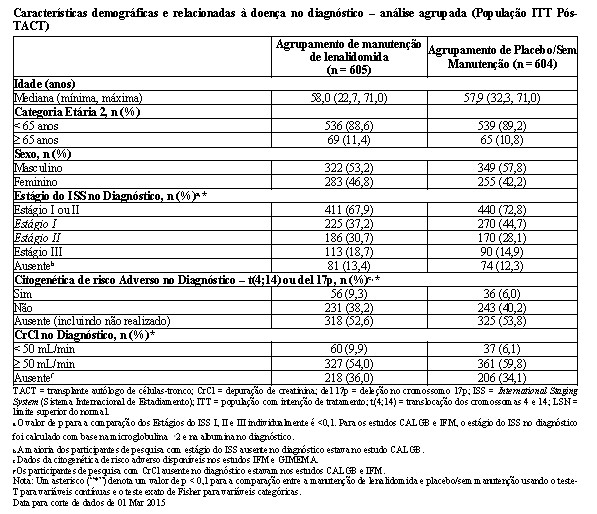
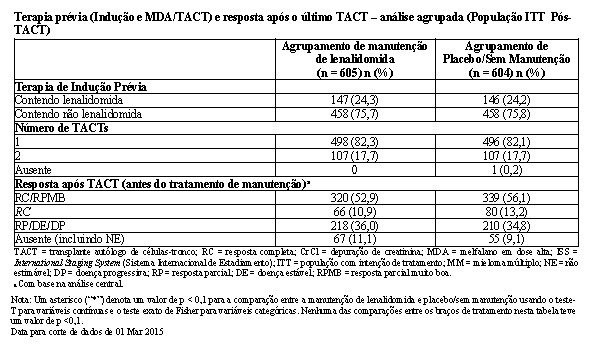
O acompanhamento mediano de todos os pacientes sobreviventes foi 6,6 anos (79,9 meses para a coorte agrupada). Houve 491 óbitos reportados para a população de ITT pós-TACT: 35,5% (215/605) dos pacientes no grupo de lenalidomida e 45,7% (276/604) dos pacientes no grupo de placebo. Para a análise da SG, a HR observada foi 0,744 para lenalidomida versus placebo (IC de 95% = 0,623, 0,890, p = 0,001), indicando uma redução de 26% no risco de óbito. A SG mediana não foi alcançada no agrupamento de manutenção de lenalidomida e foi estimada em 86,0 meses (IC de 95% = 79,8 a 96,0 meses) no agrupamento de placebo/sem manutenção. A SG mediana após a manutenção de lenalidomida pode alcançar aproximadamente 116 meses (conforme calculado a partir da SG mediana do agrupamento de placebo/sem manutenção dividido pela HR de 0,744). Isso representaria uma melhoria aproximada de 2,5 anos na SG mediana, em comparação com a ausência de tratamento de manutenção.
Todos os 3 estudos contribuíram favoravelmente para os resultados da análise agrupada, conforme demonstrado pelas HRs dos estudos individuais. Embora os resultados tenham variado entre os estudos, cada estudo favoreceu o tratamento de manutenção de lenalidomida. O benefício do tratamento de manutenção de lenalidomida na SG geralmente foi compatível nos subgrupos, incluindo os que alcançaram uma RC ou haviam alcançado uma RC ou RPMB após o TACT.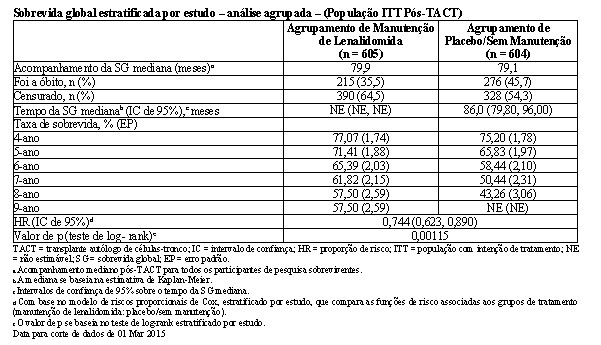
Para 4 subgrupos com valor prognóstico fortemente negativo (Estágio III do ISS, citogenética de risco adverso, CrCl < 50 mL/min e DHL > LSN - todos no diagnóstico), o efeito do tratamento parece menos favorável (com a estimativa pontual da HR ligeiramente > 1,0), embora o número de participantes de pesquisa nesses subgrupos seja pequeno e haja uma quantidade considerável de dados ausentes para essas variáveis de subgrupos. Os respectivos ICs de 95% da HR, no entanto, se sobrepõem aos do respectivo subgrupo com o valor prognóstico mais positivo (Estágios I e II do ISS, citogenética de risco-não adverso, CrCl ≥ 50 mL/min e DHL normal).
Os resultados da análise do subgrupo devem ser interpretados com cautela porque a análise não está sendo pré-especificada e devido à multiplicidade e à disponibilidade limitada dos dados, particularmente dos subgrupos de CrCl no diagnóstico, citogenética no diagnóstico e DHL no diagnóstico, que representam 3 dos subgrupos menores.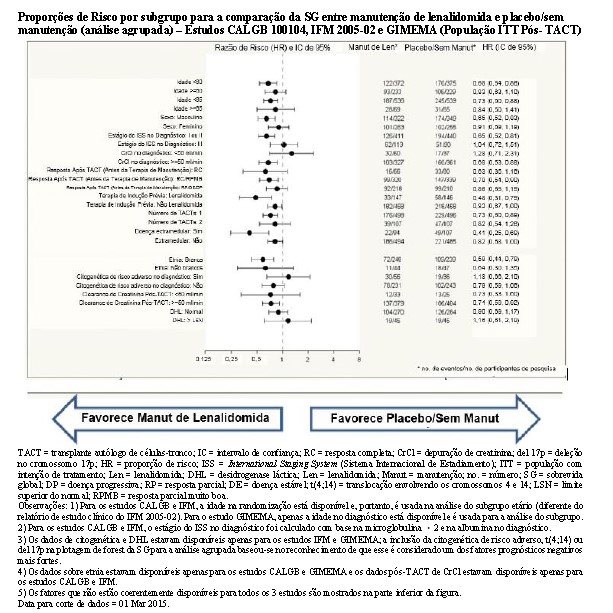
2.4 Mieloma múltiplo refratário/recidivado
Dois estudos randomizados (estudos MM-009 e MM-010) foram conduzidos para avaliar a eficácia e a segurança de lenalidomida. Estes estudos multicêntricos, multinacionais, duplo-cegos, controlados por placebo, compararam lenalidomida em combinação com altas doses de dexametasona à terapia com dexametasona isolada, em pacientes com mieloma múltiplo que receberam ao menos um esquema prévio de tratamento. Estes estudos incluíram pacientes com ANC ≥ 1000/mm3, contagens de plaquetas ≥ 75.000/mm3, creatinina sérica ≤ 2,5 mg/dL, TGO/AST ou TGP/ALT séricas ≤ 3,0 x LSN, e bilirrubina direta sérica ≤ 2,0 mg/dL.
Em ambos os estudos, os pacientes no grupo lenalidomida/dexametasona receberam 25 mg de lenalidomida via oral uma vez ao dia nos Dias 1 a 21 e uma cápsula de placebo equivalente uma vez ao dia nos Dias 22 a 28 de cada ciclo de 28 dias. Os pacientes no grupo placebo/dexametasona receberam 1 cápsula de placebo nos Dias 1 a 28 de cada ciclo de 28 dias. Os pacientes em ambos os grupos de tratamento receberam 40 mg de dexametasona via oral uma vez ao dia nos Dias 1 a 4, 9 a 12, e 17 a 20 de cada ciclo de 28 dias pelos primeiros 4 ciclos de terapia. A dose de dexametasona foi reduzida para 40 mg via oral uma vez ao dia nos Dias 1 a 4 de cada ciclo de 28 dias após os primeiros 4 ciclos de terapia. Em ambos os estudos, o tratamento deveria continuar até a progressão da doença.
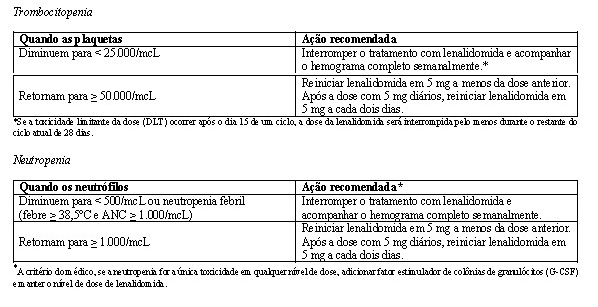
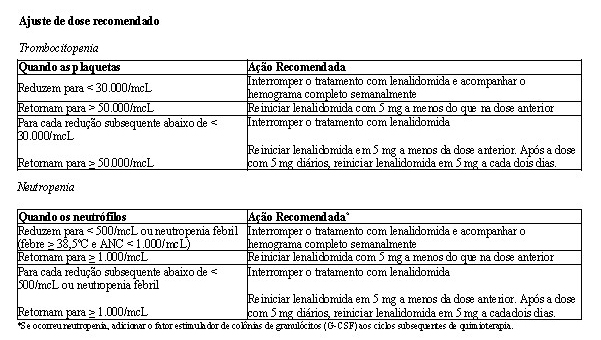
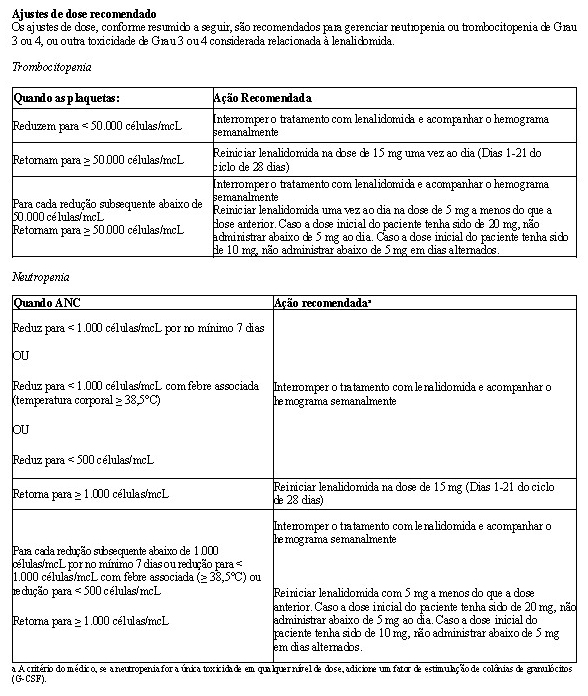
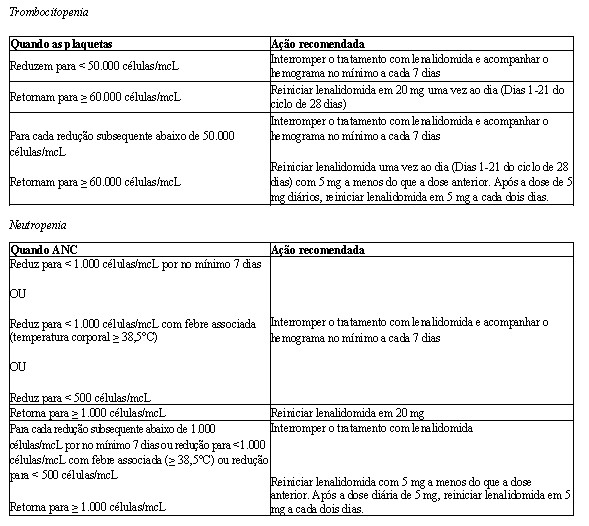
Em ambos os estudos, ajustes de dose foram permitidos com base em achados clínicos e laboratoriais. As reduções sequenciais de dose para 15 mg diariamente, 10 mg diariamente e 5 mg diariamente foram permitidas em decorrência da toxicidade (vide item Mieloma múltiplo).
A tabela abaixo resume as características basais do paciente e da doença nos dois estudos. Em ambos os estudos, as características basais demográficas e relacionadas à doença foram comparáveis entre os grupos lenalidomida/dexametasona e placebo/dexametasona.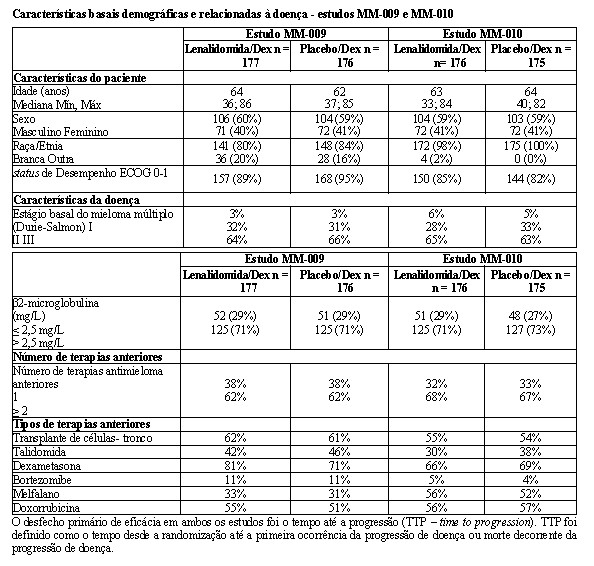
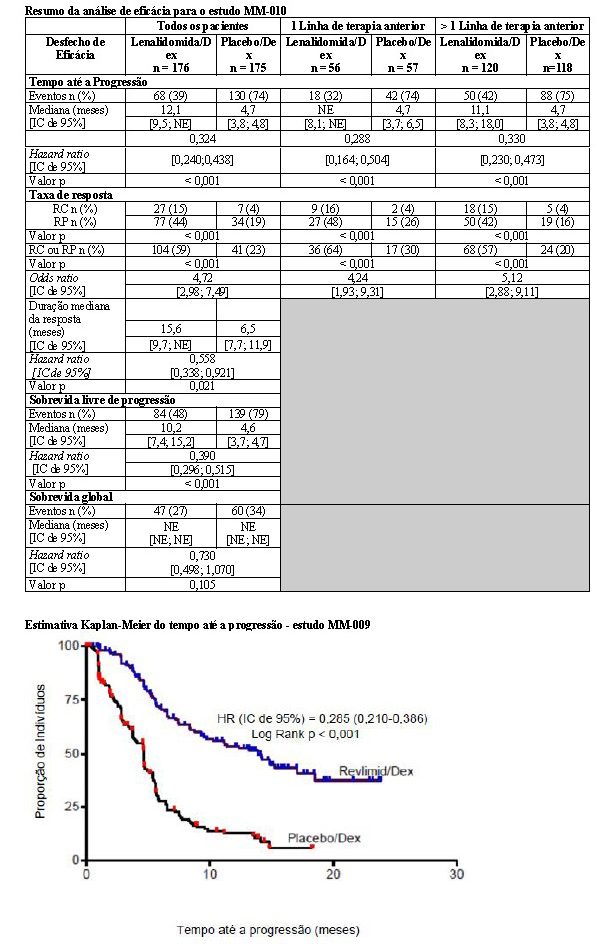
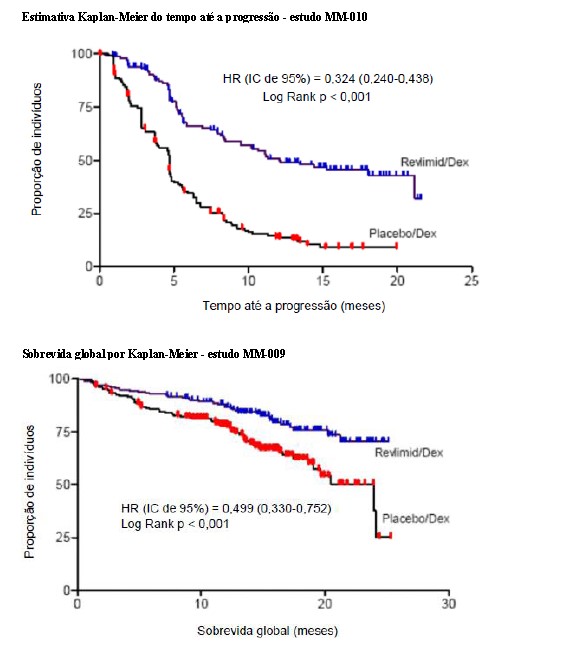
As análises interinas pré-planejadas de ambos os estudos mostraram que a combinação de lenalidomida/dexametasona foi significativamente superior à dexametasona isolada para TTP. Os estudos tiveram o caráter cego quebrado para permitir que os pacientes no grupo placebo/dexametasona recebessem tratamento com a combinação lenalidomida/dexametasona.
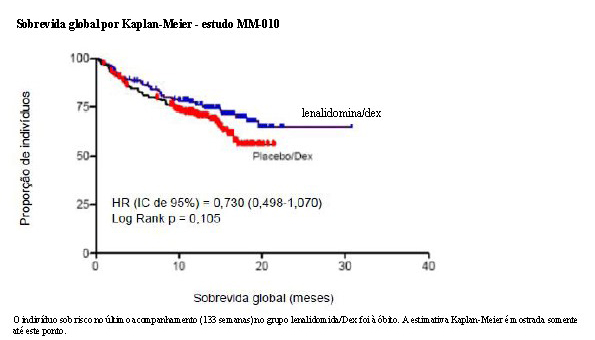
Para ambos os estudos, foram analisados os dados cruzados de sobrevida de acompanhamento prolongado. No estudo MM-009, o tempo mediano de sobrevida foi 39,4 meses (IC 95%: 32,9, 47,4) no grupo lenalidomida/dexametasona e 31,6 meses (IC 95%: 24,1, 40,9) no grupo placebo/dexametasona, com uma taxa de risco de 0,79 (IC 95%: 0,61-1,03). No estudo MM-010, o tempo mediano de sobrevida foi 37,5 meses (IC 95%: 29,9, 46,6) no grupo lenalidomida/dexametasona e 30,8 meses (IC 95%: 23,5, 40,3) no grupo placebo/dexametasona, com uma taxa de risco de 0,86 (IC 95%: 0,65-1,14).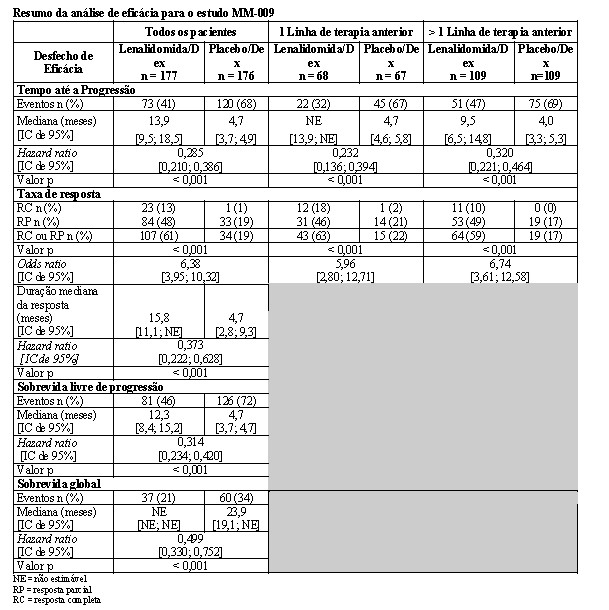
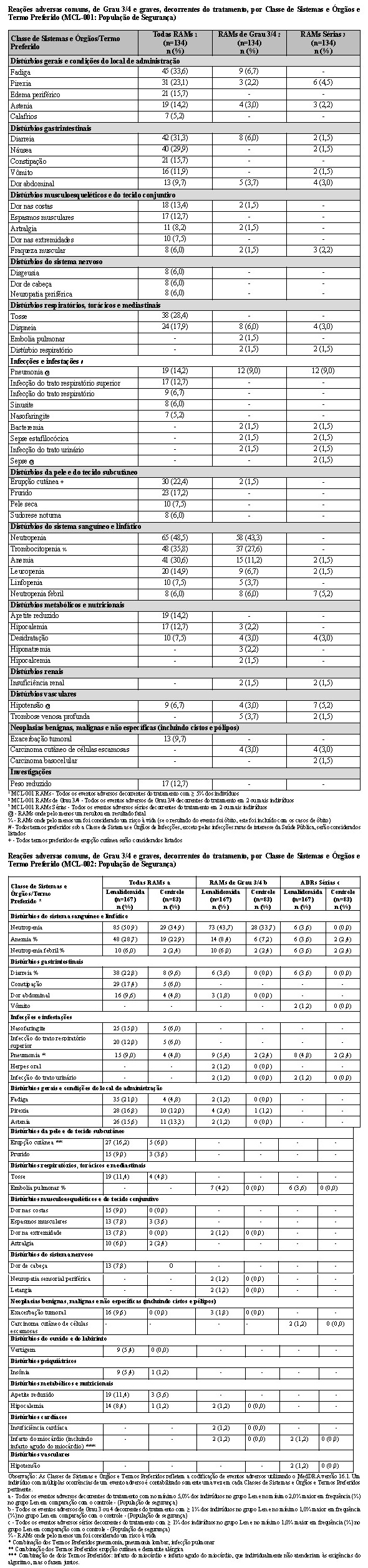
5 Linfoma folicular (LF) e linfoma de zona marginal (LZM)
Dois estudos patrocinados pela Celgene foram conduzidos para avaliar a eficácia e a segurança de lenalidomida em combinação com rituximabe (regime R2) para o tratamento de pacientes com linfoma folicular ou linfoma de zona marginal previamente tratados.
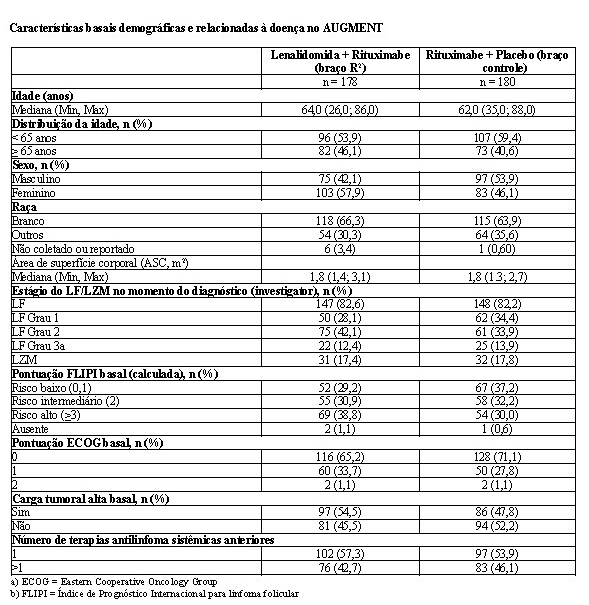
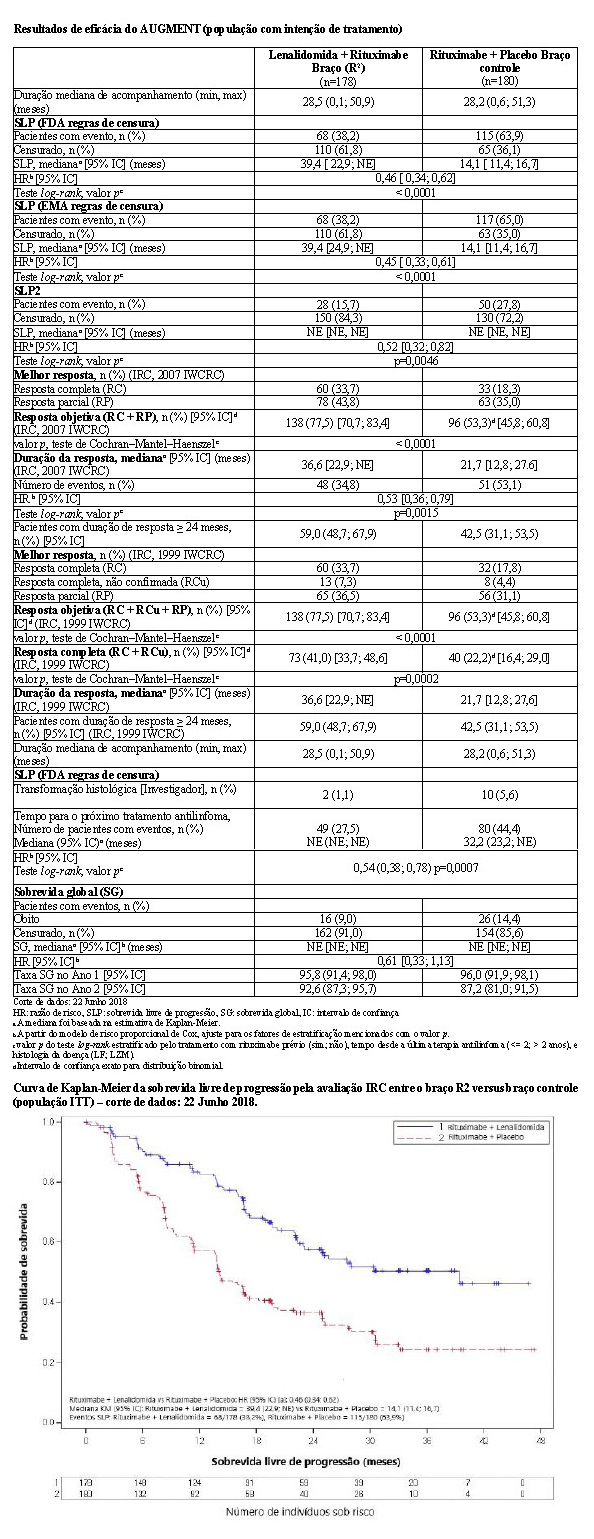
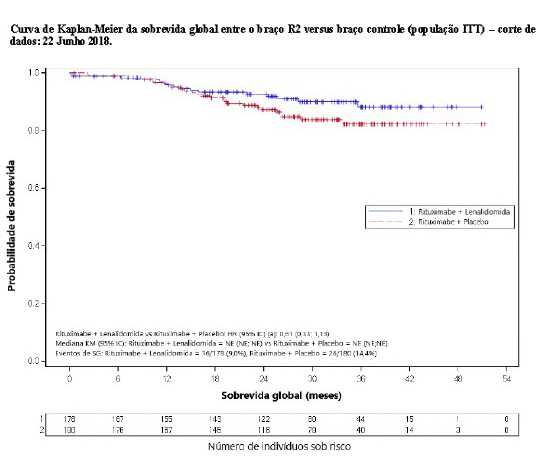
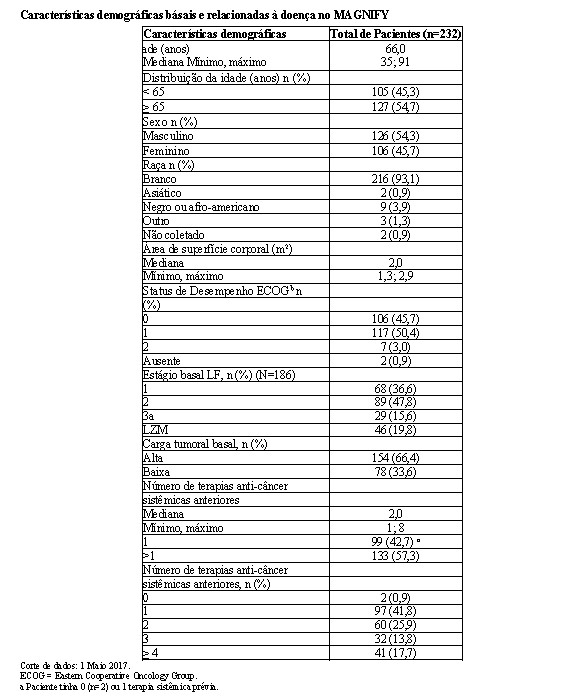
2.5.1 Estudo CC-5013-NHL-007 (AUGMENT)
O estudo CC-5013-NHL-007 (AUGMENT, NCT01938001) foi um estudo de Fase 3 controlado, multicêntrico, rand