NORVIR
ABBVIE
Comprimidos
ritonavir
Antiviral.
Apresentações.
Comprimidos revestidos de 100 mg: embalagem com 30 comprimidos revestidos.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 01 MÊS DE IDADE*
(*para crianças capazes de deglutir comprimidos)
Composição.
NORVIR® (ritonavir) comprimido revestido 100 mg:
Cada comprimido revestido contém: ritonavir 100 mg. Excipientes: copovidona, laurato de sorbitana, dióxido de silício, estearilfumarato de sódio, fosfato de cálcio dibásico, água purificada. Constituintes do revestimento do comprimido: hipromelose, dióxido de titânio, magrogol, hiprolose, talco, dióxido de silício, polissorbato 80.
Informações técnicas.
1. INDICAÇÕES
NORVIR® (ritonavir) é destinado, em combinação com outros antirretrovirais, ao tratamento de pacientes adultos e pediátricos infectados pelo HIV, quando uma terapia antirretroviral for indicada com base em evidência clínica ou imunológica de progressão da doença.
2. RESULTADOS DE EFICÁCIA
Danner et al. demonstraram a potência, segurança e eficácia do ritonavir em um estudo que avaliou regimes contendo diferentes doses de ritonavir, comparando-os com placebo. Após 32 semanas, o grupo que recebeu 600 mg a cada 12 horas, em combinação com outros agentes ARV (inibidores da transcriptase reversa análogos de nucleosídeo) apresentaram um ganho de CD4 de 230 células por mm3 e uma queda na carga viral de 0,81 log. Os eventos adversos mais comuns foram náuseas, aumento dos triglicérides e das enzimas hepáticas. Assim, o ritonavir mostrou-se seguro, eficaz e bem tolerado.1
Contemporaneamente, Markowitz et al. mostraram resultados similares, demonstrando uma resposta imunológica satisfatória, com um ganho de CD4 de 74 e 83 células por mm3 após 4 e 12 semanas, respectivamente. Na semana 12, a queda da carga viral foi de 1,1 log.2
Referências Bibliográficas
1. Danner S, Carr A, Leonard J, Leahman L, Gudiol F Gonzalez J. A short term Study of the Safety, Pharmacokinetics and Efficacy of Ritonavir, an Inhibitor of HIV-1 Protease. N Engl J Med 1995;333:1528-33.
2. Markowitz M, Saag M, Powderly W, Hurley A, Hsu A, Valdes J. A Preliminary Study of Ritonavir, an Inhibitor of HIV-1 Protease, to Treat HIV Infection. N Engl J Med 1995;333:1534-9.
3. CARACTERÍSTICAS FARMACOLÓGICAS
NORVIR® (ritonavir) é um inibidor da protease do HIV, apresentando atividade contra o Vírus da Imunodeficiência Humana (HIV).
O ritonavir é um pó branco ou com leve cor marrom, de sabor metálico amargo. É facilmente solúvel em metanol e etanol, solúvel em isopropanol e praticamente insolúvel em água.
O ritonavir é chamado quimicamente de éster 5-tiazolilmetílico, [5S-(5R*,8R*,10R*,11R*)] do ácido 10-hidroxi-2-metil-5-(1-metiletil)-1-[2-(1-metiletil)-4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraazatridecan-13-óico. Sua fórmula molecular é C37H48N6O5S2 e seu peso molecular é 720,95.
Farmacologia clínica
Mecanismo de ação: ritonavir é um inibidor peptidomimético oral ativo das aspartil-proteases do HIV-1 e HIV-2. A inibição da protease do HIV torna a enzima incapaz de processar o precursor da poliproteína gag-pol, fazendo com que as partículas virais produzidas se tornem imaturas e, portanto, incapazes de iniciar um novo ciclo de infecção. O ritonavir tem afinidade seletiva pela protease do HIV e pouca atividade inibitória diante da aspartil-protease humana. Pode ser utilizado também em conjunto com outros antirretrovirais da mesma classe com a finalidade de reduzir sua metabolização, diminuindo a dose necessária a cada tomada ou aumentando o intervalo entre as tomadas.
Atividade antiviral in vitro: dados in vitro indicam que ritonavir é ativo contra todas as cepas de HIV testadas em uma variedade de linhagens celulares humanas primárias e transformadas. A concentração da droga que inibe 50% e 90% da replicação viral in vitro é aproximadamente 0,02 mcM e 0,11 mcM, respectivamente. Potências similares foram observadas com cepas de HIV sensíveis e resistentes ao AZT. Estudos que avaliaram a citotoxicidade direta de ritonavir em diversas linhagens celulares não mostraram toxicidade direta em concentrações de até 25 mcM, com um índice terapêutico resultante in vitro de pelo menos 1000.
Resistência: isolados de HIV-1 resistentes ao ritonavir foram selecionados in vitro. Os isolados resistentes demonstraram reduzir a susceptibilidade de ritonavir e a análise genotípica desses isolados mostrou que a resistência foi primariamente atribuída a substituições específicas de aminoácidos na protease do HIV-1 nas posições 84 (Ile por Val), 82 (Val por Phe), 71 (Ala por Val) e 46 (Met por Ile). Alterações fenotípicas e genotípicas nos isolados de HIV de pacientes selecionados tratados com ritonavir foram monitoradas em ensaios clínicos de Fase I/II. A análise genotípica e fenotípica em série indicou que a sensibilidade ao ritonavir caiu de forma ordenada e gradual. As mutações iniciais ocorreram nas posições 82(Val por Ala/Phe), 54 (Ile por Val), 71 (Ala por Val/Thr) e 36 (Ile por Leu), seguidas pelas combinações de mutações em cinco posições adicionais específicas de aminoácidos. Cepas virais isoladas in vivo sem alteração na posição 82 não têm sensibilidade diminuída ao ritonavir. A mutação na posição 82 parece ser necessária, mas não suficiente para conferir resistência fenotípica. A resistência fenotípica foi definida como uma diminuição ≥ 5 vezes na sensibilidade viral in vitro em relação ao basal. A relevância clínica das alterações genotípicas e fenotípicas associadas ao ritonavir ainda não foi estabelecida.
Resistência cruzada com outros antirretrovirais: o potencial para resistência cruzada ao HIV entre inibidores de protease não foi completamente explorado. Portanto, não se conhece o efeito que ritonavir terá na atividade de outros inibidores da protease administrados subsequentemente. Isolados de HIV obtidos em série de seis pacientes tratados com ritonavir mostraram sensibilidade reduzida in vitro a este medicamento, mas não demonstraram redução correspondente da sensibilidade in vitro ao saquinavir quando comparada aos isolados basais. Entretanto, isolados de dois desses pacientes mostraram uma redução (8 vezes) na sensibilidade in vitro ao indinavir. Isolados de cinco pacientes também foram testados quanto à resistência cruzada ao amprenavir e nelfinavir; isolados de dois pacientes apresentaram diminuição da sensibilidade ao nelfinavir (12-14 vezes) e nenhuma ao amprenavir. A resistência cruzada entre ritonavir e inibidores da transcriptase reversa é improvável, já que os alvos enzimáticos envolvidos são diferentes. Um isolado de HIV resistente a zidovudina (ZDV) testado in vitro manteve total sensibilidade ao ritonavir.
Farmacocinética
Em um estudo de farmacocinética de dose única em indivíduos infectados pelo HIV do sexo masculino, em jejum, altos níveis do fármaco foram encontrados e mantidos por várias horas após administração oral de 100 mg, 200 mg, 400 mg, 600 mg, 800 mg ou 1000 mg de ritonavir. A área sob a curva (AUC) da concentração versus tempo variou de 3,92 a 123 mcg.h/mL, respectivamente e a Cmáx variou de 0,416 a 12,7 mcg/mL. A farmacocinética do ritonavir foi dose-dependente e aumentos maiores do que o proporcional na AUC e Cmáx com aumento de dose foram relatados.
O Tmáx permaneceu constante por aproximadamente 3 horas com o aumento de dose. A depuração renal foi, em média, inferior a 0,1 L/h e relativamente constante na faixa de dosagem. Como não há formulação parenteral de ritonavir, a biodisponibilidade absoluta não foi determinada.
Após uma dose única de 600 mg sob condições de plenitude gástrica, a formulação em cápsula gelatinosa mole de 100 mg (n=57) produziu AUCs de 121,7 ± 53,8 mcg.h/mL (média ± desvio padrão). A concentração plasmática de ritonavir após a administração de uma dose simples de 100 mg de comprimidos revestidos é similar à administração de 100 mg de ritonavir em cápsulas de gelatina mole sob condição alimentada. Em comparação à ingestão em jejum, a extensão de absorção da cápsula gelatinosa mole foi 12% maior quando administrada com uma refeição. A alimentação reduz ligeiramente a biodisponibilidade de ritonavir comprimidos. A administração de uma dose única de 100 mg de ritonavir comprimidos com uma alimentação moderada em gordura (857 Kcal, 31% calorias em gordura) ou uma alimentação rica em gordura (907 Kcal, 52% calorias em gordura) foi associado com uma diminuição de 20-23% na AUC e na Cmáx de ritonavir.
A farmacocinética do ritonavir durante regimes de múltiplas doses foram estudadas em voluntários adultos HIV-positivos em condições de plenitude gástrica. Sob condições de múltiplas doses, o acúmulo de ritonavir é ligeiramente menor do que o previsto a partir da dose única devido a um aumento relacionado ao tempo e à dose na depuração aparente (Cl/F). Foi observado que concentrações mínimas de ritonavir diminuem com o tempo, possivelmente devido à indução enzimática, atingindo níveis estáveis após 2 semanas. No estado de equilíbrio, com uma dose de 600 mg duas vezes ao dia, foram observados valores de Cmáx e Cmín de 11,2 e 3,7 mcg/mL, respectivamente. O t½ de ritonavir foi de aproximadamente 3 a 5 horas. A depuração aparente no estado de equilíbrio em pacientes tratados com 600 mg duas vezes ao dia foi em média 8,8 ± 3,2 L/h.
Não foram observadas diferenças clinicamente significantes na AUC ou Cmáx entre homens e mulheres. Os parâmetros farmacocinéticos do ritonavir não foram significativamente associados à perda de peso ou massa corpórea magra.
O volume aparente de distribuição (VB/F) do ritonavir é de aproximadamente 0,41 ± 0,25 L/kg após uma dose única de 600 mg. A ligação do ritonavir a proteínas no plasma humano foi de aproximadamente 98 a 99%. O ritonavir se liga à alfa-1 glicoproteína ácida (AAG) e à albumina sérica humana (HSA) com afinidades comparáveis. A ligação a proteínas plasmáticas total é constante na faixa de concentração de 1 a 100 mcg/mL.
Estudos de distribuição tecidual em ratos com ritonavir marcado com 14C demonstraram que o fígado, as supra-renais, o pâncreas, os rins e a tireoide tinham as maiores concentrações de ritonavir. Uma relação tecido-plasma de aproximadamente 1, medido nos gânglios linfáticos de ratos, sugere que o ritonavir é distribuído no tecido linfático. Ritonavir penetra de maneira mínima no cérebro.
Observou-se que o ritonavir é extensamente metabolizado pelo sistema do citocromo hepático P450, principalmente pela isoenzima CYP3A e em menor extensão pela CYP2D6. Estudos em animais, assim como experimentos in vitro com microssomos hepáticos humanos, indicam que o ritonavir sofre principalmente metabolismo oxidativo. Cinco metabólitos de ritonavir foram identificados no homem. O metabólito da oxidação isopropiltiazólico (M-2) é o principal metabólito e tem atividade antiviral similar à da substância precursora. Entretanto, a AUC do metabólito M-2 foi aproximadamente 3% da AUC da substância precursora.
Estudos em humanos com ritonavir marcado radioativamente demonstraram que sua eliminação se faz principalmente pela via hepatobiliar; aproximadamente 86% da substância marcada foi recuperada nas fezes. Nestes estudos, a via renal de eliminação não foi observada como via de eliminação importante do ritonavir.
Efeitos no Eletrocardiograma
O intervalo QTcF foi avaliado em um estudo controlado cruzado, randomizado, placebo e ativo (moxifloxacina 400 mg/uma vez ao dia), com 45 adultos sadios, com 10 medidas durante 12 horas no Dia 3. A média de diferença máxima (limite de confiança superior a 95%) no QTcF do placebo foi de 5,5 (7,6) mseg para ritonavir 400 mg duas vezes ao dia. A exposição ao ritonavir no Dia 3 foi aproximadamente 1,5 vezes maior que a observada com a dose de 600 mg duas vezes ao dia no estado de equilíbrio. Nenhum voluntário teve um aumento na QTcF ≥ 60 mseg da baseline ou um intervalo QTcF que excedesse o limite clinicamente relevante de 500 mseg.
Um discreto prolongamento no intervalo PR também foi verificado em voluntários recebendo ritonavir durante o mesmo estudo no Dia 3. O intervalo PR máximo foi de 252 mseg e não houve bloqueio cardíaco de segundo ou terceiro graus.
Populações Especiais
Insuficiência renal: atualmente não há dados específicos sobre esta população de pacientes. Entretanto, devido à alta ligação protéica de ritonavir é pouco provável que o ritonavir seja significativamente removido por hemodiálise ou diálise peritoneal.
Insuficiência hepática: em seis indivíduos adultos, com doença hepática leve, infectados pelo HIV, recebendo 400 mg de ritonavir duas vezes ao dia, a exposição ao ritonavir foi semelhante em comparação com os indivíduos controles recebendo 500 mg de ritonavir duas vezes ao dia. Os resultados indicam que não é necessário ajuste de dose em pacientes com doença hepática leve. Dados adequados de farmacocinética não estão disponíveis para pacientes com doença hepática moderada. A ligação proteica do ritonavir não foi afetada de modo estatisticamente significativo pela doença hepática leve e moderada.
Pacientes pediátricos: a farmacocinética no estado de equilíbrio foi avaliada em 37 pacientes com idades entre 02 e 14 anos infectados por HIV, que recebiam doses variando entre 250 mg/m2 de superfície corporal e 400 mg/m2 de superfície corporal duas vezes ao dia, no Grupo de Estudo de Pacientes Pediátricos com AIDS do estudo 310 (PACTG), e em 41 pacientes com idades entre 01 mês e 02 anos, infectados por HIV, em doses de 350 e 450 mg/m2 duas vezes ao dia, no estudo 345 em PACTG.
A depuração de ritonavir oral no estado de equilíbrio foi aproximadamente 1,5 a 1,7 vezes mais rápida em pacientes pediátricos do que em adultos. As concentrações de ritonavir em pacientes pediátricos, maiores de 02 anos, obtidas após 350 a 400 mg/m2 duas vezes ao dia, foram comparáveis àquelas obtidas em adultos recebendo 600 mg (aproximadamente 330 mg/m2) duas vezes ao dia. As seguintes observações foram feitas a respeito das concentrações do ritonavir após a administração de 350 ou 450 mg/m2, duas vezes ao dia, em crianças menores de 02 anos de idade. Exposições mais elevadas do ritonavir não foram evidentes com 450 mg/m2 duas vezes ao dia comparado a 350 mg/m 2 duas vezes ao dia. As concentrações mais baixas de ritonavir eram um pouco menores do que aquelas obtidas nos adultos que receberam 600 mg duas vezes ao dia.
A área sob a curva de concentração plasmática de ritonavir/tempo e menores concentrações obtidas após a administração de 350 ou 450 mg/m2, duas vezes ao dia, em crianças menores de 02 anos, foram aproximadamente 16% e 60% mais baixas, respectivamente, do que aquelas obtidas em adultos que receberam 600 mg duas vezes ao dia.
Dados de segurança pré-clinica
Toxicidade Aguda, Subaguda e Crônica
O ritonavir apresenta baixos índices de toxicidade aguda quando administrado oralmente. A DL50 foi determinada como sendo maior do que 2500 mg/kg, tanto em ratos quanto em camundongos. Os sinais de toxicidade com altas doses nas duas espécies incluíram diminuição da atividade, ataxia, dispneia e tremores. Em geral, os sinais de toxicidade foram aparentes de 1 a 3 dias após a administração. Nenhuma mudança morfológica bruta foi vista em necrópsia de ratos depois de um período de 2 semanas de observação.
Os estudos de toxicidade com doses repetidas nos animais identificaram como principais órgãos-alvo: o fígado, a glândula tireoide, retina e o rim. Alterações hepáticas envolveram elementos hepatocelular, biliar e fagocíticos e foram acompanhados de aumentos nas enzimas hepáticas. Hipertrofia no epitélio pigmentado da retina (RPE) e degeneração da retina foram observados nos roedores nos estudos conduzidos com ritonavir, mas não foram observados em cachorros. Evidências ultraestruturais sugeriram que essas alterações na retina em roedores podem ser secundárias a fosfolipidose. Entretanto, três experiências clínicas de fase II não revelaram evidências claras de alterações na retina por indução do fármaco em humanos. Alterações relacionadas à glândula tireoide incluíram hipertrofia das células foliculares, diminuição dos níveis séricos de tiroxina (T4) e/ou aumento dos níveis séricos de TSH. Todas as alterações tireoidianas foram reversíveis após a descontinuação do fármaco. Investigações clínicas em humanos não revelaram alterações clínicas significantes nos testes das funções da tireoide. Alterações renais, incluindo degeneração tubular, inflamação crônica e proteinúria, foram observadas em ratos e foram atribuídas a doenças espécie-específicas espontâneas. Além disso, não foram observadas alterações renais clinicamente significantes nas experiências clínicas.
4. CONTRAINDICAÇÕES
NORVIR® (ritonavir) é contraindicado a pacientes com conhecida hipersensibilidade ao ritonavir ou a quaisquer componentes da fórmula.
Quando ritonavir for coadministrado com outro inibidor de protease, o médico deve verificar as informações completas de prescrição destes inibidores de protease, inclusive suas contraindicações.
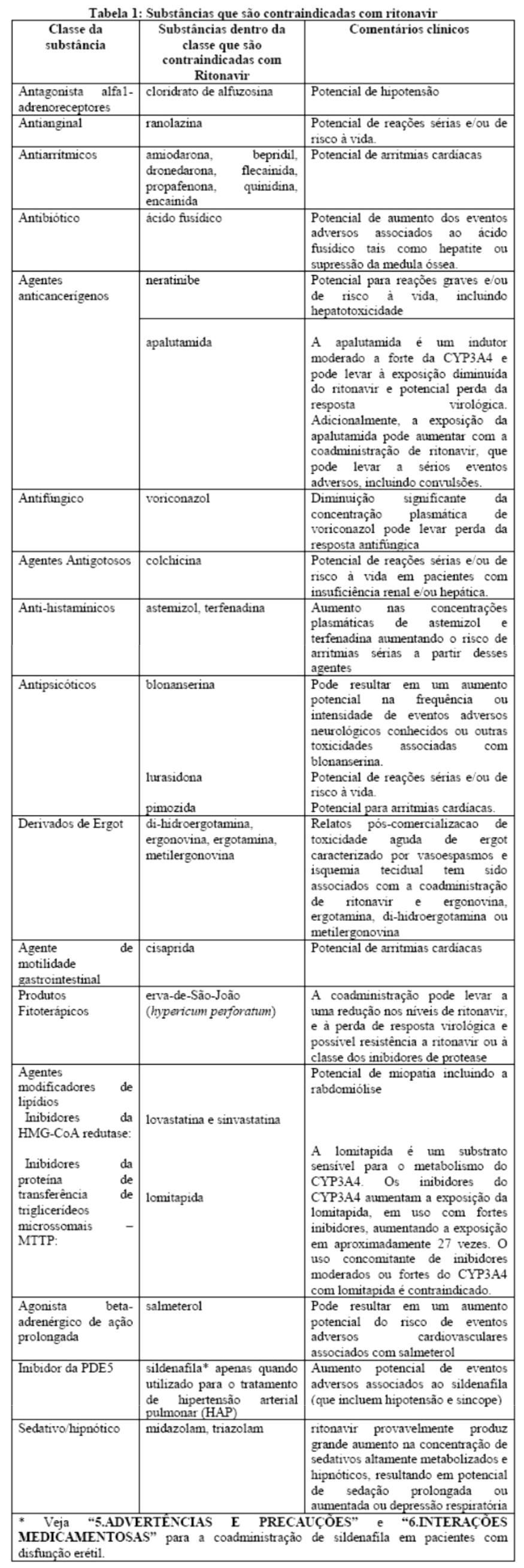
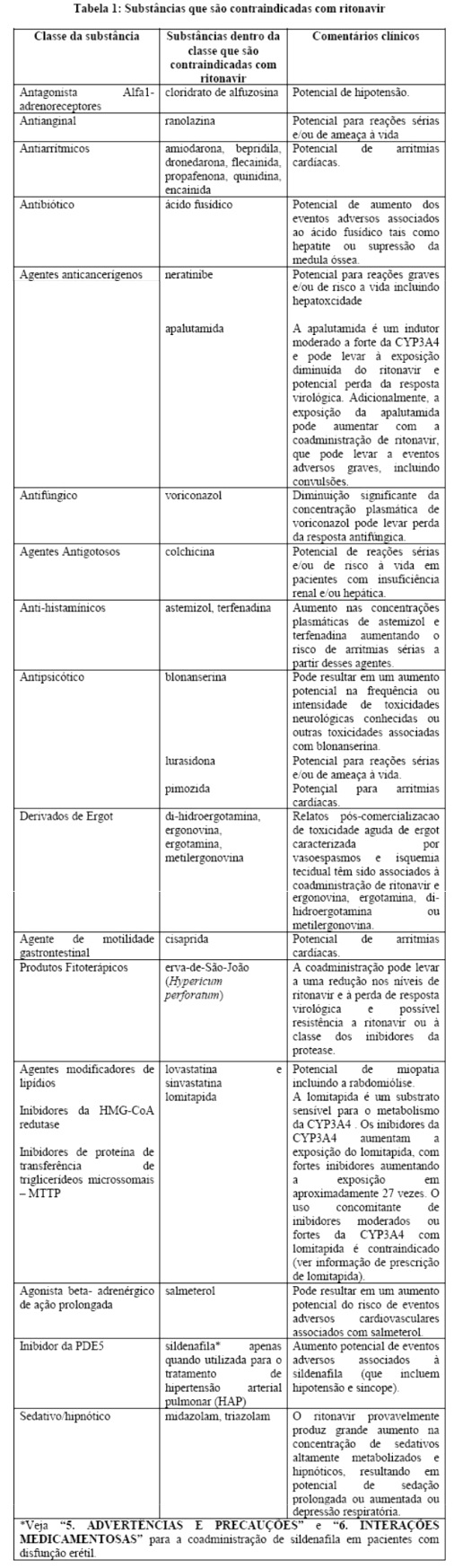
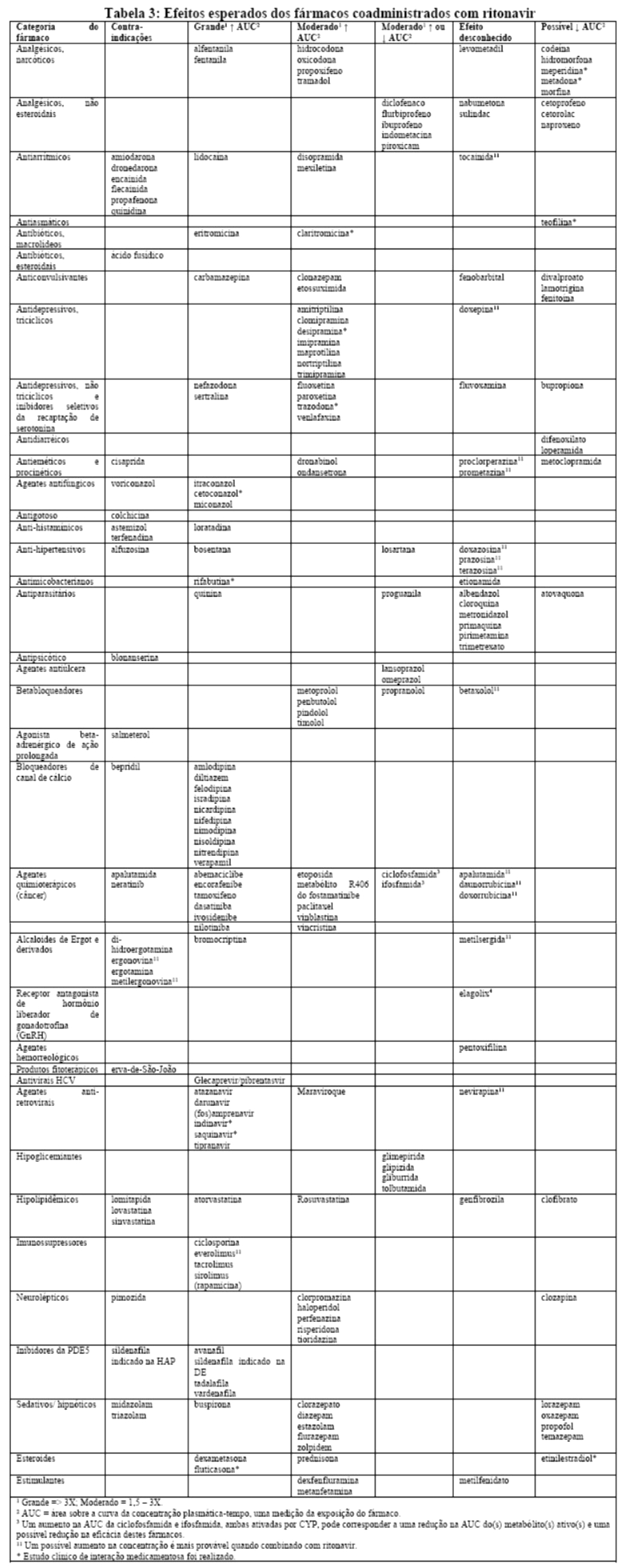
Estudos in vitro demonstraram que ritonavir é um potente inibidor de várias biotransformações mediadas pelo citocromo P450. Baseado principalmente em revisões da literatura presume-se que ritonavir produza importantes aumentos nas concentrações séricas das substâncias metabolizadas pelo citocromo P450. A coadministração de NORVIR® (ritonavir) é contraindicada com as substâncias listadas na Tabela 1:
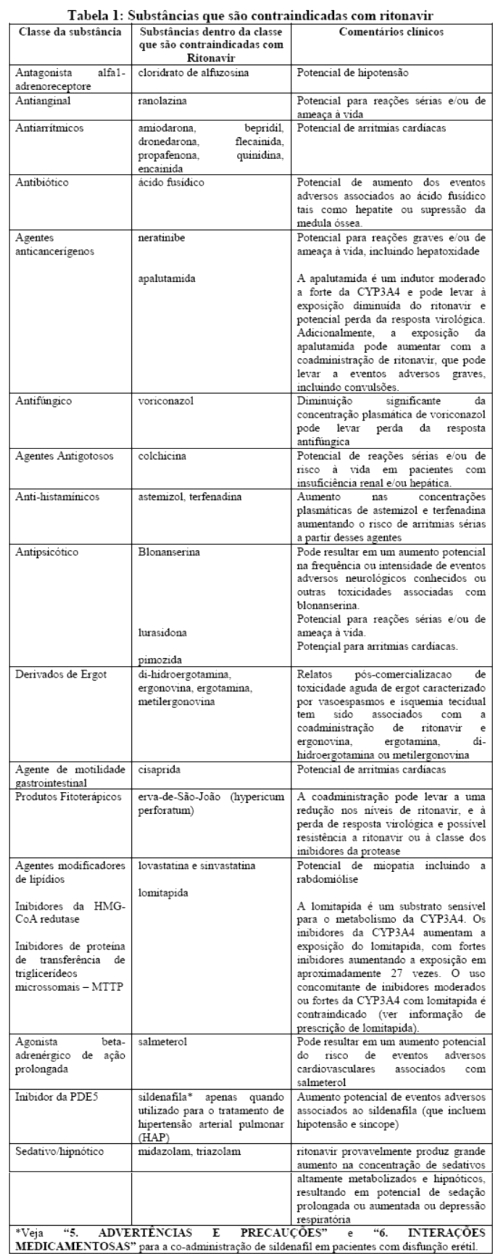
5. ADVERTÊNCIAS E PRECAUÇÕES
Quando ritonavir for coadministrado com outro inibidor de protease, o médico deve verificar as informações completas de prescrição destes inibidores de protease, inclusive suas advertências e precauções.
Reações alérgicas: foram relatadas reações alérgicas, incluindo urticária, erupções de pele, broncoespasmo, angioedema e, raramente, anafilaxia e Síndrome de Stevens-Johnson.
Reações hepáticas: o ritonavir é metabolizado e eliminado principalmente pelo fígado. Portanto, deve-se ter cautela ao administrar ritonavir a pacientes com insuficiência hepática moderada a grave. Elevações de transaminases hepáticas excedendo cinco vezes o limite superior de normalidade, hepatite clínica e icterícia ocorreram em pacientes recebendo ritonavir isoladamente ou em combinação com outros medicamentos antirretrovirais. Pode haver um risco aumentado de elevação de transaminases em pacientes com hepatite B ou C subjacente. Portanto, deve-se ter cautela quando se administra ritonavir a pacientes com doença hepática preexistente, alterações em enzimas hepáticas ou hepatite.
Houve relatos pós-comercialização de disfunção hepática, incluindo alguns óbitos. Esses casos geralmente ocorreram em pacientes tomando múltiplos medicamentos concomitantes e/ou com AIDS avançada. Uma relação causa/efeito definitiva não foi estabelecida.
Pancreatite: pancreatite foi observada em pacientes em uso de ritonavir, incluindo aqueles que desenvolveram hipertrigliceridemia. Alguns casos fatais foram relatados. Pacientes com doença avançada pelo HIV podem apresentar risco aumentado de elevação de triglicérides e pancreatite.
Pancreatite deve ser considerada se ocorrerem sinais clínicos (náusea e vômitos, dor abdominal) ou alterações laboratoriais (como valores aumentados de lipase ou amilase) sugestivos de pancreatite. Pacientes que apresentem estes sinais ou sintomas devem ser avaliados e o tratamento com ritonavir descontinuado se houver diagnóstico de pancreatite.
Diabetes mellitus /hiperglicemia: foram relatados novo aparecimento de diabetes mellitus, exacerbação de diabetes mellitus pré-existente e hiperglicemia durante a farmacovigilância de pós-comercialização em pacientes infectados por HIV que receberam tratamento com inibidores da protease. Alguns pacientes necessitaram iniciar ou ajustar as doses de insulina ou hipoglicemiantes orais para o tratamento destes eventos. Em alguns casos ocorreu cetoacidose diabética. Nos pacientes que descontinuaram o tratamento com inibidores da protease, a hiperglicemia persistiu em alguns casos. Como estes eventos foram relatados espontaneamente durante a prática clínica, não se pôde estimar a sua frequência, e uma relação causal entre o tratamento com inibidores da protease e estes eventos não foi estabelecida. Deve-se considerar a monitoração da glicemia.
Interações medicamentosas:
Agentes antigotosos: interações medicamentosas fatais e de risco à vida foram reportadas em pacientes tratados com colchicina e inibidores fortes da CYP3A como o ritonavir (veja "4. CONTRAINDICAÇÕES" e "6.INTERAÇÕES MEDICAMENTOSAS").
Antipsicóticos: deve-se ter cautela no uso concomitante de NORVIR® (ritonavir) e quetiapina. Devido à inibição da enzima CYP3A por ritonavir, espera-se um aumento das concentrações de quetiapina, podendo levar a efeitos tóxicos relacionados a este antipsicótico (veja "6. INTERAÇÕES MEDICAMENTOSAS").
Corticosteroides: o uso concomitante de ritonavir e fluticasona (inalatória, injetável, ou intranasal), budesonida, triancinolona, ou outro glicocorticoide que é metabolizado pela enzima CYP3A4 não é recomendado a menos que os benefícios potenciais do tratamento sobreponham os riscos dos efeitos sistêmicos dos corticosteroides, incluindo Síndrome de Cushing e supressão adrenal. O uso concomitante de ritonavir e propionato de fluticasona pode aumentar significativamente a concentração de propionato de fluticasona no plasma e reduzir a concentração sérica de cortisol. Efeitos sistêmicos dos corticosteroides, incluindo Síndrome de Cushing e supressão adrenal, foram relatados quando ritonavir foi administrado concomitantemente com propionato de fluticasona, budesonida, ou triancinolona injetável.
Inibidores da PDE5: a administração de ritonavir e avanafil não é recomendada. Atenção especial deve ser dada quando sildenafila, tadalafila ou vardenafila forem prescritos para o tratamento da disfunção erétil em pacientes que estejam recebendo ritonavir. Presume-se que a administração concomitante de ritonavir e de tais drogas aumente substancialmente suas concentrações e possa resultar num aumento dos eventos adversos, tais como hipotensão e ereção prolongada. O uso concomitante de ritonavir e sildenafila é contraindicado em pacientes com hipertensão arterial pulmonar (veja "4. CONTRAINDICAÇÕES" e "6. INTERAÇÕES MEDICAMENTOSAS").
Produtos fitoterápicos: pacientes utilizando ritonavir não devem usar produtos contendo erva-de-São-João (Hypericum perforatum), pois a administração concomitante pode reduzir as concentrações plasmáticas de ritonavir. Isto pode resultar em perda do efeito terapêutico e desenvolvimento de resistência (veja "4. CONTRAINDICAÇÕES" e "6. INTERAÇÕES MEDICAMENTOSAS").
Inibidores da HMG-CoA redutase: os inibidores da HMG-CoA redutase sinvastatina e lovastatina são altamente dependentes do CYP3A para seu metabolismo, de modo que o uso concomitante de ritonavir e sinvastatina ou lovastatina é contraindicado devido ao risco aumentado de miopatia, incluindo rabdomiólise (veja "4. CONTRAINDICAÇÕES"). Recomenda-se também cautela e redução das doses se ritonavir for administrado concomitantemente com atorvastatina, metabolizada em menor extensão pelo CYP3A4. Mesmo considerando que a eliminação de rosuvastatina não é dependente do CYP3A, uma elevação da exposição de rosuvastatina foi relatada com o uso concomitante com ritonavir. Se o tratamento com um inibidor da HMG-CoA redutase for indicado, recomenda-se utilizar a pravastatina ou fluvastatina.
Antagonistas alfa1-adrenoreceptores: com base em estudos de interação medicamentosa com cetoconazol, outro potente inibidor do CYP3A4, e alfuzosina, um aumento significativo de alfuzosina ocorreu quando da administração de ritonavir (600mg duas vezes ao dia). Portanto, alfuzosina não deve ser coadministrado com ritonavir.
Antimicobacterianos: saquinavir e ritonavir, não devem ser administrados concomitantemente com rifampicina, devido ao risco de hepatotoxicidade grave (aumento das transaminases hepáticas) se as três substâncias forem administradas concomitantemente.
A coadministração de bedaquilina com forte inibidor da CYP3A4 pode aumentar a exposição sistêmica da bedaquilina, que pode potencialmente aumentar o risco de reações adversas relacionadas à bedaquilina. A bedaquilina deve ser usada cautelosamente com NORVIR® (ritonavir), somente se o benefício da coadministração for superior aos riscos.
A coadministração de delamanide com um potente inibidor da CYP3A (ritonavir) pode aumentar ligeiramente a exposição ao metabólito delamanide, que tem sido associada com o prolongamento do intervalo QTc. Portanto, se a coadministração de delamanide com ritonavir é considerada necessária, é recomendada a monitoramento frequente por ECG durante todo o período de tratamento com delamanide.
Inibidor de protease: tipranavir coadministrado com 200 mg de ritonavir foi associado com relatos de hepatite clínica e descompensação hepática, incluindo algumas fatalidades. É necessária vigilância extra em pacientes com hepatite B crônica ou coinfecção por hepatite C, já que esses pacientes tem um risco aumentado de hepatotoxicidade.
Resistência/Resistência cruzada: o potencial de resistência cruzada entre inibidores de protease não foi completamente explorado. Portanto, não se sabe qual será o efeito do tratamento com ritonavir sobre a atividade de inibidores de protease administrados concomitantemente ou subsequentemente.
Exames laboratoriais: ritonavir foi associado a alterações de triglicérides, colesterol, transaminases (AST e ALT), GGT, CPK e ácido úrico. Recomenda-se realizar testes laboratoriais adequados antes do início do tratamento com ritonavir e periodicamente durante o tratamento, ou na presença de sinais ou sintomas clínicos.
Hemofilia: foram relatados sangramentos aumentados, incluindo hematomas espontâneos de pele e hemartrose, em pacientes com hemofilia tipo A e B, tratados com inibidores de protease. Em alguns casos foi administrado fator VIII adicional. Em mais da metade dos casos relatados, o tratamento com inibidores da protease foi continuado ou reiniciado. Uma relação causal foi postulada, embora não tenha sido estabelecido um mecanismo de ação.
Prolongamento do intervalo PR: anormalidades do sistema de condução pré-existente ou pacientes recebendo medicamentos conhecidos por prolongarem o intervalo PR (como verapamil ou atazanavir) foram reportados em pacientes ritonavir mostrou causar discreto e assintomático prolongamento do intervalo PR em alguns pacientes. Raros casos de bloqueio AV de segundo ou terceiro graus em pacientes com insuficiência cardíaca estrutural subjacente e recebendo ritonavir. Ritonavir deve ser utilizado com cautela nestes pacientes.
Redistribuição de gordura: a redistribuição/acúmulo de gordura corporal incluindo obesidade centrípeta, depósito de gordura dorsocervical (corcunda de búfalo), emagrecimento periférico, perda de gordura na face, aumento das mamas, e aparência cushingoide foram observados em pacientes recebendo terapia antirretroviral. O mecanismo e as consequências destas alterações por longo prazo são ainda desconhecidos. Não foi estabelecida uma relação causal.
Alterações lipídicas: o tratamento com ritonavir isoladamente ou em combinação com saquinavir resultou em aumentos substanciais na concentração de triglicérides e colesterol. Dosagens de triglicérides e colesterol devem ser realizadas antes do início e a intervalos periódicos durante o tratamento com ritonavir. Alterações lipídicas devem ser controladas de forma clinicamente apropriada.
Síndrome da Reconstituição Imunológica: tal síndrome foi relatada em pacientes infectados por HIV tratados com terapia antirretroviral combinada, incluindo ritonavir. Durante a fase inicial da terapia antirretroviral combinada, quando o sistema imunológico reage, pacientes podem desenvolver uma resposta inflamatória a infecções assintomáticas ou a infecções oportunistas latentes (como infecção causada por Mycobacterium avium, citomegalovírus, pneumonia causada por Pneumocystis jiroveci, ou tuberculose), que podem necessitar de avaliação e tratamentos adicionais.
Alterações autoimunes (como Doença de Graves, polimiosite e Síndrome de Guillain-Barré) também foram reportadas durante a fase de reconstituição imunológica, no entanto, o tempo de início é muito variável e pode ocorrer muitos meses após o início do tratamento.
Carcinogênese e mutagênese
O ritonavir não foi mutagênico ou clastogênico nos estudos in vitro e in vivo que incluíram ensaios de mutação reversa Ames usando S. typhimurium e E. coli, ensaios com linfoma de camundongos, teste de micronúcleo em camundongos e ensaio de aberração cromossômica em linfócitos humanos. Além disso, estudos de carcinogenicidade em ratos e camundongos indicaram que o ritonavir não tem ação carcinogênica direta nas dosagens testadas. Uma incidência maior de adenoma hepatocelular foi encontrada em camundongos machos recebendo alta dose de 200 mg/kg/dia. Tais respostas tumorais em fígado de camundongos associadas a compostos não-genotóxicos são consideradas de pequena relevância para a resposta do fígado humano.
Atenção: o uso incorreto causa resistência do vírus da AIDS e falha no tratamento.
Uso em idosos: Não há recomendações específicas para o uso de NORVIR® (ritonavir) em idosos.
Uso pediátrico: em pacientes com idade entre 01 mês e 21 anos, infectados por HIV, a atividade antiviral e o perfil de eventos adversos observados durante estudos clínicos e a experiência pós-comercialização foram similares aos de pacientes adultos.
Uso na gravidez, fertilidade e reprodução: ritonavir não teve efeitos sobre a fertilidade em ratos machos que receberam doses orais de até 125 mg/kg/dia (exposição média sérica de 61 mcg.h/mL) ou em ratas que receberam até 75 mg/kg/dia (91 mcg.h/mL). Não foi possível administrar doses superiores devido à toxicidade hepática.
Em ratos e coelhos, não foram observadas malformações associadas ao tratamento com ritonavir. A toxicidade observada no crescimento dos ratos (reabsorções fetais, diminuição do peso fetal, atraso da ossificação e variações no desenvolvimento) ocorreu com uma dose tóxica materna de 75 mg/kg/dia (exposição média sérica de 45 mcg.h/mL). Um aumento discreto na incidência de criptorquidia foi observado em ratos que receberam 35 mg/kg/dia (34 mcg.h/mL). A toxicidade do crescimento observada em coelhos (reabsorções, diminuição do tamanho da ninhada, diminuição do peso fetal) ocorreu com uma dose tóxica materna de 110 mg/kg/dia.
Não existem ensaios adequados e bem controlados em mulheres grávidas.
Com base em relatórios prospectivos dos Registros de Gravidez em uso de Antirretroviral (APR) de aproximadamente 6.100 nascidos vivos, após a exposição a regimes contendo ritonavir (incluindo os mais de 2.800 nascidos vivos expostos no primeiro trimestre e os mais de 3.200 nascidos vivos expostos no segundo e terceiro trimestres), não houve diferença na taxa de malformação congênita total para ritonavir em comparação com a taxa de malformação de 2,7% na população de referência dos EUA do MACDP. A prevalência de malformações congênitas em nascidos vivos foi de 2,3% (IC 95%: 1,7% - 2,9%) após a exposição aos regimes contendo ritonavir no primeiro trimestre e de 2,9% (IC 95%: 2,3% - 3,5%) após a exposição aos regimes contendo ritonavir no segundo e terceiro trimestres.
Considerando que os estudos de reprodução animal nem sempre podem predizer a resposta humana, este medicamento somente deve ser usado durante a gravidez quando, na opinião do médico, os benefícios potenciais claramente justificarem os possíveis riscos.
Categoria de risco: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação: Dados limitados e publicados reportam que ritonavir está presente no leite humano.
Não há informação sobre os efeitos de ritonavir em recém-nascidos em fase de amamentação ou sobre os efeitos do ativo sobre a produção de leite. Devido ao potencial para: 1) transmissão do HIV (em recém-nascidos HIV-negativos), 2) desenvolvimento da resistência viral (em recém-nascidos HIV-positivos) e 3) reações adversas graves em recém-nascidos amamentados, a instrução é não realizar a amamentação durante o uso de ritonavir.
6. INTERAÇÕES MEDICAMENTOSAS
Quando ritonavir for coadministrado com outro inibidor de protease, o médico deve verificar as informações completas de prescrição destes inibidores de protease, inclusive suas interações medicamentosas.
Estes exemplos são um guia e não são considerados uma lista abrangente de todos os possíveis medicamentos que podem interagir com NORVIR® (ritonavir). O profissional de saúde deve consultar referências apropriadas para informações abrangentes.
Efeitos sobre o ritonavir: presume-se que medicamentos que aumentam a atividade da CYP3A (por ex.: fenobarbital, carbamazepina, dexametasona, fenitoína, rifampicina e rifabutina) aumentem a depuração de ritonavir, reduzindo, consequentemente, as concentrações séricas deste. O uso de tabaco é associado a uma redução de 18% na AUC do ritonavir.
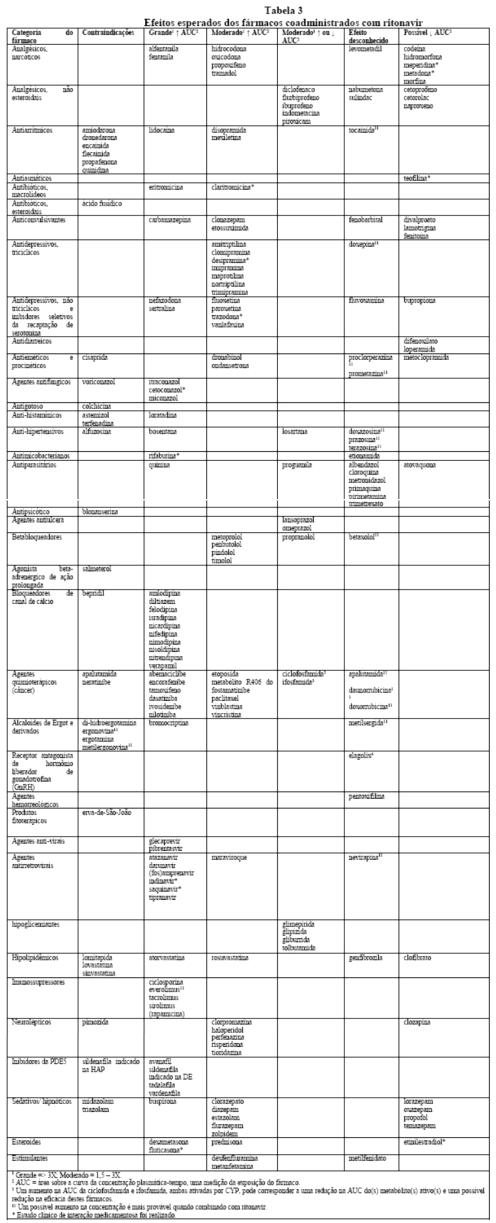
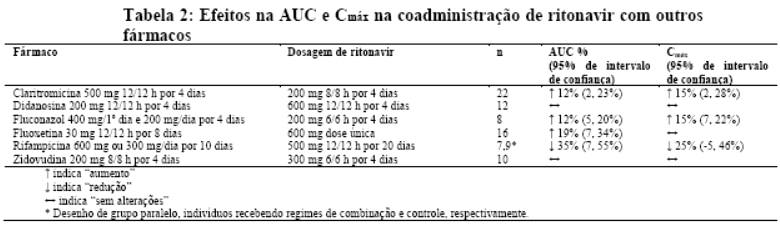
Efeitos sobre medicamentos coadministrados: ritonavir tem uma alta afinidade por várias isoformas do citocromo P450 (CYP) na seguinte ordem: CYP3A4 > CYP2D6 > CYP2C9 > CYP2C19 > > CYP2A6, CYP1A2, CYP2E1. Há evidências de que ritonavir possa induzir as enzimas glicuronil-transferase, CYP1A2, CYP2C9 e CYP2C19; assim, a redução da concentração plasmática da outra substância e a perda do efeito terapêutico durante o tratamento concomitante com ritonavir pode requerer ajuste de doses desses agentes. Além dos medicamentos citados em CONTRAINDICAÇÕES, na Tabela 1 constam alguns medicamentos comumente prescritos que possuem uma magnitude prevista de interação se administrados com o ritonavir.
A coadministração de ritonavir e substâncias metabolizadas principalmente por CYP3A pode resultar no aumento da concentração plasmática da outra substância, o que pode aumentar ou prolongar seus eventos adversos. O monitoramento cuidadoso dos efeitos adversos é recomendado quando estas substâncias forem administradas concomitantemente ao ritonavir. A redução de dose pode ser necessária para os agentes extensivamente metabolizados por CYP3A.
Interações com importantes considerações
bedaquilina: em um estudo de interação medicamentosa com pacientes saudáveis para os quais foram administradas uma dose única de 400 mg de bedaquilina e 400/100 mg de NORVIR® (ritonavir) duas vezes por dia por 24 dias, a exposição de bedaquilina (AUC) foi aumentada em 22%. A bedaquilina deve ser usada cautelosamente com NORVIR® (ritonavir), ou seja, somente se o benefício da coadministração for superior ao risco.
corticosteroides: o uso concomitante de ritonavir e fluticasona ou outro glicocorticoide que é metabolizado pela CYP3A4 não é recomendado a menos que os benefícios potenciais do tratamento sobreponham os riscos dos efeitos sistêmicos dos corticoides, incluindo Sindrome de Cushing e supressão adrenal. Considerar drogas alternativas ao propionato de fluticasona, budesonida, e triancinolona injetável, particularmente quando o uso for prolongado (veja "5. ADVERTÊNCIAS E PRECAUÇÕES").
erva-de-São-João (Hypericum perforatum): pacientes utilizando ritonavir não devem usar produtos contendo erva-de-São-João (Hypericum perforatum), pois a administração concomitante pode reduzir as concentrações plasmáticas de ritonavir. Este efeito pode ser devido à indução da CYP3A4 e pode resultar em perda do efeito terapêutico e desenvolvimento de resistência.
saquinavir/ritonavir + rifampicina: saquinavir e ritonavir não devem ser administrados concomitantemente com rifampicina devido ao risco de hepatotoxicidade grave (apresentado como aumento das transaminases) se os três medicamentos forem administrados concomitantemente.
simeprevir: um estudo de farmacocinética demonstrou que a administração concomitante de 200 mg de simeprevir uma vez ao dia com 100 mg de ritonavir duas vezes ao dia resultou em um aumento na concentração de simeprevir. Não é recomendada a coadministração de ritonavir e simeprevir.
sildenafila: o uso concomitante de sildenafila com ritonavir é contraindicado em pacientes com hipertensão arterial pulmonar.
voriconazol: um estudo mostrou que a coadministração de 400 mg de ritonavir a cada 12 horas diminuiu a AUC no estado de equilíbrio de voriconazol em uma média de 82%; assim, a coadministração dessa droga é contraindicada.
lomitapida: a lomitapida é um substrato sensível para o metabolismo do CYP3A4. Os inibidores do CYP3A4 aumentam a exposição do lomitapida, em uso com fortes inibidores aumentando a exposição aproximadamente 27 vezes. O uso concomitante de inibidores moderados ou fortes do CYP3A4 com lomitapida é contraindicado.
Interações com recomendações de alteração de dose e monitoramento
Eventos cardíacos e neurológicos foram reportados quando ritonavir foi coadministrado a disopiramida, mexiletina, nefazodona ou fluoxetina. A possibilidade de interação medicamentosa não deve ser excluída.
bosentana: a coadministração de bosentana e ritonavir pode aumentar a Cmáx e a AUC da bosentana. Consulte as informações da bula de bosentana.
buspirona: é metabolizada principalmente pelo CYP3A4. A administração concomitante de buspirona associada a outras drogas que possuam potencial inibidor ao CYP3A, como o ritonavir, produz aumento substancial nos níveis de buspirona. Quando coadministrada com ritonavir, é recomendado que se use com cautela uma dose baixa ou que se faça uma redução da dosagem.
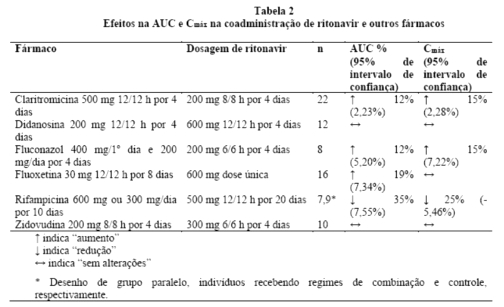
claritromicina: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 200 mg a cada 8 horas e claritromicina 500 mg a cada 12 horas resultou em inibição marcante do metabolismo da claritromicina. A Cmáx da claritromicina aumentou em 31%, a Cmín aumentou em 182% e a AUC aumentou em 77% com a administração concomitante de ritonavir. Uma inibição completa da formação de 14-[R]-hidróxi-claritromicina foi observada. Devido a uma janela terapêutica grande para a claritromicina, não é necessária uma redução na dosagem em pacientes com função renal normal. Entretanto, para pacientes com função renal comprometida (N19), os seguintes ajustes de dosagem devem ser considerados: para pacientes com depuração da creatinina entre 30 e 60 mL/min, a dose de claritromicina deve ser
reduzida em 50%; para pacientes com depuração da creatinina < 30 mL/min, reduzir a dose de claritromicina em 75%. Doses de claritromicina superiores a 1 g diário não devem ser administradas com ritonavir.
colchicina: é esperado um aumento das concentrações de colchicina quando coadministrado com ritonavir. Interações medicamentosas de risco à vida e fatais foram relatadas em pacientes tratados com colchicina e ritonavir (veja "4. CONTRAINDICAÇÕES" e "5. ADVERTÊNCIAS E PRECAUÇÕES"). Consulte as informações de prescrição de colchicina.
delamanide: não há estudo disponível de interação somente com ritonavir. Em um estudo de interação medicamentosa com voluntários saudáveis administrou-se delamanide 100 mg duas vezes ao dia e lopinavir/ritonavir 400 mg/100 mg duas vezes ao dia por 14 dias, as exposições de delamanide e um metabólito delamanide, DM-6705, aumentaram ligeiramente.
Caso a coadministração de delamanide com ritonavir for considerada necessária, devido ao risco de prolongamento do QTc associada ao DM-6705, recomenda-se a monitoramento frequente por ECG durante todo o período de tratamento com delamanide.
delavirdina: é um inibidor do metabolismo mediado por CYP3A. Em um estudo publicado, a administração de doses clínicas de delavirdina 400 mg três vezes ao dia e ritonavir 600 mg duas vezes ao dia (n = 12 pacientes infectados pelo HIV) levou a um aumento da Cmax e da AUC no estado de equilíbrio de ritonavir de aproximadamente 50% e da Cmin em torno de 75%. Com base na comparação de dados históricos, a farmacocinética de delavirdina não parece ser afetada pelo ritonavir. Quando usado juntamente com delavirdina, pode-se considerar a redução na dose de ritonavir.
desipramina: a administração concomitante de ritonavir 500 mg a cada 12 horas e uma dose única de desipramina 100 mg resultou em aumento médio de 145% na AUC da desipramina. Redução na dosagem de desipramina deve ser considerada em pacientes recebendo esta combinação.
didanosina (ddl): um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 600 mg a cada 12 horas e didanosina 200 mg a cada 12 horas resultou em um decréscimo da Cmáx e AUC da didanosina no estado de equilíbrio de 16% e 13%, respectivamente. Por outro lado, foi observado pouco ou nenhum efeito na farmacocinética do ritonavir. Não é necessário ajuste de dosagem de ddl, contudo, os dois medicamentos devem ser administrados separadamente, com 2,5 horas de intervalo, para evitar incompatibilidade das formulações.
digoxina: um relato na literatura demonstrou que a coadministração de ritonavir (300 mg a cada 12 horas) e digoxina resultou em um aumento significativo dos níveis de digoxina. Atenção especial deve ser dada quando digoxina e ritonavir forem administrados concomitantemente, com monitoramento apropriado dos níveis de digoxina sérica.elagolix: a coadministração de elagolix com ritonavir poderia aumentar a exposição do elagolix devido a inibição da CYP3A e da P-gp. Eventos adversos graves conhecidos para elagolix incluem ideação suicida e elevações na transaminase hepática. Adicionalmente, elagolix é um indutor fraco/moderado da CYP3A, o que pode diminuir a exposição do ritonavir. Consulte a bula de elagolix para informações sobre a posologia com fortes inibidores da CYP-3A4.
fentanila: ritonavir inibe o citocromo CYP3A4 resultando em aumento esperado das concentrações plasmáticas de fentanila. Ë recomendado monitoramento cuidadoso da terapêutica e dos eventos adversos (incluindo depressão respiratória) quando fentanila é coadministrada com ritonavir.
glecaprevir/pibrentasvir: A administração concomitante com ritonavir não é recomendada devido a um aumento do risco de elevações da ALT associadas a uma exposição aumentada ao glecaprevir (GLE).
indinavir: o ritonavir inibe o metabolismo do indinavir mediado pelo CYP3A. Em indivíduos saudáveis, a administração de 200 a 400 mg de ritonavir duas vezes ao dia com dose única de 400 a 600 mg de indinavir aumentou a AUC do indinavir de 185% a 475%, a Cmax de 21% a 110%, e a Cmin de 11 a 33 vezes em relação à administração isolada de 400 a 600 mg de indinavir. A administração concomitante de 400 mg de ritonavir e 400 mg de indinavir duas vezes ao dia com uma refeição produziu uma AUC do indinavir similar, um aumento de 4 vezes na Cmín e diminuição de 50 a 60% na Cmáx em comparação com os resultados da administração de indinavir 800 mg três vezes ao dia em condições de jejum. A coadministração do ritonavir com indinavir resultará em aumento das concentrações séricas do indinavir. Os dados disponíveis de segurança ou eficácia desta combinação em pacientes são limitados. O risco de nefrolitíase pode estar aumentado quando doses de indinavir ≥ 800 mg duas vezes ao dia são administradas concomitantemente com ritonavir. Nestas condições, recomenda-se adequada hidratação e monitoramento dos pacientes.
cetoconazol: a administração concomitante de ritonavir (500 mg, a cada 12 horas) e cetoconazol (200 mg por dia) resultou em um aumento na AUC24 e Cmáx de 244% e 55%, respectivamente. A meia-vida do cetoconazol aumentou de 2,7 para 13,2 horas. A AUC24 e Cmáx do ritonavir aumentaram em 18 e 10%, respectivamente. Não há necessidade de ajuste de dosagem do ritonavir; entretanto, doses de cetoconazol de 200 mg/dia ou mais em combinação com ritonavir devem ser usadas com cautela e uma redução de dosagem pode ser considerada.
maraviroque: a administração concomitante de maraviroque e ritonavir aumenta os níveis plasmáticos de maraviroque. A dose de maraviroque deve ser reduzida durante a coadministração com ritonavir. Para mais detalhes consulte as informações completas na bula de maraviroque.
metadona: a administração concomitante de ritonavir com metadona resultou em diminuição das concentrações de metadona. Um aumento na dosagem de metadona pode ser considerado.
contraceptivos orais e adesivos: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 h e uma combinação fixa de contraceptivo oral resultou em reduções da Cmáx e AUC médias de 32% e 40% de etinilestradiol, respectivamente. Aumento da dosagem de contraceptivos orais e adesivos contendo etinilestradiol ou substituição por métodos alternativos de contracepção devem ser considerados.
quetiapina: devido à inibição da enzima CYP3A por ritonavir, espera-se um aumento das concentrações de quetiapina. Para instruções de dose de quetiapina, consultar suas informações de prescrição (veja "5. ADVERTÊNCIAS E PRECAUÇÕES").
rifabutina: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 horas e rifabutina resultou em um aumento de aproximadamente 4 vezes na AUC de rifabutina e 35 vezes na AUC do seu metabólito ativo 25-O-desacetil rifabutina. A significância desta interação foi confirmada em estudos clínicos.
Uma redução na dosagem de rifabutina de pelo menos três quartos (3/4) da dose usual de 300 mg/dia é recomendada (ex.: 150 mg em dias alternados ou três vezes por semana). Uma redução adicional na dosagem também pode ser necessária.
rivaroxabana: a coadministração de ritonavir e rivaroxabana resultou em um aumento da exposição de rivaroxabana podendo aumentar o risco de sangramentos.
avanafil: um estudo farmacocinético demonstrou que a administração concomitante de 50 mg de avanafil e 600 mg de ritonavir a cada 12 horas resultou em um aumento de aproximadamente 13 vezes e 2,4 vezes nas AUCinf e Cmax de avanafil respectivamente. A coadministração de ritonavir com avanafil não é recomendada.
sildenafila: para o tratamento da disfunção erétil, use a sildenafila com atenção em doses reduzidas de 25 mg a cada 48 horas, com monitoramento dos efeitos adversos. Espera-se que a coadministração de ritonavir e sildenafila aumente substancialmente as concentrações de sildenafila (aumento de 11 vezes na AUC) e possa resultar em aumento dos eventos adversos associados à sildenafila, incluindo hipotensão, síncope, alterações visuais e ereção prolongada.
tadalafila: usar tadalafila, para o tratamento de disfunção erétil, com atenção, em doses reduzidas de, no máximo, 10 mg a cada 72 horas, com monitoramento dos efeitos adversos (veja "5. ADVERTÊNCIAS E PRECAUÇÕES"). Consulte as informações da bula de tadalafina quando esta for coadministrada com ritonavir em pacientes com hipertensão arterial pulmonar.
teofilina: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 horas e teofilina resultou em decréscimo de 43% na AUC da teofilina. Aumento da dosagem de teofilina pode ser necessário.
trazodona: o uso concomitante de ritonavir e trazodona pode aumentar a concentração de trazodona. Efeitos adversos como náuseas, vertigens, hipotensão e síncope foram observados. Se trazodona for prescrita com um inibidor de CYP3A4, como ritonavir, a combinação deve ser usada com atenção e uma dose menor de trazodona pode ser considerada.
vardenafila: usar vardenafila com atenção, em doses reduzidas de no máximo 2,5 mg a cada 72 horas, com monitoramento dos efeitos adversos.
varfarina: em um estudo farmacocinético, múltiplas doses de ritonavir (400 mg duas vezes ao dia) afetaram de modos diferentes as farmacocinéticas de dose única dos enantiômeros da varfarina. A AUC da S-varfarina foi afetada de modo variável pelo ritonavir, mas não foi estatisticamente significante. A AUC da menos potente R-varfarina foi reduzida em média 33% durante a coadministração de ritonavir. A gama de efeitos da coadministração do ritonavir sobre a ação anticoagulante da varfarina é difícil de ser prevista com base nesses resultados farmacocinéticos. Recomenda-se controle inicial frequente da Razão Normalizada Internacional (INR) durante a coadministração de ritonavir e varfarina.
agentes anticancerígenos (abemaciclibe, apalutamida, dasatinibe, encorafenibe, ibrutinibe, ivosidenibe, neratinibe, nilotinibe, venetoclax, vincristina, vimblastina): as concentrações séricas podem aumentar quando houver coadministração com ritonavir, resultando em um possível aumento na incidência de eventos adversos, dentre alguns que podem ser graves. A co-administração de venetoclax ou ibrutinibe e NORVIR® (ritonavir) poderia aumentar potencialmente a exposição à venetoclax ou ibrutinibe, resultando em um sério risco de Síndrome da Lise Tumoral. A coadministração de encorafenibe ou ivosidenibe com NORVIR® (ritonavir) poderia aumentar a exposição de encorafenibe ou ivosidenibe, aumentando potencialmente o risco de eventos adversos graves, como o prolongamento do intervalo QT. O uso concomitante de NORVIR® (ritonavir) com apalutamida é contraindicado (veja "4. CONTRAINDICAÇÕES").
Outras interações medicamentosas
alprazolam: a coadministração de alprazolam e ritonavir resultou em uma redução nos valores da Cmáx média de alprazolam (16%), mas não nos valores médios da AUC (12%). Similarmente, foi observado um efeito na curva do efeito sedativo, mas não na extensão da sedação. Discreta depressão psicomotora foi confundida com um efeito de aprendizado. Estes resultados farmacocinéticos e farmacodinâmicos são inconsistentes com o efeito farmacológico do alprazolam.
Estes resultados não foram considerados clinicamente significantes.
amprenavir: há dados na literatura mostrando que as concentrações do inibidor de protease do HIV amprenavir são aumentadas quando coadministrado com ritonavir.
bupropiona: bupropiona é originalmente metabolizada por CYP2B6. É esperado que a administração concomitante de bupropiona com doses repetidas de ritonavir reduza os níveis de bupropiona.
efavirenz: em indivíduos saudáveis recebendo 500 mg de ritonavir duas vezes ao dia concomitante com 600 mg de efavirenz uma vez ao dia, a AUC de efavirenz no estado de equilíbrio aumentou 21%. Foi observado um aumento concomitante na AUC de ritonavir em 17%.
ácido fusídico: presume-se que a administração concomitante de inibidores de proteases, incluindo ritonavir, com ácido fusídico resulte em aumento das concentrações tanto de ácido fusídico como do inibidor de protease no plasma (veja "4. CONTRAINDICAÇÕES").
inibidores de quinase: a coadministração de fostamatinibe com ritonavir poderia aumentar a exposição do metabólito R406 do fostamatinibe, resultando em eventos adversos relacionados a dose, como hepatotoxicidade e neutropenia
nelfinavir: as interações entre ritonavir e nelfinavir podem envolver tanto a inibição quanto a indução do sistema citocromo P450. A administração concomitante de 400 mg de ritonavir duas vezes ao dia aumenta significativamente as concentrações do M8 (o principal metabólito do nelfinavir) e resulta em pequenos aumentos nas concentrações do nelfinavir. Em um estudo de dez pacientes, a administração do nelfinavir na dose de 750 mg e ritonavir na dose de 400 mg duas vezes ao dia, resultou em aumentos discretos nos parâmetros do nelfinavir [AUC (160%), Cmax (121%) e Cmin (123%)] em relação aos dados disponíveis sobre a monoterapia com nelfinavir na dose de 750 mg três vezes ao dia. A AUC do M8 aumentou em 347%.
raltegravir: um estudo farmacocinético demonstrou que a coadministração de 100 mg de ritonavir duas vezes ao dia e 400 mg de raltegravir uma vez ao dia resultou em redução mínima de raltegravir C12h m, AUC0-∞ e Cmax em 1%, 16% e 24%, respectivamente.
saquinavir: um estudo farmacocinético demonstrou que ritonavir inibe extensamente o metabolismo do saquinavir resultando em concentrações plasmáticas de saquinavir muito aumentadas. Após aproximadamente 4 semanas de um regime combinado de saquinavir 400 ou 600 mg duas vezes ao dia (cápsulas de gelatina dura) com 400 ou 600 mg de ritonavir duas vezes ao dia, em pacientes infectados pelo HIV, os valores da AUC de saquinavir foram, no mínimo, 17 vezes maiores que os valores de AUC obtidos com saquinavir 600 mg três vezes ao dia sem ritonavir. Quando usados em terapia combinada por até 24 semanas, doses maiores que 400 mg duas vezes ao dia tanto de saquinavir quanto de ritonavir foram associadas com um aumento de eventos adversos. As exposições plasmáticas alcançadas com Inviraseâ (mesilato de saquinavir em cápsula gelatinosa dura, 400 mg duas vezes ao dia) e ritonavir (400 mg duas vezes ao dia) são similares às atingidas com Fortovaseâ (saquinavir cápsula gelatinosa mole, 400 mg duas vezes ao dia) e ritonavir (400 mg duas vezes ao dia).
sulfametoxazol/trimetoprima: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 horas e sulfametoxazol/ trimetoprima resultou em um decréscimo de 20% na AUC do sulfametoxazol e aumento de 20% na AUC da trimetoprima. Pode não ser necessário ajuste de dosagem.
zidovudina (AZT): um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 300 mg a cada 6 horas e zidovudina 200 mg a cada 8 horas resultou em uma diminuição na Cmáx da zidovudina de 27% e na AUC de 25%. Por outro lado, foi observado pouco ou nenhum efeito na farmacocinética do ritonavir. Pode não ser necessário ajuste de dosagem de zidovudina durante terapia concomitante com ritonavir.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (temperatura entre 15 e 30°C) e não refrigerar. É importante manter NORVIR® (ritonavir) no frasco de origem. Não transferir para outra embalagem. Manter o frasco bem fechado.
Prazo de validade: se armazenado nas condições indicadas, o medicamento se manterá próprio para consumo pelo prazo de validade de 24 meses, a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas e organolépticas
NORVIR® (ritonavir) comprimido revestido apresenta-se na forma de comprimidos ovaloides brancos.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Os médicos devem consultar as informações completas de prescrição e estudos clínicos sobre inibidores da protease para verificar se estes são coadministrados com doses reduzidas de ritonavir.
NORVIR® (ritonavir) comprimido revestido deve ser administrado por via oral, com alimentos. Deve ser engolido inteiro, sem mastigar, partir ou esmagar.
A duração do tratamento depende de orientação médica, a partir da avaliação clínica e laboratorial de cada paciente.
Posologia
Adultos
A dose recomendada de NORVIR® (ritonavir) comprimido revestido é de 600 mg (6 comprimidos) duas vezes por dia.
A utilização de um esquema de titulação de doses pode ajudar a reduzir os eventos adversos decorrentes do tratamento, mantendo níveis plasmáticos adequados de ritonavir.
NORVIR® (ritonavir) deve ser iniciado com doses de, no mínimo, 300 mg (3 comprimidos) duas vezes ao dia durante o período de três dias, com incrementos de 100 mg (1 comprimido) duas vezes ao dia, até atingir a dose máxima de 600 mg (6 comprimidos) duas vezes ao dia em um período não superior a 14 dias.
Os pacientes devem ser informados de que os eventos adversos frequentemente observados no início do tratamento, como distúrbios gastrointestinais leves a moderados e parestesia, podem diminuir com a continuidade do tratamento.
Os pacientes não devem ser mantidos com dose de 300 mg (3 comprimidos) duas vezes ao dia por mais de três dias.
A dose máxima diária é de 6 comprimidos de 100 mg duas vezes ao dia, totalizando 1200 mg ao dia.
Esquemas combinados de inibidores de protease
A experiência clínica de terapia combinada incluindo dose terapêutica de ritonavir com outro inibidor de protease é limitado.
O ritonavir inibe extensamente o metabolismo da maioria dos inibidores de protease disponíveis. Portanto, qualquer consideração de tratamento combinado com ritonavir deve levar em consideração a interação farmacocinética e os dados de segurança dos agentes envolvidos. Há extensa resistência cruzada nesta classe de agentes. Deve-se considerar a combinação de dois inibidores de protease com menor padrão superposto de resistência. O uso do ritonavir em tais esquemas deve ser orientado por esses fatores.
Para uso de ritonavir com saquinavir, uma cautelosa titulação de dose é utilizada, iniciando-se a terapia com ritonavir com doses de 300 mg (3 comprimidos) duas vezes ao dia.
Para uso de ritonavir com indinavir, uma cautelosa titulação de dose é utilizada, iniciando-se a terapia com ritonavir com doses de 200 mg (2 comprimidos) duas vezes ao dia, com aumento de 100 mg (1 comprimido) duas vezes ao dia até atingir 400 mg (4 comprimidos) duas vezes ao dia em 2 semanas.
Crianças
NORVIR® (ritonavir) pode ser utilizado por crianças acima de 01 mês de idade capazes de deglutir comprimidos.
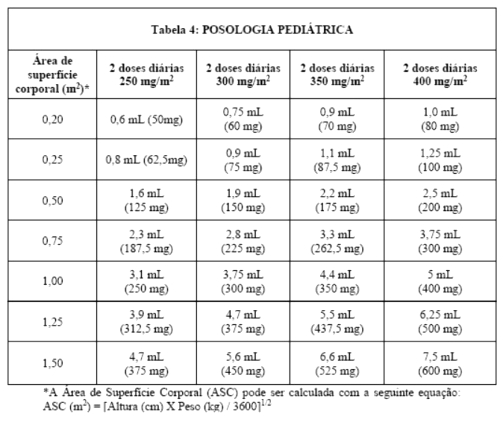
NORVIR® (ritonavir) deve ser utilizado em combinação a outros agentes antirretrovirais. A dosagem recomendada de NORVIR® (ritonavir) em crianças > 01 mês é de 350 a 400 mg/m2, baseando-se na Área de Superfície Corporal, duas vezes ao dia, não excedendo a dose de 600 mg (6 comprimidos) duas vezes ao dia. A dose deve ser iniciada em 250 mg/m2 e aumentada, em intervalos de 2 a 3 dias, em 50 mg/m2 duas vezes ao dia.
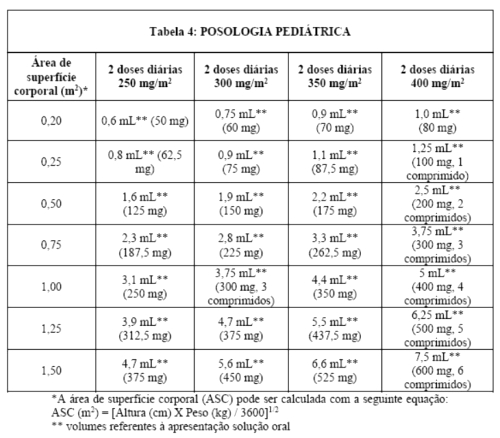
Se o paciente não tolerar a dose máxima diária devido aos eventos adversos, a dose máxima tolerada deve ser utilizada como terapia de manutenção em combinação com outros agentes antirretrovirais.
Este medicamento não deve ser partido, aberto ou mastigado.
9. REAÇÕES ADVERSAS
Quando ritonavir for coadministrado com outro inibidor de protease, o médico deve verificar as informações completas de prescrição destes inibidores de protease, inclusive suas reações adversas.
Adultos
Reação adversa muito comum ( > 1/10):
Alterações do sistema nervoso: disgeusia, cefaleia e parestesia.
Alterações gastrointestinais: diarreia, náusea, parestesia oral e vômito.
Gerais: fadiga.
Reação adversa comum ( > 1/100 e ≤1/10):
Alterações laboratoriais: aumento sanguíneo de triglicérides, teste de função hepática anormal.
Alterações sanguíneas e linfáticas: linfadenopatia.
Alterações do sistema nervoso: distúrbio de atenção, tontura, hiperestesia, hipoestesia, hiporreflexia, neuropatia periférica, sonolência e tremor.
Alterações respiratórias, torácicas e mediastinais: dispneia, tosse, dor orofaríngea, irritação na garganta.
Alterações gastrointestinais: desconforto abdominal, distensão abdominal, dor abdominal, dor abdominal superior, fezes alteradas, constipação, boca seca, dispepsia, eructação, flatulência e hipoestesia oral.
Alterações renais e urinárias: disúria.
Alterações na pele e tecido subcutâneo: hiperidrose, suores noturnos, prurido, rash, rash maculopapular, rash papular e sensação de queimação na pele.
Alterações metabólicas e nutricionais: diminuição do apetite e hipertrigliceridemia, perda de peso.
Alterações musculoesqueléticas e de articulações: artralgia, espasmos musculares e mialgia.
Infestações e infecções: faringite.
Alterações vasculares: rubor, fogachos (ondas de calor).
Gerais: astenia, calafrios, sensação de calor, indisposição, edema periférico, dor e febre.
Alterações psiquiátricas: ansiedade, depressão e insônia.
Reação adversa incomum ( > 1/1000 e ≤1/100):
Alterações laboratoriais: anormalidade de enzimas hepáticas.
Alterações cardíacas: palpitações, taquicardia sinusal e taquicardia, aumento do fluxo cardíaco.
Alterações sanguíneas e linfáticas: anemia, neutropenia e trombocitopenia.
Alterações do sistema nervoso: ageusia, amnésia, alterações de equilíbrio, descoordenação, vertigem postural, hipogeusia, danos mentais, parosmia, pré-síncope, hiperatividade psicomotora, síncope, alteração no campo visual.
Alterações visuais: visão anormal, dor ocular, uveíte, acuracidade visual diminuída, visão turva, prejuízo visual.
Alterações do ouvido e labirinto: desconforto auricular, dor de ouvido, zumbido e vertigem.
Alterações respiratórias, torácicas e mediastinais: garganta seca, soluços, dificuldade de respirar, roncos no peito, alterações respiratórias.
Alterações gastrointestinais: estomatite aftosa, quelite, colite, disfagia, desconforto epigástrico, fezes pálidas, gastrite, hipermotilidade gastrointestinal, sons gastrointestinais anormais, doença do refluxo gastroesofágico, gengivite, glossodinia, hematoquesia, hemorroida, ulcerações na boca, dor esofágica, proctalgia, tentativa de vômito sem êxito e estomatite.
Alterações renais e urinárias: noctúria, polaquiúria e poliúria.
Alterações na pele e tecido subcutâneo: acne, suor frio, pele seca, eczema, eritema, petéquia, reação de fotossensibilidade, rash eritematoso, rash macular, rash pruriginoso, seborreia, esfoliação da pele, irritação da pele, aquecimento da pele e urticária.
Alterações metabólicas e nutricionais: desidratação e diabetes mellitus.
Alterações musculoesqueléticas e de articulações: dor nas costas, dor na costela, rigidez na articulação, inchaço na articulação, espasmo muscular, fraqueza muscular, rigidez musculoesquelética, dor no pescoço e sensação de peso.
Infestações e infecções: foliculite, rinite, sinusite e infecção viral.
Alterações vasculares: frieza periférica.
Alterações do sistema imune: hipersensibilidade.
Gerais: desconforto no peito, dor torácica, desconforto, frio, nervosismo, modo de andar anormal, síndrome gripal, irritabilidade, sensibilidade e sede, aumento da temperatura corpórea.
Alterações hepatobiliares: hepatite, hepatomegalia, hepatotoxicidade e amolecimento do fígado.
Alterações do sistema reprodutivo e mamas: disfunção erétil e alterações penianas.
Alterações psiquiátricas: alterações nos sonhos, agitação, confusão, desorientação, euforia, alucinações, diminuição da libido, nervosismo e alterações de sono.
Danos, envenenamento e complicações de procedimento: contusão e queimadura de sol.
Procedimentos médicos e cirúrgicos: vasodilatação.
Reação adversa rara ( > 1/10000 e ≤1/1000):
Alterações laboratoriais e investigações: diminuição da hemoglobina e exames neurológicos anormais.
Alterações sanguíneas e linfáticas: linfadenite e linfocitose.
Alterações do sistema nervoso: ataxia, alterações cognitivas, convulsão, convulsão do tipo Grande Mal, enxaqueca, contrações musculares involuntárias, neuralgia, paralisia, sono de baixa qualidade e sedação.
Alterações visuais: blefarite, diplopia, irite, fotofobia e escotoma cintilante.
Alterações do ouvido e labirinto: hipoacusia.
Alterações respiratórias, torácicas e mediastinais: broncoespasmo, epistaxe, hipoventilação, distúrbio pulmonar, congestão nasal, edema faríngeo, congestão sinusal e espirros.
Alterações gastrointestinais: prurido anal, distúrbio retal, doença de Crohn, diarreia sanguinolenta, fezes sem cor, distúrbio gastrointestinal, hipercloridria, edema e inchaço dos lábios, ulceração labial, esofagite, pancreatite, incapacidade de defecar e ulceração lingual.
Alterações renais e urinárias: hematúria, nefrolitíase e insuficiência renal.
Alterações da pele e tecido subcutâneo: dermatite, dermatite acneiforme, dermatite de contato, dermatite esfoliativa, dermatite psoriasiforme, equimose, rash esfoliativo, edema periorbital, psoríase, rash folicular, rash vesicular, rosácea, dermatite seborreica, edema facial, telangiectasia.
Alterações metabólicas e nutricionais: hipercolesterolemia, hiperglicemia, hiperlipemia, hipovitaminose, polidipsia.
Alterações musculoesqueléticas e de articulações: artropatia, miosite e dor mandibular.
Infestações e infecções: gastroenterite, hepatite infecciosa, síndrome gripal, abscesso dentário e uretrite.
Alterações vasculares: hipotensão, hipotensão postural, distúrbio vascular periférico.
Gerais: edema.
Alterações hepatobiliares: colangite.
Alterações psiquiátricas: alterações de ciclotimia, alterações emocionais, perda de libido, depressão maior, pesadelos, inquietação, inibição sexual, terror noturno, alteração do pensamento e tiques.
Danos, envenenamento e complicações de procedimento: quedas e ferimentos acidentais.
Experiência pós-comercialização:
Os seguintes eventos foram relatados durante o período de comercialização de NORVIR® (ritonavir). A frequência das reações adversas na pós-comercialização é desconhecida.
Alterações do sistema nervoso: há relatos de convulsões. Relação de causa e efeito não foi estabelecida.
Distúrbios metabólicos e nutricionais: desidratação, geralmente associada a sintomas gastrointestinais, e algumas vezes resultando em hipotensão, síncope ou insuficiência renal. Síncope, hipotensão ortostática e insuficiência renal também foram relatadas sem evidência de desidratação.
Alterações cardíacas: há relatos de infarto do miocárdio.
Alterações do sistema reprodutor: menorragia.
Alterações de pele e tecido subcutâneo: necrólise epidérmica tóxica.
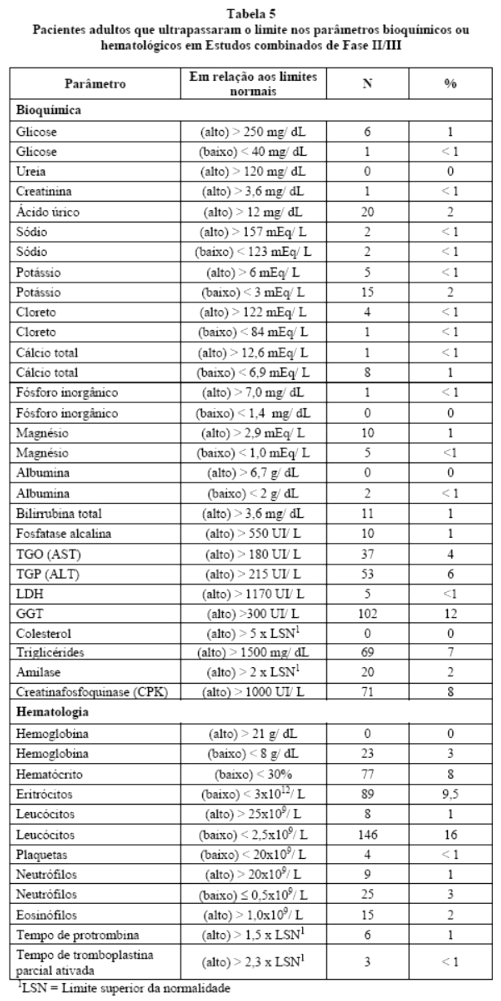
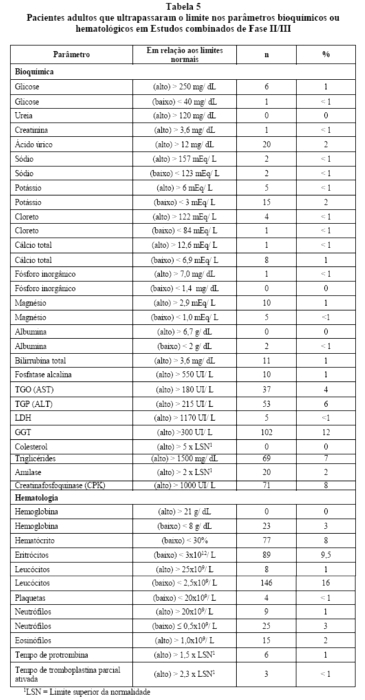
Na Tabela 5estão relacionadas às alterações de parâmetros laboratoriais ocorridas em pacientes adultos tratados com ritonavir, independentemente da comprovação de relação causa/efeito. A maioria dos pacientes recebia outros medicamentos concomitantes.
Pacientes Pediátricos
Eventos Adversos Emergentes com o Tratamento
O ritonavir foi estudado em 265 pacientes pediátricos com idades entre 01 mês e 21 anos. O perfil de eventos adversos observado durante os estudos clínicos pediátricos foi similar àquele de pacientes adultos.
Vômito, diarreia e erupção cutânea/alergia foram os únicos eventos adversos clínicos relacionados à droga de intensidade moderada a grave observados em ≥ 2% dos pacientes pediátricos registrados em estudos clínicos do ritonavir.
Anormalidades Laboratoriais
As seguintes anormalidades laboratoriais classe 3-4 ocorreram em 3% ou mais dos pacientes pediátricos que receberam tratamento com ritonavir, seja sozinho ou combinado com inibidores da transcriptase reversa: neutropenia (9%), hiperamilasemia (7%), trombocitopenia (5%), anemia (4%), e AST elevada (3%).
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em http://portal.anvisa.gov.br/notivisa ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
A experiência humana de superdosagem aguda com ritonavir é limitada. Um paciente em um ensaio clínico tomou 1500 mg/dia de ritonavir durante 2 dias e relatou parestesia, que regrediu depois que a dose foi reduzida. Insuficiência renal com eosinofilia foi relatada com superdosagem de ritonavir.
O ritonavir tem baixa ordem de toxicidade aguda quando administrado oralmente. A DL50 foi determinada como sendo maior que 2500 mg/kg, tanto em ratos quanto em camundongos. O nível de dose sem efeito foi de 200 mg/kg em camundongos e de 250 mg/kg em ratos.
Tratamento da superdosagem
Não há antídoto específico para ritonavir. O tratamento de superdosagem com ritonavir deve consistir de medidas gerais de suporte, incluindo o monitoramento de sinais vitais e observação do estado clínico do paciente. É proposto que o tratamento da superdosagem inclua também lavagem gástrica e administração de carvão ativado. Como ritonavir é extensamente metabolizado pelo fígado e altamente ligado a proteínas plasmáticas, é improvável que a diálise seja benéfica na remoção significativa do fármaco.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações sobre como proceder.
Dizeres legais.
MS: 1.9860.0009
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela ANVISA em 24/07/2019.
NORVIR
ABBVIE
Pó para Suspensão
ritonavir
Antiviral.
Apresentações.
Embalagem com 30 envelopes de pó para suspensão oral contendo 100 mg de ritonavir cada + 1 copo misturador + 2 seringas dosadoras.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 01 MÊS DE IDADE
Composição.
NORVIR® (ritonavir) pó para suspensão oral 100 mg:
Cada envelope com 666,7mg contém: ritonavir 100 mg. Excipientes: copovidona, laurato de sorbitana, dióxido de silício
Informações técnicas.
1. INDICAÇÕES
NORVIR® (ritonavir) é destinado, em combinação com outros antirretrovirais, ao tratamento de pacientes adultos e pediátricos infectados pelo HIV, quando uma terapia antirretroviral for indicada com base em evidência clínica ou imunológica de progressão da doença.
2. RESULTADOS DE EFICÁCIA
NORVIR®(ritonavir) pó para suspensão oral demonstrou ser bioequivalente à NORVIR® (ritonavir) solução oral, atualmente comercializado. Portanto, a segurança e a eficácia de NORVIR®(ritonavir) pó para suspensão oral propostas são suficientemente estabelecidas pelos dados de segurança e eficácia dos ensaios clínicos pivotais e dos estudos clínicos de suporte da apresentação aprovada NORVIR® (ritonavir) solução oral.
Danner et al. demonstraram a potência, segurança e eficácia do ritonavir em um estudo que avaliou regimes contendo diferentes doses de ritonavir, comparando-os com placebo. Após 32 semanas, o grupo que recebeu 600 mg a cada 12 horas, em combinação com outros agentes ARV (inibidores da transcriptase reversa análogos de nucleosídeo) apresentaram um ganho de CD4 de 230 células por mm3 e uma queda na carga viral de 0,81 log. Os eventos adversos mais comuns foram náuseas, aumento dos triglicérides e das enzimas hepáticas. Assim, o ritonavir mostrou-se seguro, eficaz e bem tolerado.1
Contemporaneamente, Markowitz et al. mostraram resultados similares, demonstrando uma resposta imunológica satisfatória, com um ganho de CD4 de 74 e 83 células por mm3 após 4 e 12 semanas, respectivamente. Na semana 12, a queda da carga viral foi de 1,1 log.2
Pediátrico
Em um ensaio clínico aberto, concluído em 1998, em pacientes pediátricos infectados pelo HIV clinicamente estáveis, houve uma diferença significativa (p = 0.03) nos níveis detectáveis de RNA em favor de um regime triplo (ritonavir, zidovudina e lamivudina) após 48 semanas de tratamento.
Em um estudo concluído em 2003, 50 pacientes pediátricos na faixa etária de 4 semanas a 2 anos de idade, infectados pelo HIV-1, não tratados com lamivudina ou inibidores de protease, receberam ritonavir 350 mg / m2 ou 450 mg / m2 a cada 12 horas administrados com zidovudina 160 mg / m2 a cada 8 horas e lamivudina 4 mg / kg a cada 12 horas3. Nas análises por intenção de tratar, 72% e 36% dos pacientes obtiveram redução de ≤ 400 cópias / ml de RNA de HIV-1 plasmático nas semanas 16 e 104, respectivamente. A resposta foi semelhante em ambos os regimes de dosagem e em toda faixa etária dos pacientes.
Em um estudo concluído em 2000, 76 pacientes pediátricos na faixa etária 6 meses a 12 anos de idade, infectados pelo HIV-1, não tratados com inibidores de protease ou lamivudina e/ou estavudina receberam ritonavir 350 mg / m2 ou 450 mg / m2 a cada 12 horas co-administrado com lamivudina e estavudina. Nas análises por intenção de tratar, 50% e 57% dos pacientes nos grupos de dose de 350 e 450 mg / m2, respectivamente, obtiveram redução de ≤ 400 cópias / ml de RNA de HIV-1 plasmático na Semana 48.
Referências Bibliográficas
1. Danner S, Carr A, Leonard J, Leahman L, Gudiol F Gonzalez J. A short term Study of the Safety, Pharmacokinetics and Efficacy of Ritonavir, an Inhibitor of HIV-1 Protease. N Engl J Med 1995;333:1528-33.
2. Markowitz M, Saag M, Powderly W, Hurley A, Hsu A, Valdes J. A Preliminary Study of Ritonavir, an Inhibitor of HIV-1 Protease, to Treat HIV Infection. N Engl J Med 1995;333:1534-9.
3. Chadwick EG, Rodman JH, Brito P, et al. Ritonavir-based highly active antiretroviral therapy in human immunodeficiency virus type 1-infected infants younger than 24 months of age. Pediatr Infec Dis J. 2005;24(9):793-800.
3. CARACTERÍSTICAS FARMACOLÓGICAS
NORVIR® (ritonavir) é um inibidor de protease do HIV, apresentando atividade contra o Vírus da Imunodeficiência Humana (HIV).
O ritonavir é um pó branco ou com leve cor marrom, de sabor metálico amargo. É facilmente solúvel em metanol e etanol, solúvel em isopropanol e praticamente insolúvel em água.
O ritonavir é chamado quimicamente de éster 5-tiazolilmetílico, [5S-(5R*,8R*,10R*,11R*)] do ácido 10-hidroxi-2-metil-5-(1-metiletil)-1-[2-(1-metiletil)-4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraazatridecan-13-óico. Sua fórmula molecular é C37H48N6O5S2 e seu peso molecular é 720,95.
Farmacologia clínica
Mecanismo de ação: ritonavir é um inibidor peptidomimético oral ativo das aspartil-proteases do HIV-1 e HIV-2. A inibição da protease do HIV torna a enzima incapaz de processar o precursor da poliproteína gag-pol, fazendo com que as partículas virais produzidas se tornem imaturas e, portanto, incapazes de iniciar um novo ciclo de infecção. O ritonavir tem afinidade seletiva pela protease do HIV e pouca atividade inibitória diante da aspartil-protease humana. Pode ser utilizado também em conjunto com outros antirretrovirais da mesma classe com a finalidade de reduzir sua metabolização, diminuindo a dose necessária a cada tomada ou aumentando o intervalo entre as tomadas.
Atividade antiviral in vitro: dados in vitro indicam que ritonavir é ativo contra todas as cepas de HIV testadas em uma variedade de linhagens celulares humanas primárias e transformadas. A concentração da droga que inibe 50% e 90% da replicação viral in vitro é aproximadamente 0,02 mcM e 0,11 mcM, respectivamente. Potências similares foram observadas com cepas de HIV sensíveis e resistentes ao AZT. Estudos que avaliaram a citotoxicidade direta de ritonavir em diversas linhagens celulares não mostraram toxicidade direta em concentrações de até 25 mcM, com um índice terapêutico resultante in vitro de pelo menos 1000.
Resistência: isolados de HIV-1 resistentes ao ritonavir foram selecionados in vitro. Os isolados resistentes demonstraram reduzir a susceptibilidade de ritonavir e a análise genotípica desses isolados mostrou que a resistência foi primariamente atribuída a substituições específicas de aminoácidos na protease do HIV-1 nas posições 84 (Ile por Val), 82 (Val por Phe), 71 (Ala por Val) e 46 (Met por Ile). Alterações fenotípicas e genotípicas nos isolados de HIV de pacientes selecionados tratados com ritonavir foram monitoradas em ensaios clínicos de Fase I/II. A análise genotípica e fenotípica em série indicou que a sensibilidade ao ritonavir caiu de forma ordenada e gradual. As mutações iniciais ocorreram nas posições 82(Val por Ala/Phe), 54 (Ile por Val), 71 (Ala por Val/Thr) e 36 (Ile por Leu), seguidas pelas combinações de mutações em cinco posições adicionais específicas de aminoácidos. Cepas virais isoladas in vivo sem alteração na posição 82 não têm sensibilidade diminuída ao ritonavir. A mutação na posição 82 parece ser necessária, mas não suficiente para conferir resistência fenotípica. A resistência fenotípica foi definida como uma diminuição ³ 5 vezes na sensibilidade viral in vitro em relação ao basal. A relevância clínica das alterações genotípicas e fenotípicas associadas ao ritonavir ainda não foi estabelecida.
Resistência cruzada com outros antirretrovirais: o potencial para resistência cruzada ao HIV entre inibidores de protease não foi completamente explorado. Portanto, não se conhece o efeito que ritonavir terá na atividade de outros inibidores de protease administrados subseqüentemente. Isolados de HIV obtidos em série de seis pacientes tratados com ritonavir mostraram sensibilidade reduzida in vitro a este medicamento, mas não demonstraram redução correspondente da sensibilidade in vitro ao saquinavir quando comparada aos isolados basais. Entretanto, isolados de dois desses pacientes mostraram uma redução (8 vezes) na sensibilidade in vitro ao indinavir. Isolados de cinco pacientes também foram testados q
3/4) da dose usual de 300 mg/dia é recomendada (ex. 150 mg em dias alternados ou três vezes por semana). Uma redução adicional na dosagem também pode ser necessária.
rivaroxabana: a coadministração de ritonavir e rivaroxabana resultou em um aumento da exposição de rivaroxabana podendo aumentar o risco de sangramentos.
avanafil: um estudo farmacocinético demonstrou que a administração concomitante de 50 mg de avanafil e 600 mg de ritonavir a cada 12 horas resultou em um aumento de aproximadamente 13 vezes e 2,4 vezes nas AUCinf e Cmax de avanafil respectivamente. A coadministração de ritonavir com avanafil não é recomendada.
sildenafila: para o tratamento da disfunção erétil, use a sildenafila com atenção em doses reduzidas de 25 mg a cada 48 horas, com monitoramento dos efeitos adversos. Espera-se que a coadministração de ritonavir e sildenafila aumente substancialmente as concentrações de sildenafila (aumento de 11 vezes na AUC) e possa resultar em aumento dos eventos adversos associados à sildenafila, incluindo hipotensão, síncope, alterações visuais e ereção prolongada.
tadalafila: usar tadalafila, para o tratamento de disfunção erétil, com atenção, em doses reduzidas de, no máximo, 10 mg a cada 72 horas, com monitoramento dos efeitos adversos (veja "5. ADVERTÊNCIAS E PRECAUÇÕES"). Consulte as informações da bula de tadalafina quando esta for coadministrada com ritonavir em pacientes com hipertensão arterial pulmonar.
teofilina: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 horas e teofilina resultou em decréscimo de 43% na AUC da teofilina. Aumento da dosagem de teofilina pode ser necessário.
trazodona: o uso concomitante de ritonavir e trazodona pode aumentar a concentração de trazodona. Efeitos adversos como náuseas, vertigens, hipotensão e síncope foram observados. Se trazodona for prescrita com um inibidor de CYP3A4, como ritonavir, a combinação deve ser usada com atenção e uma dose menor de trazodona pode ser considerada.
vardenafila: usar vardenafila com atenção, em doses reduzidas de, no máximo, 2,5 mg a cada 72 horas, com monitoramento dos efeitos adversos.
varfarina: em um estudo farmacocinético, múltiplas doses de ritonavir (400 mg duas vezes ao dia) afetaram de modos diferentes as farmacocinéticas de dose única dos enantiômeros da varfarina. A AUC da S-varfarina foi afetada de modo variável pelo ritonavir, mas não foi estatisticamente significante. A AUC da menos potente R-varfarina foi reduzida em média 33% durante a coadministração de ritonavir. A gama de efeitos da coadministração do ritonavir sobre a ação anticoagulante da varfarina é difícil de ser prevista com base nesses resultados farmacocinéticos. Recomenda-se controle inicial frequente da Razão Normalizada Internacional (INR) durante a coadministração de ritonavir e varfarina.
agentes anticancerígenos (abemaciclibe, apalutamida, dasatinibe, encorafenibe, ibrutinibe, ivosidenibe, neratinibe, nilotinibe, venetoclax, vincristina, vimblastina) as concentrações séricas podem aumentar quando houver coadministração com ritonavir, resultando em um possível aumento na incidência de eventos adversos, dentre alguns que podem ser graves. A coadministração de venetoclax ou ibrutinibe e NORVIR® (ritonavir) poderia aumentar potencialmente a exposição à venetoclax ou ibrutinibe resultando em um sério risco de Síndrome da Lise Tumoral. A coadministração de encorafenibe ou ivosidenibe com ritonavir poderia aumentar a exposição de encorafenibe ou ivosidenibe, aumentando potencialmente o risco de eventos adversos sérios, como o prolongamento do intervalo QT. O uso concomitante de NORVIR® com apalutamida é contraindicado (veja "4. CONTRAINDICAÇÕES").
Outras interações medicamentosas:
alprazolam: a coadministração de alprazolam e ritonavir resultou em uma redução nos valores da Cmáx média de alprazolam (16%), mas não nos valores médios da AUC (12%). Similarmente, foi observado um efeito na curva do efeito sedativo, mas não na extensão da sedação. Discreta depressão psicomotora foi confundida com um efeito de aprendizado. Estes resultados farmacocinéticos e farmacodinâmicos são inconsistentes com o efeito farmacológico do alprazolam. Estes resultados não foram considerados clinicamente significantes.
amprenavir: há dados na literatura mostrando que as concentrações do inibidor de protease do HIV amprenavir são aumentadas quando coadministrado com ritonavir.
bupropiona: bupropiona é originalmente metabolizada por CYP2B6. É esperado que a administração concomitante de bupropiona com doses repetidas de ritonavir reduza os níveis de bupropiona.
efavirenz: em indivíduos saudáveis recebendo 500 mg de ritonavir duas vezes ao dia concomitante com 600 mg de efavirenz uma vez ao dia, a AUC de efavirenz no estado de equilíbrio aumentou 21%. Foi observado um aumento concomitante na AUC de ritonavir em 17%.
ácido fusídico: presume-se que a administração concomitante de inibidores de proteases, incluindo ritonavir, com ácido fusídico resulte em aumento das concentrações tanto de ácido fusídico como do inibidor de protease no plasma (veja "4.CONTRAINDICAÇÕES").
fostamatinibe: a coadministração de fostamatinibe com ritonavir poderia aumentar a exposição do metabólito R406 do fostamatinibe, resultando em eventos adversos relacionados a dose, como hepatotoxicidade e neutropenia.
nelfinavir: as interações entre ritonavir e nelfinavir podem envolver tanto a inibição quanto a indução do sistema citocromo P450. A administração concomitante de 400 mg de ritonavir duas vezes ao dia aumenta significativamente as concentrações do M8 (o principal metabólito do nelfinavir) e resulta em pequenos aumentos nas concentrações do nelfinavir. Em um estudo de dez pacientes, a administração do nelfinavir na dose de 750 mg e ritonavir na dose de 400 mg duas vezes ao dia, resultou em aumentos discretos nos parâmetros do nelfinavir [AUC (160%), Cmax (121%) e Cmin (123%)] em relação aos dados disponíveis sobre a monoterapia com nelfinavir na dose de 750 mg três vezes ao dia. A AUC do M8 aumentou em 347%.
raltegravir: um estudo farmacocinético demonstrou que a coadministração de 100 mg de ritonavir duas vezes ao dia e 400 mg de raltegravir uma vez ao dia resultou em redução mínima de raltegravir C12h m, AUC e Cmax em 1%, 16% e 24%, respectivamente.
saquinavir: um estudo farmacocinético demonstrou que ritonavir inibe extensamente o metabolismo do saquinavir resultando em concentrações plasmáticas de saquinavir muito aumentadas. Após aproximadamente 4 semanas de um regime combinado de saquinavir 400 ou 600 mg duas vezes ao dia (cápsulas de gelatina dura) com 400 ou 600 mg de ritonavir duas vezes ao dia, em pacientes infectados pelo HIV, os valores da AUC de saquinavir foram, no mínimo, 17 vezes maiores que os valores de AUC obtidos com saquinavir 600 mg três vezes ao dia sem ritonavir. Quando usados em terapia combinada por até 24 semanas, doses maiores que 400 mg duas vezes ao dia tanto de saquinavir quanto de ritonavir foram associadas com um aumento de eventos adversos. As exposições plasmáticas alcançadas com Inviraseâ (mesilato de saquinavir em cápsula gelatinosa dura, 400 mg duas vezes ao dia) e ritonavir (400 mg duas vezes ao dia) são similares às atingidas com Fortovaseâ (saquinavir cápsula gelatinosa mole, 400 mg duas vezes ao dia) e ritonavir (400 mg duas vezes ao dia).
sulfametoxazol/trimetoprima: um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 500 mg a cada 12 horas e sulfametoxazol/ trimetoprima resultou em diminuição de 20% na AUC do sulfametoxazol e aumento de 20% na AUC da trimetoprima. Pode não ser necessário ajuste de dosagem.
zidovudina (AZT): um estudo farmacocinético demonstrou que a administração concomitante de ritonavir 300 mg a cada 6 horas e zidovudina 200 mg a cada 8 horas resultou em uma diminuição na Cmáx da zidovudina de 27% e na AUC de 25%. Por outro lado, foi observado pouco ou nenhum efeito na farmacocinética do ritonavir. Pode não ser necessário ajuste de dosagem de zidovudina durante terapia concomitante com ritonavir.
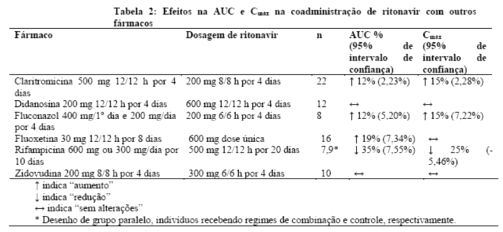
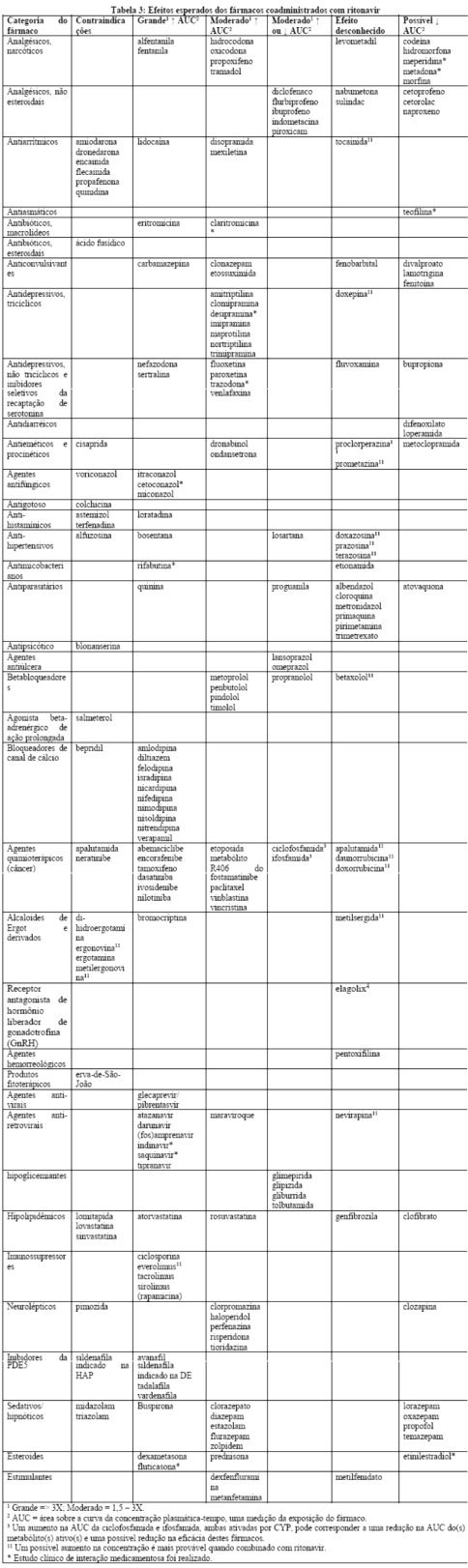
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (temperatura entre 15 e 30°C). Após preparo, manter em temperatura ambiente (temperatura entre 15°C e 30°C) por até 2 horas.
Prazo de validade: se armazenado nas condições indicadas, o medicamento se manterá próprio para consumo pelo prazo de validade de 36 meses, a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas e organolépticas
NORVIR® (ritonavir) pó para suspensão oral apresenta-se na forma de pó de cor bege a levemente amarelado a amarelo..
Após reconstituição, NORVIR® (ritonavir) deve apresentar-se como uma suspensão turva e livre de grumos. Caso haja formação de grumos, agitar vigorosamente até que todo o pó seja diluído.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Diretrizes Gerais de Dosagem: Os médicos devem consultar as informações completas de prescrição e estudos clínicos sobre os inibidores de protease para verificar se estes são coadministrados com doses reduzidas de ritonavir.
NORVIR® (ritonavir) pó para suspensão oral deve ser administrado por via oral, misturado com água.
NORVIR® (ritonavir) pó para suspensão oral pode deixar um sabor desagradável na boca. Para diminuí-lo, NORVIR® (ritonavir) pó para suspensão oral pode ser misturado com leite achocolatado, fórmula infantil ou alimentos leves (papinha de maçã ou pudim).
A duração do tratamento depende de orientação médica, a partir da avaliação clínica e laboratorial de cada paciente.
Posologia
Adultos
A dose recomendada de NORVIR® (ritonavir) pó para suspensão oral é de 600 mg (6 envelopes) duas vezes por dia misturado com alimentos leves (como papinha de maçã ou pudim) ou líquido adequado como água, leite achocolatado ou fórmula infantil. NORVIR® (ritonavir) pó para suspensão oral deve ser administrado com alimentos
Para doses de 100, 200, 300, 400, 500, 600 mg:
- Polvilhar todo o conteúdo de cada sachê sobre uma pequena quantidade de alimentos leves (como papinha de maçã ou pudim) ou misturar com uma pequena quantidade líquido adequado (como água, leite achocolatado ou fórmula infantil) e consumir todo o conteúdo.
- Uma vez preparada, a dose deve ser consumida em até 2 horas.
Para doses inferiores a 100 mg ou doses parciais entre incrementos de 100 mg:
- Misturar 01 envelope do pó para suspensão oral (100 mg) com 9,4 ml de líquido (como água, leite achocolatado ou fórmula infantil) em um copo misturador.
- Uma vez misturado, usar a seringa dosadora para mensurar e administrar o volume prescrito (Ver Tabela 4).
- Após o preparo, a dose deve ser consumida em até 2 horas.
- Descartar qualquer porção remanescente no copo misturador.
Titulação de dose
A utilização de um esquema de titulação de doses pode ajudar a reduzir os eventos adversos decorrentes do tratamento, mantendo níveis plasmáticos adequados de ritonavir. NORVIR® (ritonavir) deve ser iniciado com doses de, no mínimo, 300 mg (3 envelopes) duas vezes ao dia durante o período de três dias, com incrementos de 100 mg (1 envelopes) duas vezes ao dia, até atingir a dose máxima de 600 mg (6 envelopes) duas vezes ao dia em um período não superior a 14 dias.
Os pacientes devem ser informados de que os eventos adversos frequentemente observados no início do tratamento, como distúrbios gastrointestinais leves a moderados e parestesia, podem diminuir com a continuidade do tratamento.
Os pacientes não devem ser mantidos com dose de 300 mg (3 envelopes) duas vezes ao dia por mais de três dias.
A dose máxima diária é de 6 envelopes de 100 mg duas vezes ao dia, totalizando 1.200 mg ao dia.
Esquemas combinados de inibidores de protease
A experiência clínica de terapia combinada incluindo dose terapêutica de ritonavir com outro inibidor de protease é limitado.
O ritonavir inibe extensamente o metabolismo da maioria dos inibidores de protease disponíveis. Portanto, qualquer consideração de tratamento combinado com ritonavir deve levar em consideração a interação farmacocinética e os dados de segurança dos agentes envolvidos. Há extensa resistência cruzada nesta classe de agentes. Deve-se considerar a combinação de dois inibidores de protease com menor padrão superposto de resistência. O uso do ritonavir em tais esquemas deve ser orientado por esses fatores.
Para uso de ritonavir com saquinavir, uma cautelosa titulação de dose é utilizada, iniciando-se a terapia com ritonavir com doses de 300 mg (3 envelopes) duas vezes ao dia. Para uso de ritonavir com indinavir, uma cautelosa titulação de dose é utilizada, iniciando-se a terapia com ritonavir com doses de 200 mg (2 envelopes) duas vezes ao dia, com aumento de 100 mg (1 envelopes) duas vezes ao dia até atingir 400 mg (4 envelopes) duas vezes ao dia em duas semanas.
Crianças
NORVIR® (ritonavir) pode ser utilizado por crianças acima de 01 mês de idade.
NORVIR® (ritonavir) pode ser utilizado em combinação a outros agentes antirretrovirais. A dosagem recomendada de NORVIR® (ritonavir) em crianças > 01 mês é de 350 a 400 mg/m2, baseando-se na Área de Superfície Corporal, duas vezes ao dia, não excedendo a dose de 600 mg (6 envelopes) duas vezes ao dia.
A dose deve ser iniciada em 250 mg/m2 e aumentada, em intervalos de 2 a 3 dias, em 50 mg/m2 duas vezes ao dia.
Se o paciente não tolerar a dose máxima diária devido aos eventos adversos, a dose máxima tolerada deve ser utilizada como terapia de manutenção em combinação com outros agentes antirretrovirais. Quando possível, a dose deve ser administrada utilizando uma seringa dosadora calibrada.
Fica a critério do médico prescritor a escolha pela apresentação que mais se adequa ao paciente pediátrico.
A tabela abaixo apresenta o guia para doses pediátricas de NORVIR® (ritonavir) baseando-se na Área de Superfície Corporal:
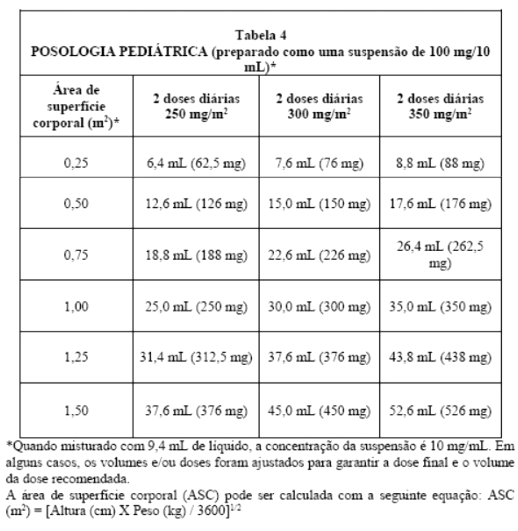
Para calcular o volume a ser administrado (em mL) para áreas de superfície corporal intermediárias não incluídas na tabela acima, a área de superfície corporal deve ser multiplicado por um fator de: 25 para uma dose de 250 mg/m2; 30 para 300 mg/m2; e 35 para 350 mg/m2.
Instruções de preparo para dose correta de NORVIR® (ritonavir) pó para suspensão oral com líquidos:
Siga as instruções abaixo:
Antes de preparar a dose necessária de NORVIR® (ritonavir), colete os itens mostrados na Figura 1. Pode ser necessário mais de 1
envelope de NORVIR® (ritonavir) para cada dose. Verifique a dosagem prescrita pelo médico. Se for necessário mais do que 1 envelope para a dose, repita todas as etapas de preparação para cada envelope.
Instruções da seringa
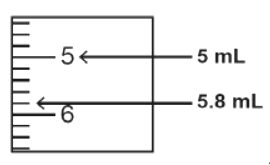
Leitura da escala
a. Cada mililitro (mL) é mostrado como um número em um traço longo.
b. Cada 0.2 mL é mostrado como um traço curto.
Verificação da seringa dosadora antes de cada uso
Será necessário utilizar uma nova seringa se:
• Não for possível a limpeza da seringa.
• Não for possível a leitura da escala.
• Não for possível movimentar o êmbolo.
• A seringa estiver danificada ou com vazamento.
Passo 1. Encha a seringa
a. Empurre completamente o êmbolo da seringa.
b. Coloque a ponta da seringa dentro do líquido.
c. Puxe lentamente o êmbolo até a marcação de 10 mL da seringa (ver Figura 2).
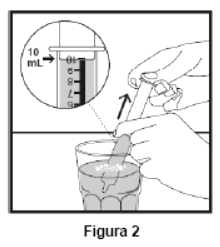
Passo 2. Mover as bolhas para a ponta da seringa
a. Segurar a seringa com a ponta apontada para cima.
b. Bater levemente a seringa utilizando a outra mão. Isso irá mover as bolhas para a ponta da seringa.
c. Puxar o êmbolo para baixo. Tomar cuidado para não remover o êmbolo.
d. Novamente bater levemente na seringa. Isso irá ajudar a estourar as bolhas e garantir que estejam todas na ponta (ver Figura 3).
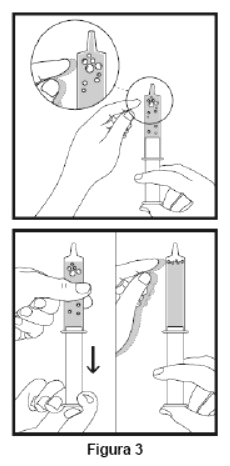
Passo 3. Mensurar o líquido
a. Manter a seringa apontada para cima.
b. Empurrar lentamente o êmbolo para cima até que o topo do êmbolo esteja marcando 9,4 mL - isso irá remover qualquer bolha da seringa (ver Figura 4).
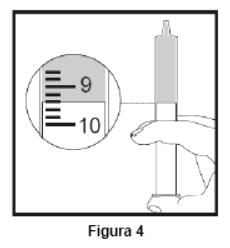
Passo 4. Esvaziar a seringa
a. Empurrar lentamente o êmbolo para esvaziar o líquido da seringa para o copo misturador (ver Figura 5).
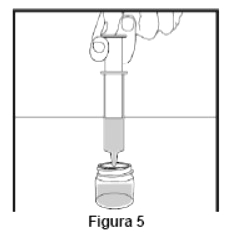
Passo 5. Colocar o pó no copo misturador
a. Abrir um envelope contendo NORVIR® (ritonavir).
b. Colocar todo o conteúdo do pó no copo misturador (ver Figura 6).
c. Verificar se o envelope está vazio.
Tomar o cuidado para não derramar qualquer quantidade de pó fora do copo misturador (ver Figura 6).
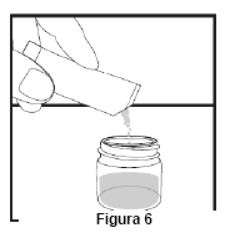
Passo 6. Misturar o pó e o líquido
a. Fechar bem a tampa do frasco misturador e agitar vigorosamente por pelo menos 90 segundos até que todos os grumos sejam diluídos.
b. Verificar a presença de qualquer grumo do pó. Se ainda houver grumos, continuar agitando até que sejam completamente diluídos.
c. O líquido pode ficar turvo após a mistura - esta situação é aceitável.
d. Deixar o líquido em descanso por 10 minutos e a maioria das bolhas irá sumir.
e. Algumas bolhas poderão ser observadas na superfície do líquido - esta situação é aceitável (ver Figura 7).
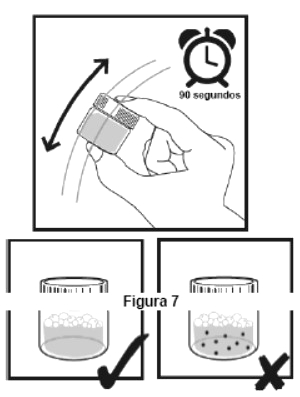
Passo 7. Encher a seringa
a. Empurrar completamente o êmbolo da seringa.
b. Colocar a ponta da seringa no fundo do copo misturador.
c. Puxar lentamente o êmbolo da seringa até a marcação de 10 mL - Tentar não puxar qualquer bolha para dentro da seringa (ver Figura 8).
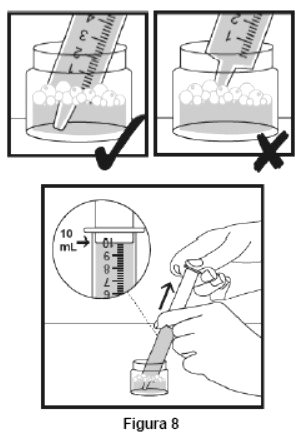
Passo 8. Remover as bolhas
a. Segurar a seringa com a ponta para cima
b. Bater levemente com a outra mão. Isso irá mover as bolhas para a ponta da seringa.
c. Puxar o êmbolo para baixo. Tomar cuidado para não remover o êmbolo.
d. Novamente bater levemente na seringa para estourar as bolhas e garantir que estejam todas na ponta (ver Figura 9).
e. Empurrar lentamente o êmbolo até que seja visto uma pequena quantidade de líquido na ponta da seringa.
f. Se houver qualquer bolha de ar grande, esvaziar o líquido da seringa no copo misturador e começar novamente do Passo 7.
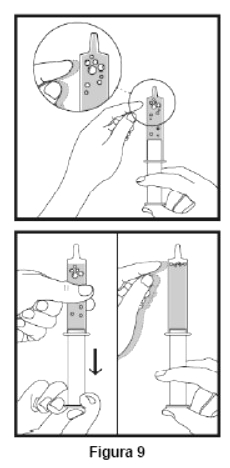
Passo 9. Mensurar a dose
a. Verificar a dose em mL prescrita pelo médico.
b. Apontar a seringa no copo misturador e empurrar o êmbolo lentamente até o volume indicado para a dose (ver Figura 10).
c. Se houver excesso de líquido retornado ao copo misturador, é necessário iniciar novamente do Passo 7. Tomar cuidado para não derramar líquido fora do copo misturador.
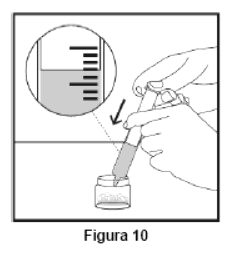
Passo 10. Administrar o medicamento ao paciente
a. Posicionar a ponta da seringa no interior da bochecha do paciente.
b. Empurrar lentamente o êmbolo para administrar toda a dose ao paciente (ver Figura 11).
c. Administrar uma dose completa ao paciente em até 2 horas após a abertura do envelope de NORVIR® (ritonavir).
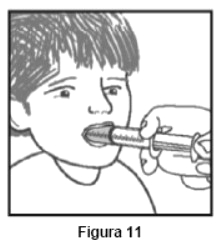
Passo 11. (Se necessário)
Se for necessário administrar mais de um envelope de NORVIR® (ritonavir), repetir todos os passos desde o início.
Passo 12. Após o fim da administração da dose ao paciente
a. Jogar no lixo o envelope de NORVIR® (ritonavir) vazio e qualquer resquício de medicamento do copo misturador.
b. Remover o êmbolo da seringa.
c. Lavar a seringa, o êmbolo, o copo misturador e tampa em água morna e detergente. Enxaguar com água e deixar secar ao ar livre. Não lavar estes aparatos em máquina de lavar louça.
d. Lavar e secar a área utilizada para o preparo do medicamento.
Instruções de preparo para dose correta de NORVIR® (ritonavir) pó para suspensão oral com alimentos leves (quando a dose de NORVIR® (ritonavir) é equivamente a, no mínimo um sachê).
Siga as instruções abaixo:
Passo 01: Antes de preparar a dose necessária de NORVIR® (ritonavir), colete os itens mostrados na Figura 12.
Passo 02: Verifique a dosagem prescrita pelo médico e o número de sachês.
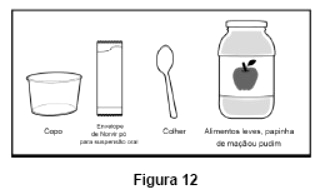
Passo 03: Coloque uma pequena quantidade de alimento (como papinha de maçã ou pudim) em um copo (ver Figura 13).
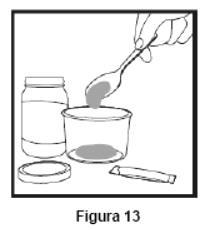
Passo 04: Abra o sachê (ver Figura 14).
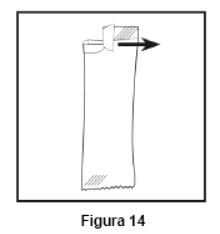
Passo 05: Coloque TODO o conteúdo do sachê no alimento (ver Figura 15).
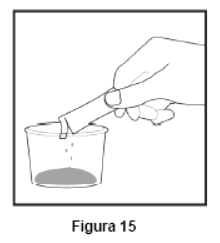
Passo 06: Homogenize bem (ver Figura 16).
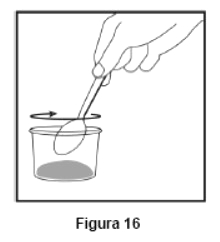
Passo 07: Administre essa preparação ao paciente.
Passo 08: TODO o conteúdo deve administrado ao paciente (ver Figura 17). Se houver qualquer resíduo de pó, adicione mais colheres do alimento utilizado no Passo 03 e administre ao paciente. Utilize em até 2 horas.
Passo 09: Coloque o sachê vazio no lixo. Lavar e secar a área de preparação. Lave imediatamente a colher e o copo com água morna e detergente (ver Figura 18). Enxágue e deixe secar.
9. REAÇÕES ADVERSAS
Quando ritonavir for coadministrado com outro inibidor de protease, o médico deve verificar as informações completas de prescrição destes inibidores de protease, inclusive suas reações adversas.
Adultos
As reações adversas mais frequentemente relatadas entre os pacientes recebendo NORVIR® (ritonavir) isoladamente ou em combinação com outros medicamentos antirretrovirais foram alterações gastrointestinais (incluindo diarreia, náusea, vômito, dor abdominal (superior e inferior), distúrbios neurológicos (incluindo parestesia e parestesia oral), e fadiga/astenia.
Reações adversas muito comuns ( > 1/10):
Alterações do sistema nervoso: disgeusia, parestesia, incluindo parestesia oral, tontura, neuropatia periférica
Alterações respiratórias, torácicas e do mediastino: dor orofaríngea e tosse.
Alterações gastrointestinais: dor abdominal (superior e inferior) diarreia, incluindo grave desequilíbrio eletrolítico, náusea, vômito e dispepsia..
Alterações de pele e tecido subcutâneo: prurido, rash, incluindo eritomatoso e maculopapular.
Alterações musculares e tecido conectivo: artralgia e dor nas costas.
Alterações Gerais e condições do local de administração: fadiga, incluindo astenia, rubor e sensação de calor.
Reações adversas comuns ( > 1/100 e ≤ 1/10):
Alterações do sistema imune: hipersensibilidade, incluindo urticária e edema de face.
Alterações do sistema nervoso: síncope.
Alterações gastrointestinais: flatulência, hemorragia gastrointestinal e doença do distúrbio gastroesofágico.
Alterações hepatobiliares: hepatite, incluindo aumento de AST, ALT e GGT, aumento da bilirrubina no sangue, incluindo icterícia.
Alterações renais e urinárias: aumento da micção
Alterações na pele e tecido subcutâneo: acne. Alterações metabólicas e nutricionais: hipercolesterolemia, hipertrigliceridemia, gota, edema e edema periférico. Alterações musculoesqueléticas e do tecido conectivo: mialgia. e miopatia/aumento de CPK.
Alterações visuais: visão embaçada.
Alterações vasculares: hipertensão, hipotensão incluindo hipotensão ortostática, extremidades frias.
Alterações psiquiátricas: confusão e distúrbio de atenção.
Experiência pós-comercialização:
Os seguintes eventos foram relatados durante o período de comercialização de NORVIR® (ritonavir). A frequência das reações adversas na pós-comercialização é desconhecida.
Alterações do sistema nervoso: há relatos de convulsões. Relação de causa e efeito não foi estabelecida.
Distúrbios metabólicos e nutricionais: desidratação, geralmente associada a sintomas gastrointestinais, e algumas vezes resultando em hipotensão, síncope ou insuficiência renal. Síncope, hipotensão ortostática e insuficiência renal também foram relatadas sem evidência de desidratação.
Alterações cardíacas: há relatos de infarto do miocárdio.
Alterações do sistema reprodutor: menorragia.
Alterações de pele e tecido subcutâneo: necrólise epidérmica tóxica.
Na Tabela 4 estão relacionadas às alterações de parâmetros laboratoriais ocorridas em pacientes adultos tratados com ritonavir, independentemente da comprovação de relação causa/efeito. A maioria dos pacientes recebia outros medicamentos concomitantes.
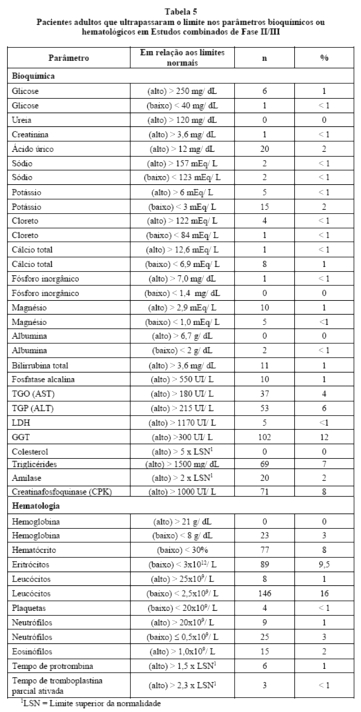
Pacientes Pediátricos
Eventos Adversos Emergentes com o Tratamento
O ritonavir foi estudado em 265 pacientes pediátricos com idades entre 01 mês e 21 anos. O perfil de eventos adversos observado durante os estudos clínicos pediátricos foi similar àquele de pacientes adultos.
Vômito, diarreia e erupção cutânea/alergia foram os únicos eventos adversos clínicos relacionados à droga de intensidade moderada a grave observados em ≥2% dos pacientes pediátricos registrados em estudos clínicos do ritonavir.
Anormalidades Laboratoriais
As seguintes anormalidades laboratoriais classe 3-4 ocorreram em 3% ou mais dos pacientes pediátricos que receberam tratamento com ritonavir, seja sozinho ou combinado com inibidores da transcriptase reversa: neutropenia (9%), hiperamilasemia (7%), trombocitopenia (5%), anemia (4%), e AST elevada (3%).
Atenção: este produto é um medicamento que possui nova forma farmacêutica no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Neste caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
A experiência humana de superdosagem aguda com ritonavir é limitada. Um paciente em um ensaio clínico tomou 1500 mg/dia de ritonavir durante 2 dias e relatou parestesia, que regrediu depois que a dose foi reduzida. Insuficiência renal com eosinofilia foi relatada com superdosagem de ritonavir.
O ritonavir tem baixa ordem de toxicidade aguda quando administrado oralmente. A DL50 foi determinada como sendo maior que 2500 mg/kg, tanto em ratos quanto em camundongos. O nível de dose sem efeito foi de 200 mg/kg em camundongos e de 250 mg/kg em ratos.
Tratamento da superdosagem
Não há antídoto específico para ritonavir. O tratamento de superdosagem com ritonavir deve consistir de medidas gerais de suporte, incluindo o monitoramento de sinais vitais e observação do estado clínico do paciente. É proposto que o tratamento da superdosagem inclua também lavagem gástrica e administração de carvão ativado. Como ritonavir é extensamente metabolizado pelo fígado e altamente ligado a proteínas plasmáticas, é improvável que a diálise seja benéfica na remoção significativa do fármaco.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações sobre como proceder.
Dizeres legais.
MS: 1.9860.0015
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela ANVISA em 24/07/2019.
NORVIR
ABBVIE
Solução Oral
ritonavir
Antiviral.
Apresentações.
Solução oral de 80 mg/mL: embalagens com 1 frasco de 240 mL.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 01 MÊS DE IDADE
Composição.
NORVIR® (ritonavir) solução oral 80 mg/mL:
Cada mL contém: ritonavir 80 mg. Excipientes: álcool etílico, água deionizada (14,9% v/v), óleo de rícino (10,5% p/v), propilenoglicol, sacarina sódica (1,0% p/v), ácido cítrico para ajuste do pH (0,277% p/v), óleo de hortelã, sabor de caramelo e corante amarelo FD&C n° 6 (E110). NORVIR® (ritonavir) solução oral contém 43,2% de álcool etílico (v/v) e 26,57% de propilenoglicol (p/v).
Informações técnicas.
1. INDICAÇÕES
NORVIR® (ritonavir) é destinado, em combinação com outros antirretrovirais, ao tratamento de pacientes adultos e pediátricos infectados pelo HIV-1, quando uma terapia antirretroviral for indicada com base em evidência clínica e/ou imunológica de progressão da doença.
2. RESULTADOS DE EFICÁCIA
A atividade de NORVIR® (ritonavir) como monoterapia ou em combinação com inibidores da transcriptase reversa foi avaliada em 1.446 pacientes em dois estudos duplo-cegos randomizados.
Pacientes Avançados com Terapia Antirretroviral Prévia
O Estudo 247, randomizado, duplo-cego (com abertura de seguimento) foi conduzido em pacientes HIV infectados em terapia antirretroviral nos últimos 09 meses que apresentavam contagem celular média basal de CD4 menor ou igual a 100 células/mm3. Em cada paciente em regime de terapia antirretroviral (com até dois agentes antirretrovirais aprovados) foi administrado NORVIR® (ritonavir) 600 mg duas vezes ao dia ou placebo.
O estudo ampliou-se com 1.090 pacientes, com contagem celular média basal de CD4 de 32 células/mm3. Após a demonstração dos benefícios clínicos da terapia com NORVIR® (ritonavir), todos os pacientes foram elegíveis a optar por NORVIR® (ritonavir) durante o período de acompanhamento. A duração média da terapia duplo-cega com NORVIR® (ritonavir) e placebo foi de 06 meses. A duração média do acompanhamento até a fase final do estudo aberto foi de 13,5 meses para pacientes randomizados com NORVIR® (ritonavir) e 14 meses para os pacientes randomizados com placebo.
A incidência acumulada de progressão da doença ou morte durante a fase duplo-cega do Estudo 247 foi de 26% para os pacientes inicialmente randomizados com NORVIR® (ritonavir) contra 42% dos pacientes inicialmente randomizados com o placebo. Essa diferença de porcentagem foi estatisticamente relevante. (veja Figura 01)
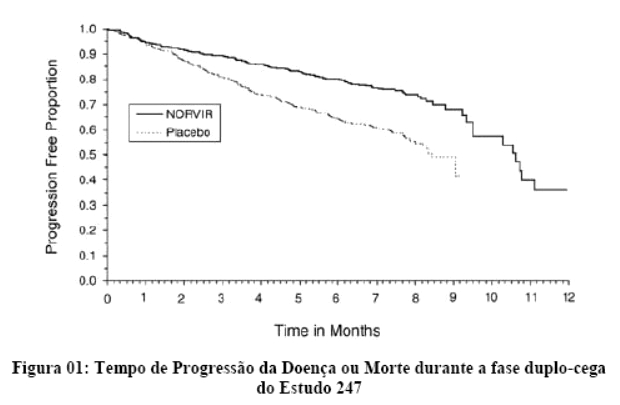
O acumulado de mortalidade no final da fase aberta de seguimento do Estudo 247 resultou em 18% para os pacientes inicialmente randomizados com NORVIR® (ritonavir) e 26% para os pacientes inicialmente randomizados com placebo. Essa diferença na porcentagem foi estatisticamente relevante (veja Figura 02). Como a análise no final da fase aberta de seguimento incluiu pacientes que inicialmente estavam no grupo placebo e passaram a utilizar NORVIR® (ritonavir), os benefícios na sobrevida com NORVIR® (ritonavir) não foram estimados com precisão.
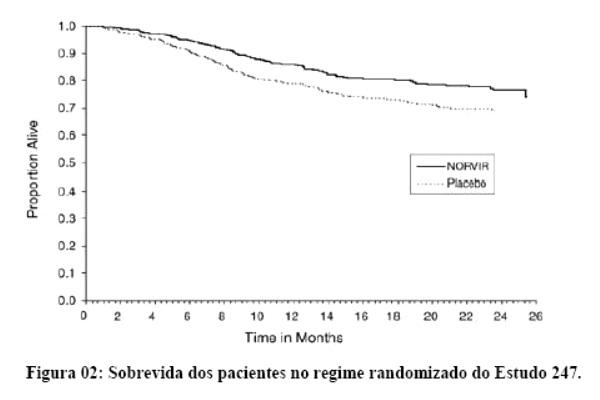
As Figuras 3 e 4 resumem as diferenças de contagem celular média basal de CD4 e de níveis de HIV RNA plasmático (cópias/mL) respectivamente, durante as primeiras 24 semanas da fase duplo-cega do Estudo 247.
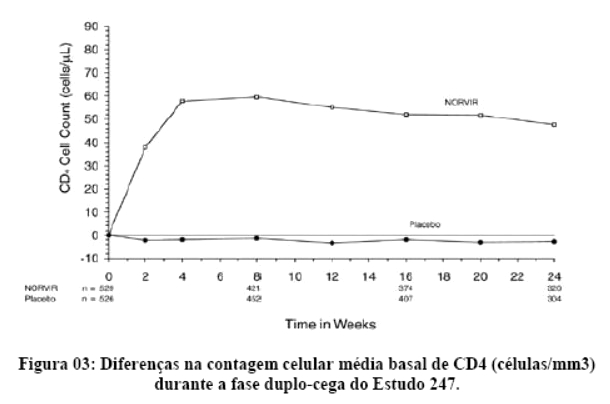
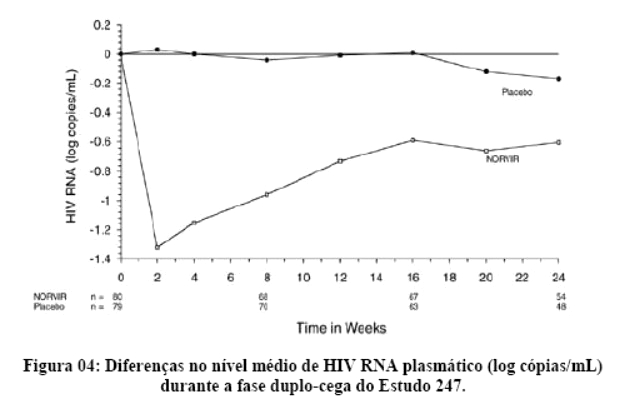
Pacientes sem Terapia com antirretrovirais prévia
No Estudo 245, 356 pacientes HIV infectados sem terapia antirretroviral prévia - naive (média basal de CD4 = 364 células/mm3) foram randomizados para receber NORVIR® (ritonavir) 600 mg duas vezes ao dia, zidovudina 200 mg três vezes ao dia ou uma combinação dessas drogas. A Figura 05 e 06 resumem a contagem celular média basal de CD4 e os níveis plasmáticos de HIV RNA (cópias/mL), respectivamente durante as primeiras 24 semanas da fase duplo-cega do Estudo 245.
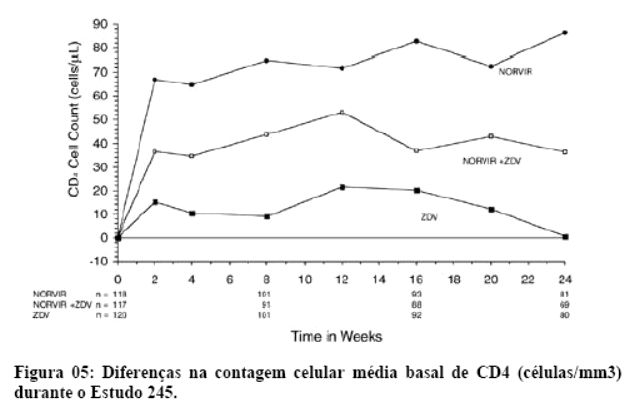
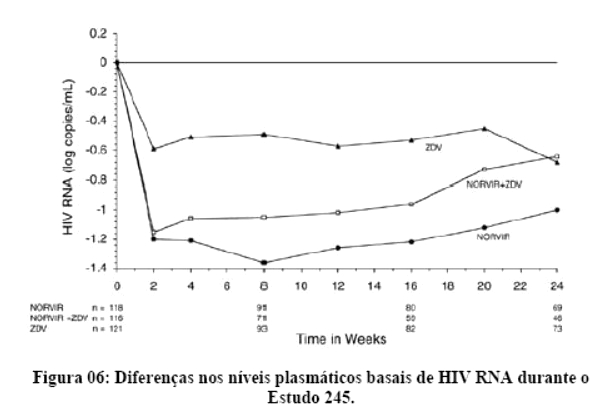
Referências Bibliográficas
Caso haja interesse em conhecer as referências bibliográficas e/ou estudos clínicos disponíveis para este medicamento, por favor, entre em contato com a Central de Relacionamento AbbVie Line através do telefone 0800 022 2843.
3. CARACTERÍSTICAS FARMACOLÓGICAS
NORVIR® (ritonavir) é um inibidor da protease do HIV, apresentando atividade contra o Vírus da Imunodeficiência Humana (HIV).
Farmacologia clínica
Mecanismo de ação: ritonavir é um inibidor peptidomimético oral ativo das aspartil-proteases do HIV-1 e HIV-2. A inibição da protease do HIV torna a enzima incapaz de processar o precursor da poliproteína gag-pol, fazendo com que as partículas virais produzidas se tornem imaturas e, portanto, incapazes de iniciar um novo ciclo de infecção. O ritonavir tem afinidade seletiva pela protease do HIV e pouca atividade inibitória diante da aspartil-protease humana. Pode ser utilizado também em conjunto com outros antirretrovirais da mesma classe com a finalidade de reduzir sua metabolização, diminuindo a dose necessária a cada tomada ou aumentando o intervalo entre as tomadas.
Atividade antiviral in vitro: dados in vitro indicam que ritonavir é ativo contra todas as cepas de HIV testadas em uma variedade de linhagens celulares humanas primárias e transformadas. A concentração da droga que inibe 50% e 90% da replicação viral in vitro é aproximadamente 0,02 mcM e 0,11 mcM, respectivamente. Potências similares foram observadas com cepas de HIV sensíveis e resistentes ao AZT. Estudos que avaliaram a citotoxicidade direta de ritonavir em diversas linhagens celulares não mostraram toxicidade direta em concentrações de até 25 mcM, com um índice terapêutico resultante in vitro de pelo menos 1000.
Resistência: isolados de HIV-1 resistentes ao ritonavir foram selecionados in vitro. A análise genotípica desses isolados mostrou que a resistência foi primariamente atribuída a substituições específicas de aminoácidos na protease do HIV-1 nas posições 84 (Ile por Val), 82 (Val por Phe), 71 (Ala por Val) e 46 (Met por Ile). Alterações fenotípicas e genotípicas nos isolados de HIV de pacientes selecionados tratados com ritonavir foram monitoradas em ensaios clínicos de Fase I/II. A análise genotípica e fenotípica em série indicou que a sensibilidade ao ritonavir caiu de forma ordenada e gradual. As mutações iniciais ocorreram nas posições 82 (Val por Ala/Phe), 54 (Ile por Val), 71 (Ala por Val/Thr) e 36 (Ile por Leu), seguidas pelas combinações de mutações em cinco posições adicionais específicas de aminoácidos. Cepas virais isoladas in vivo sem alteração na posição 82 não têm sensibilidade diminuída ao ritonavir. A mutação na posição 82 parece ser necessária, mas não suficiente para conferir resistência fenotípica. A resistência fenotípica foi definida como uma diminuição ³ 5 vezes na sensibilidade viral in vitro em relação ao basal. A relevância clínica das alterações genotípicas e fenotípicas associadas ao ritonavir ainda não foi estabelecida.
Resistência cruzada com outros antirretrovirais: o potencial para resistência cruzada ao HIV entre inibidores de protease não foi completamente explorado. Portanto, não se conhece o efeito que ritonavir terá na atividade de outros inibidores da protease administrados subsequentemente. Isolados de HIV obtidos em série de seis pacientes tratados com ritonavir mostraram sensibilidade reduzida in vitro a este medicamento, mas não demonstraram redução correspondente da sensibilidade in vitro ao saquinavir quando comparada aos isolados basais. Entretanto, isolados de dois desses pacientes mostraram uma redução (8 vezes) na sensibilidade in vitro ao indinavir. Isolados de cinco pacientes também foram testados quanto à resistência cruzada ao amprenavir e nelfinavir; isolados de dois pacientes apresentaram diminuição da sensibilidade ao nelfinavir (12-14 vezes) e nenhuma ao amprenavir. A resistência cruzada entre ritonavir e inibidores da transcriptase reversa é improvável, já que os alvos enzimáticos envolvidos são diferentes. Um isolado de HIV resistente a zidovudina (ZDV) testado in vitro manteve total sensibilidade ao ritonavir.
Farmacocinética
Em um estudo de farmacocinética de dose única em indivíduos infectados pelo HIV do sexo masculino, em jejum, altos níveis do fármaco foram encontrados e mantidos por várias horas após administração oral de 100 mg, 200 mg, 400 mg, 600 mg, 800 mg ou 1000 mg de ritonavir. A área sob a curva (AUC) da concentração versus tempo variou de 3,92 a 123 mcg.h/mL, respectivamente, e a Cmáx variou de 0,416 a 12,7 mcg/mL. A farmacocinética do ritonavir foi dose-dependente e aumentos maiores do que o proporcional na AUC e Cmáx com aumento de dose foram relatados.
O Tmáx permaneceu constante por aproximadamente 3 horas com o aumento de dose. A depuração renal foi, em média, inferior a 0,1 L/h e relativamente constante na faixa de dosagem. Como não há formulação parenteral de ritonavir, a biodisponibilidade absoluta não foi determinada.
Após uma dose única de 600 mg sob condições de plenitude gástrica, as formulações em cápsula gelatinosa mole de 100 mg (n=57) e em solução oral (n=18) produziram AUCs de 121,7 ± 53,8 mcg.h/mL e 129,0 ± 39,3 mcg.h/mL (média ± desvio padrão), respectivamente. Em comparação à ingestão em jejum, a extensão de absorção da cápsula gelatinosa mole foi 12% maior quando administrada com uma refeição. Quando a formulação líquida foi dada sob condições de jejum, as concentrações de pico de ritonavir aumentaram 28% em relação às condições de plenitude.
A farmacocinética do ritonavir durante regimes de múltiplas doses foi estudada em voluntários adultos HIV-positivos em condições de plenitude gástrica. Sob condições de múltiplas doses, o acúmulo de ritonavir é ligeiramente menor do que o previsto a partir da dose única devido a um aumento relacionado ao tempo e à dose na depuração aparente (Cl/F). Foi observado que concentrações mínimas de ritonavir diminuem com o tempo, possivelmente devido à indução enzimática, atingindo níveis estáveis após 2 semanas. No estado de equilíbrio, com uma dose de 600 mg duas vezes ao dia, foram observados valores de Cmáx e Cmín de 11,2 e 3,7 mcg/mL, respectivamente. O t½ de ritonavir foi de aproximadamente 3 a 5 horas. A depuração aparente no estado de equilíbrio em pacientes tratad