NUCALA
GLAXOSMITHKLINE
mepolizumabe
Antiasmático.
Apresentações.
Pó liofilizado para solução injetável. Nucala® é apresentado em embalagem com 1 frasco-ampola contendo 100 mg de mepolizumabe (100 mg/mL após a reconstituição).
USO SUBCUTÂNEO.
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS DE IDADE.
Composição.
Cada frasco-ampola contém: mepolizumabe 100 mg (100 mg/mL após a reconstituição) excipientes* q.s.p para 1 frasco-ampola
*Excipientes: sacarose, fosfato de sódio dibásico heptaidratado, polissorbato 80 e ácido clorídrico.
Informações técnicas.
1. INDICAÇÕES
Asma eosinofílica grave Nucala® (mepolizumabe) é indicado como tratamento complementar de manutenção da asma eosinofílica grave em pacientes adultos e pediátricos a partir de 6 anos de idade.
Granulomatose Eosinofílica com Poliangeíte (GEPA)
Nucala® é indicado como tratamento complementar aos corticosteroides em pacientes adultos com granulomatose eosinofílica com poliangeíte (GEPA) recidivante ou refratária.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
Asma Grave
A eficácia de mepolizumabe no tratamento de um grupo-alvo de indivíduos com asma eosinofílica grave foi avaliada em 3 estudos clínicos randomizados, duplo-cegos e em grupos paralelos, com duração de 24 a 52 semanas em pacientes a partir de 12 anos de idade. Esses estudos foram delineados para avaliar a eficácia da administração de mepolizumabe 1 vez a cada 4 semanas, por injeção subcutânea ou intravenosa, em pacientes com asma eosinofílica grave não controlada com os tratamentos padrões [p. ex. corticosteroides inalatórios (CI), corticosteroides orais (CO), combinação de CI com agonistas beta-2 adrenérgicos de ação prolongada (LABA), modificadores de leucotrienos, agonistas beta-2 adrenérgicos de curta ação (SABA)].
A segurança e eficácia em pacientes pediátricos com idade inferior a 6 anos não foram estabelecidas. Um total de 28 adolescentes entre 12 e 17 anos com asma foram inscritos nos estudos de fase 3. Desses, 25 foram inscritos num estudo de exacerbação de 32 semanas (Estudo 2) e tinham uma idade média de 14,8 anos. Os indivíduos tinham um histórico de 2 ou mais exacerbações no ano anterior apesar do uso regular de altas doses de corticosteroides inalatórios mais controlador(es) adicional(ais) com ou sem corticosteroides orais e tinham eosinófilos sanguíneos superior ou igual a 150 células/mL na triagem ou maior ou igual a 300 células/mL dentro de 12 meses anteriores a inscrição. Indivíduos tiveram uma redução da taxa de exacerbação que tendiam a favor do mepolizumabe. Dos 19 adolescentes que receberam mepolizumabe, 9 receberam Nucala® e o clearance médio aparente nesses indivíduos era 35% menor que nos adultos. O perfil de eventos adversos em adolescentes foi geralmente similar ao da população global no estudo de fase 3.
Estudos controlados com placebo
No MEA112997, um estudo randomizado, duplo-cego, placebo-controlado, de grupos paralelos, multicêntrico, com 52 semanas de duração e 616 pacientes, os resultados demonstraram que mepolizumabe (nas doses de 75mg, 250mg e 750mg) reduziu significativamente as exacerbações de asma quando administrado por via intravenosa, em comparação ao placebo. Não se observou nenhuma diferença estatisticamente significativa quanto ao efeito das 3 doses estudadas. Por meio das contagens de eosinófilos sanguíneos ≥150 células/mL na triagem, ou de eosinófilos sanguíneos ≥300 células/mL nos 12 meses anteriores, antecipou-se quais seriam os indivíduos que mais se beneficiariam do tratamento com mepolizumabe. Os resultados desse estudo foram usados para determinar a seleção de doses para os estudos com administração subcutânea de mepolizumabe. O mepolizumabe não é indicado para uso por via intravenosa e somente deve ser administrado por via subcutânea.
Desfechos Primários MEA112997
Avaliação da relação dose-resposta baseada na eficácia e segurança das três dosagens de mepolizumabe (75 mg, 250 mg e 750 mg) durante o período de tratamento de 52 semanas em pacientes adultos e adolescentes com asma refratária grave não controlada.
Desfechos secundários MEA112997
Avaliação do efeito de farmacodinâmica de mepolizumabe no número de eosinófilos no sangue, níveis séricos de IL-5 e o número de eosinófilos na expectoração induzida.
A proporção de eosinófilos sanguíneos na linha de base era consistentemente menor em todos os grupos de mepolizumabe comparado ao grupo placebo [p < 0.001 para todas as doses e em todos os pontos de tempo medidos (a cada 4 semanas, da semana 4 à semana 52)] (Fonte de dados nas Tabelas 6.67 e 6.68).
Esses dados sugerem que houve efeito dose-dependente; houve diferença maior entre placebo com o aumento da dose de mepolizumabe (Fonte de dados -Tabela 6.68). Na semana 52 as proporções dos grupos de mepolizumabe para placebo (ajustado para covariáveis) da razão de eosinófilos sanguíneos para a linha de base para mepolizumabe a 75 mg, 250 mg e 750 mg foram 0,22 (95% IC: 0,18 a 0,27); 0,14 (95% IC: 0,12 a 0,18); e 0,12 (95% IC: 0,09 a 0,14), respectivamente. Uma análise da razão comparada da linha de base dos eosinófilos no sangue na semana 56 (8 semanas após a última dose de mepolizumabe) não demonstra o retorno de eosinófilos além da linha de base em qualquer um dos grupos de tratamento por mepolizumabe.
O resumo dos dados sobre as concentrações séricas totais de IL-5 (IL-5 e complexo mepolizumabe/IL-5) é apresentado na Tabela de Fonte de Dados 6.75 (em testes de laboratórios: Interleucina 5 pg/mL), com análise estatística do total de IL-5 apresentada na Tabela de Fonte de Dados 6.76. Houve aumento dos níveis médios de IL-5 sérica total comparado ao placebo para todas as doses de mepolizumabe e em todos os pontos de tempo medidos (semana 16 e 48; Fonte de Dados - Tabela 6.76)
Os indivíduos do subgrupo que passou por análise do escarro (N=94) exibiram uma redução consistente similar em todos os grupos de mepolizumabe em comparação ao placebo, quando foi avaliada a proporção de eosinófilos em relação a linha de base.
Devido ao menor número de pacientes neste subgrupo, o poder de detectar a alteração entre cada grupo de tratamento de mepolizumabe e placebo foi reduzido. Houve um efeito dose-dependente, pois as diferenças entre o grupo placebo e os grupos de mepolizumabe aumentaram a medida que a dose de mepolizumabe aumentava. Na semana 52, quando comparada a linha de base, as razões entre o número de eosinófilos no escarro dos pacientes em uso de mepolizumabe 75 mg, 250 mg e 750 mg e placebo (ajustada para covariáveis) era respectivamente de 0,68 (IC 95%: 0,13 a 3,52); 0,35 (95% CI: 0,08 a 1,52); e 0,12 (IC 95%: 0,02 a 0,56), respectivamente (Tabela da Fonte de Dados 6.70).
Redução das exacerbações (MEA115588)
O MEA115588 foi um estudo randomizado, duplo-cego, placebo-controlado, de grupos paralelos e multicêntricos que avaliou a eficácia e a segurança de mepolizumabe como terapia adjuvante em 576 pacientes com asma eosinofílica grave. Este estudo analisou a frequência das exacerbações de asma clinicamente significativas, considerando-se os seguintes aspectos: piora da asma que requer uso de corticosteroides orais ou sistêmicos e/ou hospitalização e/ou consulta no setor de emergência.
Os pacientes tinham 12 anos de idade ou mais, história de duas ou mais exacerbações de asma nos 12 meses anteriores e asma não controlada com os tratamentos medicamentosos atuais [isto é, altas doses de corticosteroides inalatórios (CI), combinados com pelo menos outro agente de controle, como os agonistas beta-2 adrenérgicos de ação prolongada (LABA) ou os modificadores de leucotrienos]. Permitiu-se que os pacientes estivessem em corticoterapia oral e continuassem a receber a sua medicação existente para asma durante o estudo. A asma eosinofílica grave foi definida como contagem de eosinófilos do sangue periférico ≥150 células/mL dentro de 6 semanas após a randomização (primeira dose), ou contagem de eosinófilos sanguíneos ≥ 300 células/mL dentro dos 12 meses anteriores à randomização.
Os pacientes receberam 100 mg de mepolizumabe por via subcutânea (SC), ou 75 mg por via intravenosa (IV), ou placebo, 1 vez a cada 4 semanas, ao longo de 32 semanas.
O desfecho primário, isto é, a redução da frequência de exacerbações de asma clinicamente significativas, foi estatisticamente significativo (p < 0,001). A Tabela 2 mostra os resultados do desfecho primário e dos desfechos secundários do MEA115588.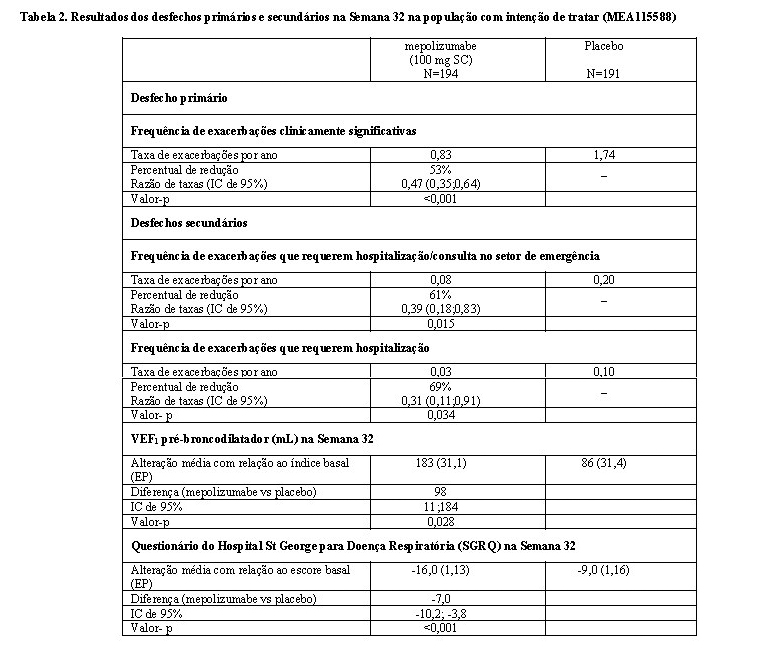
O tempo até a primeira exacerbação foi mais longo para os grupos que receberam Nucala® e mepolizumabe 75 mg IV em comparação ao grupo placebo no Estudo 2 (Figura 1).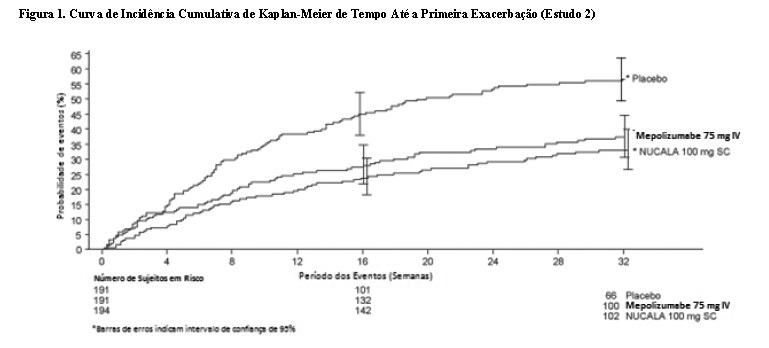
Os dados do Estudo 1 foram explorados para determinar um critério que pudesse identificar indivíduos que poderiam se beneficiar do tratamento com Nucala®. A análise exploratória sugeriu que a contagem de eosinófilos sanguíneos acima de 150 células/mL era um potencial indicador do benefício ao tratamento.
A análise exploratória dos dados do Estudo 2 também sugeriu que a contagem de eosinófilos sanguíneos (obtidos dentro de 6 semanas após o início da dosagem) de 150 células/mL na linha de base era um potencial indicador da eficácia e mostraram uma tendência de maior benefício de exacerbação com o aumento da contagem de eosinófilos no sangue. No Estudo 2, os indivíduos inscritos apenas com base na contagem histórica de eosinófilos no sangue de 300 células/mL ou mais nos últimos 12 meses, mas que tiveram uma contagem basal sanguínea de eosinófilos menor que 150 células/mL, praticamente tiveram nenhum benefício de exarcebação após o tratamento com Nucala® comparado com placebo.
Uma análise exploratória foi conduzida no subgrupo de 29 indivíduos no Estudo 3 que tiveram uma contagem média de eosinófilos sanguíneos na linha de base inferior a 150 células/mL. Cinco (29%) indivíduos no grupo recebendo Nucala® contra zero (0%) no grupo placebo tiveram redução de 90% a 100% em suas doses. Quatro (24%) indivíduos no grupo recebendo Nucala® contra 8 (67%) no grupo placebo foram classificados como não tendo melhora para dose oral de corticosteroide.
Redução do uso de corticosteroides orais (MEA115575)
O MEA115575 avaliou o efeito de 100 mg de mepolizumabe SC sobre a redução do uso de corticosteroides orais (CO) de manutenção, com a doença sob controle, em indivíduos com asma eosinofílica grave dependentes de corticosteroides sistêmicos. Os pacientes apresentavam contagem de eosinófilos no sangue periférico ≥ 300 células/mL nos 12 meses anteriores à triagem ou ≥ 150 células/mL ao entrar no estudo. Os pacientes receberam mepolizumabe ou placebo 1 vez a cada 4 semanas ao longo do período de tratamento. Na fase de redução do uso de CO, reduziu-se a dose desse fármaco a cada 4 semanas (Semanas 4-20), desde que a asma se mantivesse sob controle. Durante o estudo, os pacientes continuaram com o tratamento de asma que usavam ao entrar no estudo [isto é, altas doses de corticosteroides inalatórios (CI), combinados com pelo menos outro agente de controle, como os agonistas beta-2 adrenérgicos de ação prolongada (LABA) ou os modificadores de leucotrienos].
Esse estudo incluiu um total de 135 pacientes, com idade média de 50 anos: 55% eram do sexo feminino, 48% estavam em tratamento com corticoides orais havia pelo menos 5 anos e recebiam na triagem o equivalente a uma dose média de prednisona de 13 mg por dia no início do estudo.
O desfecho primário foi a redução da dose diária de CO (semanas 20-24), com manutenção do controle da asma, em comparação a pacientes tratados com placebo (ver Tabela 3).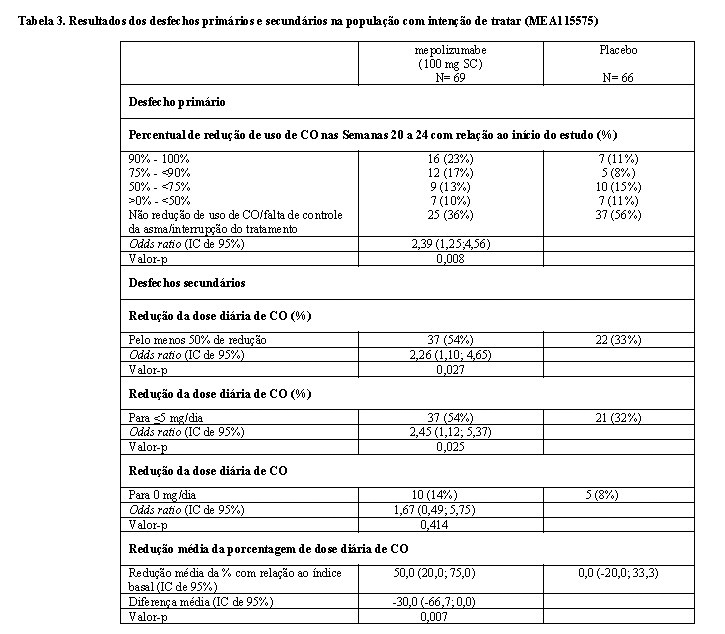
Além disso, mediu-se a qualidade de vida relacionada à saúde usando-se o SGRQ. Na Semana 24 houve melhora estatisticamente significativa do escore médio do SGRQ com mepolizumabe em comparação ao placebo: -5,8 (IC de 95%: -10,6; -1,0; p=0,019). Na Semana 24 a proporção de indivíduos que apresentaram uma redução clinicamente significativa do escore SGRQ (definida como redução de pelo menos 4 unidades em relação ao basal) foi maior com mepolizumabe (58%; 40/69) em comparação ao placebo (41%; 27/66).
O perfil de eficácia a longo prazo de mepolizumabe em pacientes com asma severa (n=998) tratados por em média 2.8 anos (intervalos de 4 semanas a 4.5 anos) nos estudos abertos MEA115666, MEA115661 e 201312 foi geralmente consistente com 3 estudos controlado com placebo.
Granulomatose Eosinofílica com Poliangeíte (GEPA)
MEA115921 foi um estudo randomizado, duplo-cego, controlado por placebo, de 52 semanas, que avaliou 136 pacientes ≥ 18 anos com GEPA reincidente ou refratária e que estavam em uso estável de corticosteroides orais (CO; ≥7,5 a ≤50 mg / dia prednisolona / prednisona). Cinquenta e três por cento (n = 72) também estavam em terapia imunossupressora estável concomitante.
Os pacientes receberam uma dose de 300 mg de mepolizumabe ou placebo administrados por via subcutânea uma vez a cada 4 semanas, além da sua terapia base prednisolona / prednisona, com ou sem terapia imunossupressora. A dose de CO foi reduzida a critério do investigador. Os desfechos co-primários foram o tempo total de remissão acumulado, definido como Escore de Atividade de Vasculite de Birmingham (BVAS) = 0 (sem vasculite ativa) mais a dose de prednisolona / prednisona ≤ 4 mg / dia e a proporção de indivíduos em remissão em ambas 36 e 48 semanas de tratamento.
Remissão
Em comparação com o placebo, pacientes tratados com mepolizumabe 300 mg atingiram um tempo acumulado de remissão significativamente maior. Além disso, em comparação com o placebo, uma proporção significativamente maior de indivíduos recebendo mepolizumabe 300 mg atingiu a remissão tanto na Semana 36 como na Semana 48 (Tabela 5).
Pacientes que receberam mepolizumabe 300 mg atingiram um tempo acumulado de remissão significativamente maior (p < 0,001) e uma proporção mais elevada de pacientes que receberam mepolizumabe 300 mg esteve em remissão tanto na Semana 36 como na Semana 48 (p < 0,001), em comparação ao placebo utilizando a definição de remissão do desfecho secundário do BVAS = 0 mais prednisolona / prednisona ≤ 7,5 mg / dia.
Reincidência
Comparado ao placebo, o tempo até à primeira reincidência (definido como agravamento relacionado com vasculite, asma ou sintomas sinusais requerendo um aumento na dose de corticosteroides ou terapia imunossupressora ou hospitalização) foi significativamente mais longo nos indivíduos que receberam mepolizumabe 300 mg (p < 0,001). Além disso, os indivíduos que receberam mepolizumabe tiveram uma redução de 50% na taxa anual de recidiva em comparação com o placebo: 1,14 vs 2,27, respectivamente.
Redução do uso de corticosteroides orais
Comparado ao placebo, os indivíduos que receberam mepolizumabe 300 mg apresentaram uma dose média diária de corticosteroide oral mais baixa entre as semanas 48 e 52 (p < 0,001). No grupo mepolizumabe 300 mg, 12 indivíduos (18%) foram capazes de reduzir completamente o tratamento com CO em comparação com 2 indivíduos (3%) no grupo placebo.
Questionário de Controle da Asma-6 (ACQ-6)
O ACQ-6, um questionário de 6 itens preenchido pelo indivíduo, foi desenvolvido para medir a adequação do controle da asma e a mudança no controle da
asma. A taxa de resposta ACQ-6 no tratamento durante as semanas 48 a 52 (definida como uma diminuição na pontuação de 0,5 ou mais em comparação com o valor basal) foi de 22% para 300 mg de Nucala® e 16% para placebo (OR 1,56; IC 95%: 0,63, 3,88 para 300 mg de Nucala® em comparação com placebo).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Classificação ATC
Grupo farmacoterapêutico: medicamentos para doenças obstrutivas das vias respiratórias, outros medicamentos sistêmicos para doenças obstrutivas das vias respiratórias. R03DX09
Mecanismo de ação
O mepolizumabe é um anticorpo monoclonal humanizado (IgG1, kappa) que tem como alvo a interleucina 5 humana (IL-5) com alta afinidade e especificidade. A IL-5 é a principal citocina responsável pelo crescimento e diferenciação, recrutamento, ativação e sobrevivência dos eosinófilos. O mepolizumabe inibe a bioatividade da IL-5 com potência nanomolar ao bloquear a ligação da IL-5 à cadeia alfa do complexo receptor dessa citocina expressa na superfície celular do eosinófilo, de maneira a inibir a sinalização da IL-5 e reduzir a produção e a sobrevivência dos eosinófilos.
Efeitos farmacodinâmicos
Em ensaios clínicos a redução dos eosinófilos sanguíneos após o tratamento com mepolizumabe foi consistentemente observada. A magnitude da redução nas populações indicadas descritas a seguir foram observadas dentro de 4 semanas de tratamento e foram mantidas durante todo o período de tratamento.
Em pacientes com asma grave (adultos / adolescentes) após uma dose de 100 mg administrada por via subcutânea a cada 4 semanas durante 32 e 52 semanas, respectivamente, os eosinófilos sanguíneos foram reduzidos para uma contagem média geométrica de 40 células / mL. Isso corresponde a uma redução média geométrica de 84% e 79% em comparação ao placebo, respectivamente. Esta magnitude na redução de eosinófilos sanguíneos foi mantida em pacientes com asma grave (n=998) tratados por em média 2.8 anos (intervalos de 4 semanas a 4.5 anos) nos estudos abertos.
Em crianças de 6 a 11 anos com asma grave, seguindo administração subcutânea de 40 mg (para peso abaixo de 40kg) ou 100 mg (para peso igual ou acima de 40 kg) a cada 4 semanas durante 52 semanas, os eosinófilos sanguíneos foram reduzidos para uma contagem média geométrica de 48 e 44 células/mL respectivamente, com uma redução em relação ao nível basal de 85% e 87% respetivamente.
Em pacientes com GEPA, após uma dose de 300 mg administrada SC a cada 4 semanas durante 52 semanas, os eosinófilos sanguíneos foram reduzidos para uma contagem média geométrica de 38 células/mL. Houve uma redução média geométrica de 83% comparada ao placebo.
Imunogenicidade
Em indivíduos com asma que receberam 100 mg de Nucala®, 15/260 (6%) desenvolveram anticorpos anti mepolizumabe. Anticorpos neutralizantes foram detectados em 1 indivíduo com asma recebendo 100 mg de Nucala®. Os anticorpos anti mepolizumabe aumentaram ligeiramente (aproximadamente 20%) a depuração do mepolizumabe. Não houve evidência de uma correlação entre os títulos de anticorpos anti mepolizumabe e a alteração no nível de eosinófilos. A relevância clínica da presença de anticorpos anti mepolizumabe não é conhecida.
O perfil de imunogenicidade do mepolizumabe em pacientes com asma grave (n=998) tratados por em média 2.8 anos (intervalos de 4 semanas a 4.5 anos) em estudos abertos foi similar ao observado nos estudos controlados com placebo.
Em crianças de 6 a 11 anos de idade com asma grave recebendo 40 mg de Nucala® subcutâneo (para peso abaixo de 40 kg) ou 100 mg de Nucala® subcutâneo (para peso igual ou acima de 40 kg), 2/35 (6%) tiveram anticorpos anti mepolizumabe detectáveis durante a fase inicial do estudo. Nenhuma criança teve anticorpo anti mepolizumabe detectável durante a fase de longo prazo do estudo.
Em indivíduos com GEPA recebendo 300 mg de Nucala®, 1/68 ( < 2%) tinham anticorpos anti mepolizumabe detectáveis. Não foram detectados anticorpos neutralizantes em nenhum indivíduo com GEPA.
A frequência relatada de anticorpos anti mepolizumabe pode subestimar a frequência real devido à menor sensibilidade do ensaio na presença de alta concentração de fármaco. Os dados refletem a porcentagem de pacientes cujos resultados foram positivos para anticorpos para mepolizumabe em ensaios específicos. A incidência observada de positividade de anticorpos em um ensaio é altamente dependente de vários fatores, incluindo sensibilidade e especificidade do ensaio, metodologia do ensaio, manipulação da amostra, tempo de coleta da amostra, medicações concomitantes e doença subjacente.
Farmacocinética
Após a administração subcutânea a indivíduos com asma moderada ou grave, o mepolizumabe exibiu farmacocinética aproximadamente proporcional à dose na faixa de 12,5 a 250 mg. A farmacocinética do mepolizumabe foi consistente em indivíduos com asma grave ou GEPA. A exposição a 300 mg em indivíduos com GEPA foi aproximadamente três vezes a observada em 100 mg em indivíduos com asma grave.
Absorção
Após administração subcutânea a indivíduos saudáveis ou pacientes com asma, o mepolizumabe foi absorvido lentamente com um tempo médio para atingir a concentração plasmática máxima (Tmáx) de 4 a 8 dias.
Com a administração única por via subcutânea no abdome, coxa ou braço de indivíduos saudáveis, a biodisponibilidade absoluta de mepolizumabe foi de 64%, 71% e 75%, respectivamente. Em pacientes com asma, a biodisponibilidade absoluta de mepolizumabe administrado por via subcutânea no braço variou de 74% -80%. Após a administração subcutânea repetida a cada 4 semanas, a acumulação é quase duplicada em estado de equilíbrio.
Distribuição
Após uma única administração de mepolizumabe por via intravenosa a pacientes com asma, o volume médio de distribuição é de 55 a 85 mL/kg.
Biotransformação
O mepolizumabe é um anticorpo monoclonal IgG1 humanizado e degradado por enzimas proteolíticas que são amplamente distribuídas no corpo e não restritas ao tecido hepático.
Eliminação
Após uma única administração por via intravenosa a pacientes com asma, a média do clearance sistêmico variou de 1,9 a 3,3 mL/dia/kg, com média de meia-vida terminal de aproximadamente 20 dias. Após a administração de mepolizumabe por via subcutânea, a média de meia-vida terminal (t1/2) variou entre 16 e 22 dias. Na análise farmacocinética da população, a taxa estimada do clearance sistêmico de mepolizumabe foi de 3,1 mL/dia/kg.
População pediátrica
Existem dados farmacocinéticos limitados disponíveis na população pediátrica (59 indivíduos com esofagite eosinofílica, 55 indivíduos com asma eosinofílica refratária grave). A farmacocinética de mepolizumabe por via intravenosa foi avaliada por análise farmacocinética populacional num estudo pediátrico realizado em indivíduos com idades entre os 2 e os 17 anos com esofagite eosinofílica. A farmacocinética pediátrica foi amplamente previsível em adultos, após levar em consideração o peso corporal. A farmacocinética do mepolizumabe em adolescentes com asma grave eosinofílica refratária incluída nos estudos de fase 3 foi consistente com os adultos.
A farmacocinética pediátrica após administração subcutânea em indivíduos dos 6 aos 11 anos de idade com asma eosinofílica refratária foi investigada num estudo aberto, não controlado, com a duração de 12 semanas. A farmacocinética pediátrica foi amplamente consistente com adultos e adolescentes após contabilizar o peso corporal e a biodisponibilidade. A biodisponibilidade subcutânea absoluta parece completa em comparação àquela observada em adultos e adolescentes de 76%. A exposição após administração subcutânea de 40 mg (para um peso < 40 kg) ou 100 mg (para um peso ≥ 40 kg) foi de 1,32 e 1,97 vezes a observada em adultos a 100 mg.
A investigação de um esquema posológico subcutâneo de 40 mg administrado a cada 4 semanas em crianças de 6 a 11 anos de idade com peso entre 15 e 70 kg por modelagem farmacocinética e simulação prediz que a exposição desse regime de dosagem permaneceria em média dentro de 38% dos adultos em 100 mg. Este regime posológico é considerado aceitável devido ao amplo índice terapêutico do mepolizumabe.
Populações especiais
Pacientes idosos (≥65 anos)
Existem dados farmacocinéticos limitados disponíveis em pacientes idosos (≥ 65 anos de idade) em todos os estudos clínicos (N = 90). No entanto, na análise farmacocinética populacional, não houve indicações de um efeito da idade na farmacocinética do mepolizumabe na faixa etária de 12 a 82 anos.
Insuficiência renal
Não foram conduzidos estudos formais para investigar o efeito da insuficiência renal sobre a farmacocinética de mepolizumabe. Com base na análise farmacocinética da população, não são necessários ajustes de dose para pacientes com valores do clearance de creatinina entre 50-80 mL/min. Há poucos dados disponíveis a respeito de pacientes com valores do clearance da creatinina inferiores a 50 mL/min.
Insuficiência hepática
Não foram conduzidos estudos formais para investigar o efeito da insuficiência hepática sobre a farmacocinética de mepolizumabe. Considerando-se que o mepolizumabe é degradado por enzimas proteolíticas amplamente distribuídas, não restritas ao tecido hepático, é provável que as alterações da função hepática não tenham nenhum efeito sobre a eliminação do mepolizumabe.
4. CONTRAINDICAÇÕES
Nucala® é contraindicado para pacientes que apresentam hipersensibilidade ao mepolizumabe ou a qualquer excipiente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Nucala® não deve ser usado para tratar exacerbações agudas de asma.
Podem ocorrer exacerbações ou eventos adversos relacionados à asma durante o tratamento com Nucala®. Os pacientes devem ser instruídos a procurar aconselhamento médico caso a asma permaneça descontrolada ou piore após o início do tratamento com mepolizumabe.
Não se recomenda a descontinuação abrupta do uso de corticosteroides após o início da terapia com Nucala®. As reduções das doses de corticosteroides, se necessárias, devem ser graduais e realizadas sob supervisão médica.
Reações de hipersensibilidade associadas à administração
Ocorreram reações sistêmicas agudas e tardias, inclusive de hipersensibilidade (p. ex. anafilaxia, urticária, angioedema, rash (erupção cutânea), broncoespasmo, hipotensão), após a administração de Nucala®. Essas reações geralmente ocorreram horas após a administração, mas em alguns casos tiveram início tardio (isto é, após dias). Em caso de ocorrência de reações de hipersensibilidade, Nucala® deve ser descontinuado.
Infecções parasitárias
Eosinófilos podem estar envolvidos na resposta imunológica a algumas infecções helmínticas. Pacientes com infecções por helmintos pré-existentes foram excluídos da participação no programa clínico. Pacientes com infecções por helmintos pré-existentes devem ser tratados para a infecção antes da terapia com Nucala®. Caso os pacientes se infectem durante o tratamento com Nucala® e não respondam ao tratamento anti-helmíntico, deve-se considerar a interrupção temporária do uso de Nucala®.
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas
Não foram realizados estudos para investigar o efeito de Nucala® sobre a capacidade de dirigir veículos ou operar máquinas. Com base na farmacologia ou no perfil de reações adversas de Nucala®, não se espera um efeito prejudicial sobre essas atividades.
Gravidez e lactação
Fertilidade
Não há dados sobre fertilidade em seres humanos. Nos estudos com animais não se observaram efeitos adversos do tratamento com anti-IL-5 sobre a fertilidade (ver tópico Toxicologia Reprodutiva -Fertilidade).
Gravidez
Não se conhecem os efeitos de Nucala® na gravidez humana. Nos estudos com animais, não se observaram efeitos decorrentes do tratamento sobre o desenvolvimento embriofetal nem pós-parto (ver tópico Toxicologia Reprodutiva -Gravidez).
Deve-se utilizar mepolizumabe durante a gravidez somente se o benefício esperado para a mãe justificar o risco potencial para o feto.
Lactação
Não existem dados relativos à excreção de mepolizumabe no leite materno. Entretanto, houve excreção de Nucala® no leite de macacas cynomolgus em concentrações menores que 0,5% das detectadas no plasma.
A decisão de interromper a lactação ou descontinuar o uso de Nucala® deve levar em consideração a importância da amamentação para o lactente e da medicação para a mãe.
Carcinogênese/mutagênese
Não foram realizados estudos de longa duração em animais para avaliar o potencial carcinogênico de mepolizumabe. O potencial mutagênico de mepolizumabe não foi avaliado. O papel da IL-5 e dos eosinófilos na vigilância tumoral é pouco caracterizado. Entretanto, não há evidências de defeitos na vigilância de tumores em camundongos deficientes em IL-5 ou eosinófilos.
Toxicologia reprodutiva
Fertilidade
Não foram observados danos à fertilidade em um estudo de fertilidade e toxicidade reprodutiva geral conduzido em camundongos com um anticorpo análogo que inibe a IL-5 em camundongos. Esse estudo não incluiu a avaliação funcional de ninhada ou geração F1.
Gravidez
Em macacos, o mepolizumabe não teve efeito sobre a gravidez nem sobre o desenvolvimento embrionário/fetal e pós-parto (incluindo a função imunológica) dos filhotes. Não foram realizados exames para investigar malformações internas ou esqueléticas. Os dados de macacos cynomolgus demonstraram que o mepolizumabe atravessa a placenta. As concentrações de mepolizumabe foram aproximadamente 2,4 vezes mais altas nos neonatos que nas mães por diversos meses no período pós-parto e não afetaram o sistema imunológico dos neonatos.
Dados de segurança pré-clínicos
Os dados não clínicos não revelaram riscos especiais para seres humanos com base em estudos convencionais de segurança farmacológica ou de toxicidade de doses repetidas em macacos. A administração por via intravenosa ou subcutânea em macacos foi associada a reduções das contagens de eosinófilos periféricos e pulmonares, sem achados toxicológicos.
Os eosinófilos foram associados a respostas do sistema imunológico a algumas infecções parasitárias. Os estudos conduzidos em camundongos tratados com anticorpos anti-IL-5 ou com deficiência genética de IL-5 ou eosinófilos não mostraram falta de capacidade de remover infecções parasitárias.
Categoria B de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Atenção diabéticos: este medicamento contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos formais sobre interações com Nucala®.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de armazenamento
Frasco-ampola fechado
Armazenar entre 2°C e 8°C. Não congelar.
Proteger da luz. Armazenar na embalagem original até o uso.
Solução reconstituída
Após a reconstituição com água para injeção, o produto permanecerá estável por até 8 horas se armazenado no frasco ampola, a temperaturas abaixo de 30°C.
Não congele.
Durante a administração, não é necessário proteger da luz.
Nucala® é apresentado como pó liofilizado estéril, em frasco-ampola de vidro tipo I de 10 mL com rolha de borracha de bromobutilo (sem látex), selo de alumínio cinza e uma tampa de levantar (flip-cap) de plástico. O medicamento é fornecido em frasco-ampola para uso único e sem conservantes.
O prazo de validade é de 48 meses a contar da data de fabricação.
Número do lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Depois de preparado, este medicamento pode ser utilizado em até 8 horas se armazenado no frasco ampola, a temperaturas abaixo de 30°C.
Aspectos físicos/características organolépticas
Nucala® se apresenta como um pó liofilizado branco.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Posologia
Asma Eosinofílica Grave
Adultos e adolescentes a partir de 12 anos
A dose recomendada é de 100 mg de Nucala® administradas por injeção subcutânea (SC) uma vez a cada 4 semanas.
Crianças com idade entre 6 anos e 11 anos
A dose recomendada é de 40 mg de Nucala® administradas por injeção subcutânea (SC) uma vez a cada 4 semanas. O pó liofilizado para solução injetável deve ser utilizado para administração em crianças com idade entre 6-11 anos de idade. Cada frasco-ampola de Nucala® deve ser usado para um único paciente e qualquer resíduo do frasco-ampola deve ser descartado. A segurança e eficácia do mepolizumabe não foram estabelecidas em crianças com idade inferior a 6 anos.
Granulomatose Eosinofílica com Poliangeíte (GEPA)
Recomenda-se que os locais de cada injeção sejam separados por pelo menos 5 cm (ver seção Modo de usar).
Adultos A dose recomendada é de 300 mg de Nucala® administradas por injeção subcutânea (SC) uma vez a cada 4 semanas
Populações especiais
Idosos (65 anos ou mais)
Não há recomendação de ajuste de dose em pacientes de 65 anos ou mais (ver seção Características Farmacológicas -Populações Especiais).
Insuficiência renal
Não é provável que haja necessidade de ajustes de dose em pacientes com insuficiência renal (ver seção Características Farmacológicas -Populações Especiais).
Insuficiência hepática
Não é provável que haja necessidade de ajustes de dose em pacientes com insuficiência hepática (ver seção Características Farmacológicas -Populações Especiais).
Incompatibilidades
Não misture a solução reconstituída para injeção com outros medicamentos.
Modo de usar
Nucala® deve ser administrado por um profissional de saúde.
Após reconstituição, Nucala® deve ser administrado somente por injeção subcutânea - por exemplo, na parte superior do braço, na coxa ou no abdome.
Nucala® é fornecido como um pó liofilizado, em frasco-ampola para uso único somente para injeção subcutânea e deve ser reconstituído por um profissional de saúde usando-se as seguintes técnicas assépticas:
Instruções de reconstituição
1. Reconstitua o mepolizumabe em pó no frasco-ampola com 1,2 mL de água para injeção estéril, preferencialmente com seringa de 2 a 3 mL e agulha de 21 gauge. A solução reconstituída contém uma concentração de 100 mg/mL de mepolizumabe.
2. O jato de água para injeção estéril deve ser direcionado verticalmente para o centro da massa liofilizada. Permita que o frasco-ampola descanse em temperatura ambiente durante a reconstituição, girando-o gentilmente com movimentos circulares por 10 segundos, em intervalos de 15 segundos, até que o pó esteja dissolvido.
Observação: não agite a solução reconstituída durante o procedimento, porque isso pode causar precipitação ou fazer com que o produto crie espuma. Geralmente, a reconstituição é concluída em 5 minutos depois da adição da água estéril, mas o processo pode ser mais demorado.
3. Em caso de uso de um dispositivo mecânico (swirler) para reconstituição, o processo pode ser realizado girando-se o frasco-ampola a 450 rpm por não mais que 10 minutos. Como alternativa, também é possível girar o frasco-ampola a 1.000 rpm por não mais que 5 minutos.
4. Antes de usar inspecione visualmente a solução reconstituída para checar a presença de material particulado e a limpidez. A solução deve ser de transparente a opalescente, de incolor a amarelo pálido ou marrom pálido e estar livre de partículas visíveis. Pode, entretanto, ocorrer a presença de pequenas bolhas de ar, o que é aceitável. Se forem observados materiais particulados na solução ou ela parecer turva ou leitosa, não deve ser usada.
5. Se não utilizada imediatamente após o preparo, a solução reconstituída de mepolizumabe:
• Deve ser armazenada no frasco ampola a uma temperatura abaixo de 30°C;
• Deve ser descartada se não for utilizada no prazo de 8 horas após a reconstituição;
• Não deve ser misturada com outros medicamentos;
• Não deve ser congelada.
Instruções para administração de cada dose de 100 mg
1. Para administração subcutânea, deve-se preferencialmente utilizar uma seringa de polipropileno de 1 mL, com agulha de 21 gauge a 27 gauge x 0,5 polegada (13 mm).
2. Logo antes da administração, remova 1 mL de mepolizumabe reconstituído do frasco-ampola. Não agite a solução reconstituída durante o procedimento, porque isso pode causar precipitação ou fazer com que o produto crie espuma.
3. Administre a injeção de 1 mL (equivalente a 100 mg de mepolizumabe) por via subcutânea na parte superior do braço, coxa ou abdome.
Se for necessário mais do que um frasco para administração da dose prescrita, repita os passos 1 a 3. Recomenda-se que os locais de injeção individuais sejam separados por pelo menos 5 cm.
Instruções para administração de cada dose de 40 mg
1. Para administração subcutânea, deve-se preferencialmente utilizar uma seringa de polipropileno de 1 mL, com agulha de 21 gauge a 27 gauge x 0,5 polegada (13 mm).
2. Logo antes da administração, remova 0,4 mL de mepolizumabe reconstituído do frasco-ampola. Não agite a solução reconstituída durante o procedimento, porque isso pode causar precipitação ou fazer com que o produto crie espuma.
3. Administre a injeção de 0,4 mL (equivalente a 40 mg de mepolizumabe) por via subcutânea na parte superior do braço, coxa ou abdome.
Descarte
Todo medicamento não utilizado ou resíduo deve ser descartado de acordo com as exigências locais.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
Adultos e adolescentes
Em estudos clínicos em indivíduos com asma eosinofílica refratária grave, as reações adversas mais frequentemente notificadas durante o tratamento foram dores de cabeça, reações no local da injeção e dor nas costas.
Lista tabelada de reações adversas
Um total de 896 adultos e 19 adolescentes com asma eosinofílica refratária grave recebeu uma dose subcutânea ou intravenosa de mepolizumabe durante três estudos clínicos controlados com placebo com 24 a 52 semanas de duração. A tabela abaixo apresenta as reações adversas dos dois estudos controlados com placebo em doentes tratados com mepolizumabe 100 mg por via subcutânea (n = 263).
O perfil de segurança do mepolizumabe em doentes com asma eosinofílica refratária grave (n = 998) tratados numa mediana de 2,8 anos (intervalo de 4 semanas a 4,5 anos) em estudos de extensão abertos foi semelhante ao observado nos estudos controlados por placebo.
A frequência das reações adversas é definida utilizand