NEPEXTO
VIATRIS
etanercepte
Anti-reumático. Antipsoriásico.
Apresentações.
Cartucho contendo:
-4 seringas preenchidas com solução injetável contendo 25 mg de etanercepte + 4 agulhas + 4 lenços umedecidos com álcool.
-4 seringas preenchidas com solução injetável contendo 50 mg de etanercepte + 4 agulhas + 4 lenços umedecidos com álcool.
-4 seringas preenchidas em canetas aplicadoras plásticas com solução injetável contendo 50 mg de etanercepte + 4 lenços umedecidos com álcool.
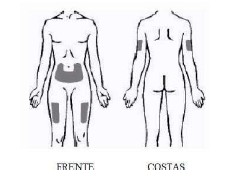
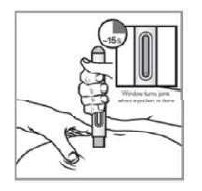
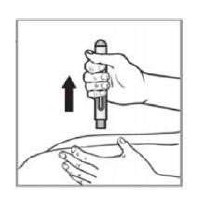
VIA DE ADMINISTRAÇÃO: SOMENTE PARA USO SUBCUTÂNEO USO ADULTO E PEDIÁTRICO ACIMA DE 8 ANOS DE IDADE
Composição.
Cada seringa preenchida de 0,5 ml de Nepexto® contém 25 mg de etanercepte (25 mg/0,5 ml).
Cada seringa preenchida de 1 ml de Nepexto® contém 50 mg de etanercepte (50 mg/ml).
Cada seringa preenchida de 1 ml em caneta aplicadora plástica de Nepexto® contém 50 mg de etanercepte (50 mg/ml).
Excipientes: glicina, citrato de sódio di-hidratado, fosfato de sódio monobásico di-hidratado, sacarose, cloreto de sódio, água para injetáveis, hidróxido de sódio e ácido clorídrico.
Não contém conservante.
Informações técnicas.
1. INDICAÇÕES
• Adulto com Artrite reumatoide
Nepexto® (etanercepte) está indicado para redução dos sinais e sintomas e inibição da progressão do dano estrutural em pacientes com artrite reumatoide ativa moderada a grave.
Nepexto® pode ser iniciado em associação ao metotrexato ou em monoterapia.
Nepexto® está indicado no tratamento da artrite reumatoide ativa moderada a grave, quando a resposta a um ou mais DMARDs (drogas modificadoras da doença artrite reumatoide) se mostrar insatisfatória.
• Adulto com Artrite psoriásica
Nepexto® é indicado na inibição do dano estrutural e na redução de sinais e sintomas de pacientes com artrite psoriásica.
• Adulto com Espondilite anquilosante
Nepexto® é indicado para redução dos sinais e sintomas em pacientes com espondilite anquilosante ativa.
• Adulto com Espondiloartrite axial não radiográfica
Nepexto® é indicado para o tratamento de adultos com espondiloartrite axial não radiográfica grave com sinais de inflamação, conforme indicado pela elevação de proteína C reativa (PCR) e/ou alteração à ressonância magnética, que tenham apresentado uma resposta inadequada ou que são intolerantes à terapia convencional.
• Adulto com Psoríase em placas
Nepexto® é indicado para o tratamento de pacientes adultos (18 anos ou mais) com psoríase crônica em placas moderada a grave que são candidatos a terapia sistêmica ou fototerapia.
• Psoríase em placas pediátrica
Nepexto® é indicado para o tratamento de psoríase crônica grave em placas em crianças e adolescentes a partir de 8 anos de idade que estão inadequadamente controlados ou são intolerantes a outra terapia sistêmica ou fototerapia.
2. RESULTADOS DE EFICÁCIA
Nepexto® é um produto biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre
o Nepexto® e o Enbrel PFS®.
-Resultados de eficácia do produto biológico comparador (Enbrel PFS®)
Pacientes adultos com artrite reumatoide
A eficácia de etanercepte foi avaliada em um estudo randomizado, duplo cego, controlado por placebo. O estudo avaliou 234 pacientes adultos com artrite reumatoide ativa, que apresentaram falhas na terapia com, pelo menos uma, mas não mais do que quatro DMARDs. Doses de 10 mg ou 25 mg de etanercepte ou placebo foram administradas por via subcutânea duas vezes por semana, durante 6 meses consecutivos. Os resultados desse estudo controlado foram expressos em porcentagem de melhora na artrite reumatoide usando os critérios de resposta do Colégio Americano de Reumatologia (ACR). Respostas ACR 20 e 50 foram superiores em pacientes tratados com etanercepte no mês 3 e no mês 6 comparado aos pacientes tratados com placebo (ACR 20: etanercepte 62% e 59%, placebo 23% e 11%, no mês 3 e no mês 6, respectivamente; ACR 50: etanercepte 41% e 40%, placebo 8% e 5% em 3 e 6 meses, respectivamente, p < 0,01 etanercepte vs. placebo em todos os pontos temporais para ambas as respostas, ACR 20 e ACR 50).
Cerca de 15% dos indivíduos que receberam etanercepte atingiram uma resposta ACR 70, no mês 3 e no mês 6, em comparação com menos de 5% dos indivíduos que receberam placebo. Entre os pacientes que receberam tratamento com etanercepte, as respostas clínicas surgiram geralmente dentro de 1 a 2 semanas após o início do tratamento e quase sempre duraram por 3 meses. Etanercepte foi significativamente melhor que placebo em todos os componentes dos critérios do ACR, bem como outras medidas de atividade de artrite reumatoide não incluídas nos critérios de resposta ACR, tal como a rigidez matinal. O Health Assessment Questionnaire (HAQ), que incluiu incapacidade, vitalidade, saúde mental, estado geral de saúde, e status dos subdomínios de saúde associado a artrite foi administrado a cada 3 meses durante o estudo. Todos os subdomínios do HAQ mostraram melhora no grupo tratado com etanercepte comparativamente ao grupo controle, em 3 e 6 meses.
Após a interrupção do uso de etanercepte, os sintomas da artrite geralmente reapareceram dentro de um mês. Baseado em resultados de estudos abertos, observou-se que a reintrodução do tratamento com etanercepte após períodos de descontinuações de, no máximo, 24 meses resultou na mesma magnitude das respostas que em pacientes que receberam etanercepte sem interrupção. Respostas duráveis continuadas foram observadas por até 10 anos em estudos de extensão abertos, quando os pacientes receberam tratamento com etanercepte sem interrupção.
A eficácia de etanercepte foi comparada ao metotrexato em um segundo estudo randomizado, controlado, com avaliações radiográficas cegas como desfecho primário em 632 pacientes adultos com artrite reumatoide ativa ( < 3 anos de duração), que nunca tinham recebido tratamento com metotrexato. Doses de 10 mg ou 25 mg de etanercepte foram administradas por via subcutânea duas vezes por semana durante um período máximo de 24 meses. As doses de metotrexato foram escalonadas a partir de 7,5 mg/semana até um máximo de 20 mg/semana durante as primeiras 8 semanas do estudo e mantida até um máximo de 24 meses. A melhora clínica, incluindo início da ação do etanercepte 25 mg em 2 semanas, foi semelhante ao observado nos estudos anteriores, e foi mantida durante um período máximo de 24 meses. No início do estudo, os pacientes tinham um grau de incapacidade moderado, com média de pontuação de 1,4 a 1,5 no HAQ. O tratamento com etanercepte 25 mg resultou numa melhoria substancial em 12 meses, com cerca de 44% dos pacientes alcançando uma pontuação normal no HAQ (menor que 0,5). Esta melhora manteve-se no Ano 2 do estudo.
Neste estudo, a lesão articular estrutural foi avaliada por radiografia e expressa como alteração na Pontuação Total de Sharp (TSS) e seus componentes, o grau de erosão e o estreitamento do espaço articular (EEA). Foram analisadas radiografias das mãos / punhos e pés no início do estudo, em 6, 12 e 24 meses. A dose de 10 mg de etanercepte apresentou consistentemente menor efeito sobre danos estruturais do que a dose de 25 mg. Etanercepte 25 mg foi significativamente superior ao metotrexato no grau de erosão para 12 e 24 meses. As diferenças entre metotrexato e etanercepte 25 mg no TSS e no EEA não foram estatisticamente significativas. Os resultados são mostrados na figura abaixo.
PROGRESSÃO RADIOGRÁFICA: COMPARAÇÃO ENTRE ETANERCEPTE vs. METOTREXATO EM PACIENTES COM ARTRITE REUMATOIDE COM < 3 ANOS DE DURAÇÃO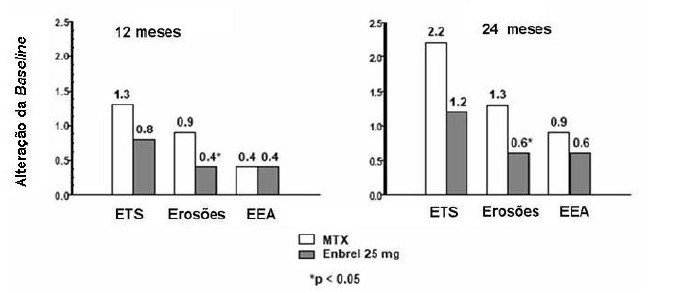
Em outro estudo controlado, duplo cego, randomizado, a eficácia clínica, a segurança e a progressão radiográfica na artrite reumatoide em pacientes tratados somente com etanercepte (25 mg duas vezes por semana), somente com metotrexato (7,5 a 20 mg semanalmente, mediana de 20 mg) e com a associação de etanercepte e metotrexato iniciados concomitantemente foram comparados a 682 pacientes adultos com artrite reumatoide ativa de 6 meses a 20 anos de duração (mediana de 5 anos) que tiveram pelo menos uma resposta satisfatória à DMARD, com exceção ao metotrexato.
Pacientes do grupo tratado com a associação de etanercepte e metotrexato tiveram respostas significativamente maiores de ACR 20, ACR 50 E ACR 70 e melhoria nas pontuações DAS E HAQ nas semanas 24 e 52 comparados aos pacientes dos grupos que receberam etanercepte isolado ou metotrexato isolado (resultados apresentados na tabela abaixo). A associação de etanercepte com metotrexato também apresentou vantagens em relação à monoterapia com etanercepte e à monoterapia com metotrexato após 24 meses.
A progressão radiográfica em 12 meses foi significativamente menor no grupo tratado com etanercepte que no grupo tratado com metotrexato, enquanto que o grupo que recebeu a associação foi significativamente melhor na desaceleração da progressão radiográfica que os grupos em monoterapia.
Vantagens significativas para o uso de etanercepte em associação ao metotrexato em comparação ao uso isolado de etanercepte e ao uso isolado de metotrexato também foram observadas após 24 meses. Do mesmo modo, também foram observadas vantagens significativas para etanercepte em monoterapia comparativamente a metotrexato em monoterapia após 24 meses.
Em uma análise na qual todos os pacientes que foram retirados do estudo por qualquer razão foram considerados como tendo progredido, a porcentagem de pacientes que não apresentaram progressão da doença (alteração ETS ≤ 0,5) aos 24 meses foi superior no grupo tratado com etanercepte associado ao metotrexato comparado aos grupos que receberam ou somente etanercepte ou somente metotrexato (62%, 50% e 36%, respectivamente, p < 0,05).
A diferença entre somente etanercepte e somente metotrexato também foi significante (p < 0,05). Entre os pacientes que completaram a terapia total de 24 meses no estudo, as taxas de não progressão foram 78%, 70% e 61%, respectivamente.
A eficácia e a segurança de etanercepte 50 mg (2 injeções SC de 25 mg) administrado 1 vez por semana foram avaliadas em um estudo duplo cego, controlado por placebo com 420 pacientes com artrite reumatoide ativa. Neste estudo, 53 pacientes receberam placebo, 214 pacientes receberam 50 mg de etanercepte uma vez por semana e 153 pacientes receberam 25 mg de etanercepte duas vezes por semana. Os perfis de segurança e eficácia dos dois regimes de tratamento com etanercepte foram comparáveis em seus efeitos sobre os sinais e sintomas da artrite reumatoide na Semana 8. Os dados da Semana 16 não apresentaram comparabilidade (não inferioridade) entre os dois regimes.
Pacientes adultos com artrite psoriásica:
A eficácia de etanercepte foi avaliada em um estudo randomizado, duplo-cego, controlado por placebo em 205 pacientes com artrite psoriásica. Os pacientes tinham entre 18 e 70 anos de idade e eram portadores de artrite psoriásica ativa (≥ 3 articulações edemaciadas e ≥ 3 articulações doloridas) em pelo menos umas das seguintes formas: (1) envolvimento interfalangiano distal (EID); (2) artrite poliarticular (ausência de nódulos reumatoides e presença de psoríase); (3) artrite mutilante; (4) artrite psoriásica assimétrica; ou (5) espondilite anquilosante.
Os pacientes também tinham psoríase em placa com lesão ≥ 2 cm de diâmetro. Tinham sido tratados com AINEs (86%), DMARDs (80%) e corticoesteroides (24%). Os pacientes que estavam em uso de metotrexato no momento (estável por ≥ 2 meses) poderiam continuar recebendo uma dose estável de 25mg/semana de metotrexato. Doses de 25 mg de etanercepte (baseado em estudos de dose em pacientes com artrite reumatoide) ou placebo foram administrados por via subcutânea duas vezes por semana durante 6 meses. No fim do estudo duplo-cego, os pacientes passaram para um estudo aberto, de longo prazo de duração total de até 2 anos.
As respostas clínicas foram expressas em porcentagem de pacientes que alcançaram a resposta de ACR 20, 50, 70 e porcentagens com melhoria no Critério de Resposta de Artrite Psoriásica (PsARC).
Entre os pacientes com artrite psoriásica que receberam etanercepte, as respostas clínicas foram evidentes na primeira visita (4 semanas) e mantidas durante 6 meses de terapia. O etanercepte foi significativamente melhor do que o placebo em todas as medidas da atividade da doença (p < 0,001), e as respostas foram semelhantes com e sem a terapia concomitante com metotrexato. A qualidade de vida em pacientes com artrite psoriásica foi avaliada utilizando o índice de incapacidade do HAQ. A pontuação do índice de incapacidade melhorou significativamente em pacientes com artrite psoriásica tratados com etanercepte, comparativamente com o placebo (p < 0,001).
Alterações radiográficas foram avaliadas no estudo de artrite psoriásica. Radiografias das mãos e dos pulsos foram obtidas no início do estudo e em 6, 12 e 24 meses. Em uma análise na qual todos os pacientes que saíram do estudo por qualquer razão foram considerados como tendo progredido, a porcentagem de pacientes sem progressão (alteração ETS ≤ 0,5) aos 12 meses foi superior no grupo etanercepte, comparativamente com o grupo do placebo (73 % vs. 47%, respectivamente, p ≤ 0,001). O efeito de etanercepte na progressão radiográfica foi mantido em pacientes que continuaram o tratamento durante o segundo ano. O abrandamento dos danos articularesperiféricos foi observado em pacientes com envolvimento poliarticular simétrico.
O tratamento com etanercepte resultou em melhoria da função física durante o estudo duplo-cego, e este benefício foi mantido durante a exposição a longo prazo de até 2 anos. Não há evidência suficiente da eficácia de etanercepte em pacientes com espondilite anquilosantesímile e artrite mutilante nas artropatias psoriásicas devido ao pequeno número de pacientes estudados. Não foi realizado nenhum estudo em pacientes com artrite psoriásica usando o esquema posológico de 50 mg uma vez por semana. A evidência de eficácia para este esquema posológico nessa população de pacientes tem sido baseada nos dados de estudos realizados em pacientes com espondilite anquilosante.
Pacientes adultos com espondilite anquilosante:
A eficácia de etanercepte na espondilite anquilosante foi avaliada em 3 estudos randomizados, duplo-cegos comparando a administração de 25 mg de etanercepte com placebo. Num total, 401 pacientes foram incluídos, dos quais 203 receberam o tratamento com etanercepte. O maior destes ensaios (n = 277) incluiu pacientes com idade entre 18 e 70 anos com espondilite anquilosante ativa definida com a pontuação da escala visual análoga (VAS) ≥ 30 para a média de duração e intensidade da rigidez matinal mais a pontuação VAS ≥ 30 para pelo menos 2 dos 3 parâmetros seguintes: avaliação global do paciente; média dos valores VAS para dor nas costas noturna e dor nas costas total; média de 10 perguntas sobre o Índice Funcional na Espondilite Anquilosante de Bath (BASDAI).
Os pacientes que receberam DMARDs, AINEs ou corticosteroides puderam continuar com doses estáveis. Pacientes com anquilose completa da coluna vertebral não foram incluídos no estudo. Doses de 25 mg de etanercepte (baseado em estudos de dose em pacientes com artrite reumatoide) ou placebo foram administrados por via subcutânea duas vezes por semana durante 6 meses em 138 pacientes.
A medida primária de eficácia (ASAS 20) foi uma melhoria ≥20% em pelo menos 3 dos 4 domínios da Avaliação da Espondilite Anquilosante (ASAS) (avaliações globais do paciente, dor nas costas, BASDAI e inflamação) e ausência de deterioração no domínio restante. Para as respostas de ASAS 50 e 70 utilizou-se os mesmos critérios, com uma melhoria de 50% ou 70%, respectivamente.
Comparado ao placebo, o tratamento com etanercepte resultou em melhora significativa no ASAS 20, ASAS 50 e ASAS 70 após 2 semanas do início da terapia. Entre os pacientes com espondilite anquilosante que receberam etanercepte as respostas clínicas foram evidentes no momento da primeira visita (Semana 2) e foram mantidas através de 6 meses de terapia. As respostas foram semelhantes em pacientes que receberam ou não terapias concomitantes no início do estudo.
Resultados similares foram obtidos em 2 estudos menores de espondilite anquilosante. Num quarto estudo duplo-cego, controlado por placebo com 356 pacientes com espondilite anquilosante ativa, avaliaram-se a eficácia e a segurança de 50 mg de etanercepte (2 injeções subcutâneas de 25 mg) administrados uma vez por semana versus 25 mg de etanercepte administrados duas vezes por semana. Os perfis de segurança e eficácia para os regimes de 50 mg uma vez por semana e 25 mg duas vezes por semana foram semelhantes.
Pacientes adultos com espondiloartrite axial não radiográfica
Estudo 1
A eficácia de etanercepte em pacientes com espondiloartrite axial não radiográfica (EANR) foi avaliada em um estudo randomizado, placebo controlado, duplo cego com duração de 12 semanas. O estudo avaliou 215 pacientes adultos (população por intenção de tratar modificada) com espondiloartrite axial não radiográfica ativa -EANR (18 a 49 anos), definida pelos pacientes que cumprem com os critérios de classificação da ASAS (Avaliação da Sociedade Internacional de Espondiloartrite) para espondiloartrite axial, mas que não cumprem os critérios modificados de Nova York para espondiloartrite axial. Os pacientes deveriam também apresentar uma resposta inadequada para dois ou mais AINEs. No período de duplo cego, os pacientes receberam 50 mg de etanercepte semanalmente ou placebo por 12 semanas. A primeira medida de eficácia (ASAS 40) foi 40% de melhora em pelo menos três dos quatro domínios de ASAS e ausência de deterioração na remissão dos domínios. Ressonâncias magnéticas da articulação sacro-ilíaca e da coluna, foram realizadas para se avaliar a inflamação no início do estudo na semana 12.
O período de duplo cego foi seguido pelo período aberto no qual os pacientes receberam 50 mg de etanercepte semanalmente por um período adicional de até 92 semanas. A comparação do tratamento com placebo e etanercepte resultou em uma melhora estatisticamente significante no ASAS 40, ASAS 20 e ASAS 5/6. Uma melhora significante também foi observada para a remissão parcial de ASAS e BASDAI 50. Os resultados de 12 semanas são apresentados na tabela abaixo: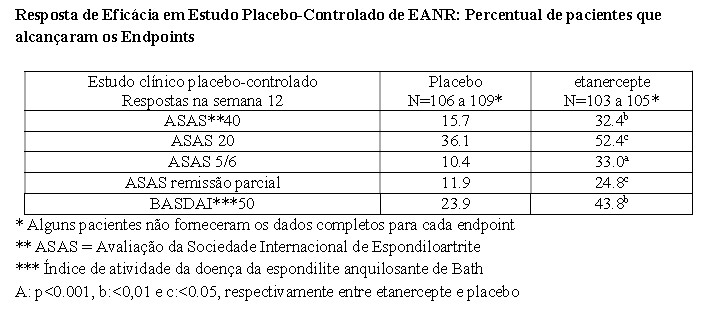
Na semana 12, houve uma melhora significante estatisticamente na pontuação (Consórcio de Pesquisa de Espondiloartrite do Canadá) para articulação sacro-ilíaca medida pela ressonância magnética para pacientes tratados com etanercepte. A variação média ajustada a partir da linha de base foi de 3.8 para pacientes tratados com etanercepte (n=95) versus 0.8 para pacientes tratados com placebo (n=105) p < 0.001.
A saúde, qualidade de vida e capacidade física foram avaliadas utilizando o BASFI (Índice funcional da espondilite anquilosante de Bath), EuroQol 5D e questionários SF-36. Etanercepte apresentou uma grande melhora estatisticamente significante na BASFI, EQ5D na Contagem Global de Estado de Saúde e do SF-36 Contagem de Componente Físico desde o início até a semana 12 comparado com o placebo.
As respostas clínicas entre pacientes com espondiloartrite axial não radiográfica que receberam etanercepte foram evidentes no momento da primeira visita (2 semanas) e foram mantidas até 2 anos da terapia. Melhorias na saúde relacionadas com qualidade de vida e função física também foram mantidas até 2 anos de terapia. Os dados de 2 anos não revelaram quaisquer novas descobertas de segurança.
Estudo 2
Este estudo multicêntrico, aberto, de fase 4, de 3 períodos avaliou a retirada e o retratamento de Enbrel® PFS em pacientes com EANR ativa que obtiveram uma resposta adequada [doença inativa definida como Pontuação de Atividade da Doença Espondilite Anquilosante (ASDAS) proteína C reativa (PCR) inferior a 1,3] após 24 semanas de tratamento.
209 pacientes adultos com EANR ativa (18 a 49 anos de idade), definidos como aqueles pacientes que atendem aos critérios de classificação de espondiloartrite axial da Sociedade Internacional de Avaliação de Espondiloartrite (ASAS) (mas não atendem aos critérios modificados de Nova Iorque para EA), tendo achados de ressonância magnética positivos (inflamação ativa na ressonância magnética altamente sugestiva de sacroileíte associada com espondiloartrite axial) e/ou PCR positiva (definida como proteína C reativa de alta sensibilidade [PCR] > 3 mg/L) e sintomas ativos definidos por um ASDAS-PCR maior ou igual a 2,1 na visita de triagem receberam abertamente Enbrel® PFS 50 mg semanalmente mais AINE usado de forma estável na dosagem anti-inflamatória ideal tolerada por 24 semanas no Período 1. Também era necessário que os pacientes tivessem uma resposta inadequada ou intolerância a dois ou mais AINEs. Na semana 24, 119 (57%) pacientes atingiram a doença inativa e entraram no Período 2, fase de retirada de 40 semanas, em que os indivíduos interromperam o etanercepte, mas mantiveram o AINE de base. A principal medida de eficácia foi a ocorrência de exacerbação (definida como ASDAS-VHS (velocidade de hemossedimentação) superior ou igual a 2,1) dentro de 40 semanas após a suspensão do Enbrel® PFS. Os pacientes com exacerbação foram tratados novamente com Enbrel® PFS 50 mg semanalmente por 12 semanas (Período 3).
No Período 2, a proporção de pacientes com ≥1 exacerbação aumentou de 22% (25/112) na semana 4 para 67% (77/115) na semana 40. No geral, 75% (86/115) dos pacientes apresentaram exacerbação em qualquer momento dentro de 40 semanas após a retirada de Enbrel® PFS.
O principal objetivo secundário do Estudo 2 foi estimar o tempo de exacerbação após a retirada do Enbrel® PFS e, adicionalmente, comparar o tempo de exacerbação para os pacientes do Estudo 1 que cumpriram os requisitos de entrada na fase de retirada do Estudo 2 e a continuação da terapia com Enbrel® PFS. O tempo médio para a exacerbação após a suspensão do Enbrel® PFS foi de 16 semanas (IC de 95%: 13-24 semanas). Menos de 25% dos pacientes no Estudo 1 que não tiveram o tratamento interrompido experimentaram uma exacerbação durante as 40 semanas equivalentes ao Período 2 do Estudo 2. O tempo para exacerbação foi estatisticamente significativamente menor em indivíduos que descontinuaram o tratamento com Enbrel® PFS (Estudo 2) em comparação com indivíduos que receberam tratamento contínuo com etanercepte (Estudo 1), p < 0,0001.
Dos 87 pacientes que entraram no Período 3 e foram tratados novamente com Enbrel® PFS 50 mg semanalmente por 12 semanas, 62% (54/87) atingiram a doença inativa, com 50% deles atingindo-a em 5 semanas (IC de 95%: 4-8 semanas).
Pacientes adultos com psoríase em placas:
A segurança e a eficácia de etanercepte nos pacientes com psoríase em placas foram avaliadas em três estudos randomizados, duplo-cegos e controlados por placebo. A avaliação final primária de eficácia nos três estudos foi a proporção de pacientes em cada grupo de tratamento que atingiu o PASI 75 (ou seja, pelo menos uma melhora de 75% na pontuação do Índice de Gravidade e Área da Psoríase [PASI] em relação ao basal) em 12 semanas.
O Estudo 1 foi um estudo de fase 2 em pacientes com psoríase em placas ativa, mas clinicamente estável, envolvendo ≥ 10% da área de superfície corpórea e com ≥ 18 anos de idade. Cento e doze
(112) pacientes foram randomizados para receber uma dose de 25 mg de etanercepte (n=57) ou placebo (n=55) duas vezes por semana por 24 semanas.
O Estudo 2 foi um estudo de fase 3 e avaliou 652 pacientes com psoríase crônica em placas utilizando os mesmos critérios de inclusão do Estudo 1 com a adição de um PASI mínimo de 10 na seleção. Etanercepte foi administrado nas doses de 25 mg uma vez por semana, 25 mg duas vezes por semana ou 50 mg duas vezes por semana por 6 meses consecutivos. Durante as 12 primeiras semanas do período de tratamento duplo-cego, os pacientes receberam placebo ou uma das três doses de etanercepte acima mencionadas. Após 12 semanas de tratamento, os pacientes do grupo placebo iniciaram o tratamento com etanercepte em regime cego (25 mg duas vezes por semana); os pacientes nos grupos de tratamento ativo continuaram até a semana 24 na dose para a qual foram originalmente randomizados.
O Estudo 3 foi um estudo de fase 3 e avaliou 583 pacientes e utilizou os mesmos critérios de inclusão do Estudo 2. Os pacientes desse estudo receberam uma dose de 25 mg ou 50 mg de etanercepte ou placebo duas vezes por semana por 12 semanas e, em seguida, todos receberam etanercepte 25 mg em regime aberto duas vezes por semana por mais 24 semanas.
No Estudo 1, o grupo tratado com etanercepte apresentou uma proporção significativamente maior de pacientes com resposta PASI 75 na Semana 12 (30%) em comparação ao grupo placebo (2%) (p < 0,0001). Em 24 semanas, 56% dos pacientes do grupo etanercepte haviam atingido PASI 75 em comparação a 5% dos que receberam placebo. Os principais resultados dos Estudos 2 e 3 são apresentados a seguir.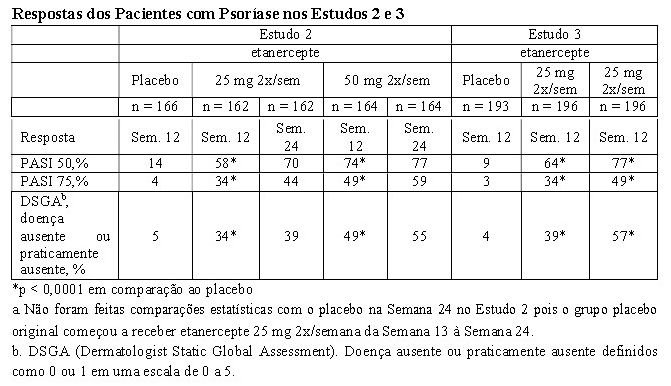
Entre os pacientes com psoríase em placas que receberam etanercepte, as respostas significativas em relação ao placebo ficaram aparentes na primeira visita (2 semanas) e foram mantidas durante as 24 semanas de terapia.
O Estudo 2 também teve um período de descontinuação do medicamento durante o qual foi interrompido o tratamento dos pacientes que atingiram uma melhora do PASI de no mínimo 50% na Semana 24. Os pacientes foram observados fora do tratamento para ocorrência de rebote (PASI ≥ 150% do basal) e tempo para recorrência (definida como perda de no mínimo metade da melhora obtida entre o basal e a Semana 24). Durante o período de descontinuação, os sintomas da psoríase retornaram gradativamente com uma mediana do tempo para recorrência da doença de 3 meses. Não foi observado rebote da doença nem eventos adversos sérios relacionados à psoríase. Houve algumas evidências que confirmaram o benefício do retratamento com etanercepte nos pacientes que responderam inicialmente ao tratamento.
No estudo 3, a maioria dos pacientes (77%) inicialmente randomizados para 50 mg duas vezes por semana e cuja dose do etanercepte foi reduzida na semana 12 para 25 mg duas vezes por semana mantiveram a resposta PASI 75 até a semana 36. Nos pacientes que receberam 25 mg duas vezes por semana durante todo o estudo, a resposta PASI 75 continuou a melhorar entre as semanas 12 e 36.
Em estudos de extensão abertos de longo prazo (até 34 meses) nos quais etanercepte foi administrado sem interrupção, as respostas clínicas foram mantidas e a segurança foi comparável a dos estudos de curto prazo.
Pacientes pediátricos com psoríase em placas:
A eficácia de etanercepte foi avaliada em um estudo randomizado, duplo-cego e controlado por placebo, com 211 pacientes pediátricos, com idade entre 4 a 17 anos, com psoríase em placas moderada a grave (conforme definido pela pontuação sPGA ≥ 3, envolvendo ≥ 10% da área de superfície corpórea, e PASI ≥ 12). Os pacientes tinham histórico de tratamento por fototerapia ou terapia sistêmica, ou estavam inadequadamente controlados pela terapia tópica.
Os pacientes receberam 0,8 mg/kg (até 50 mg) de etanercepte ou placebo uma vez por semana durante 12 semanas. Na Semana 12, um maior número de pacientes randomizados para tratamento com etanercepte apresentou respostas positivas para a eficácia (por exemplo, PASI 75) do que aqueles randomizados para receberem placebo.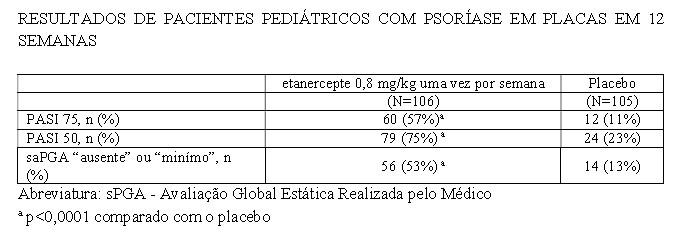
Após um período de 12 semanas de tratamento duplo-cego, todos os pacientes entraram em um estudo aberto e receberam 0,8 mg/kg (até 50 mg) de etanercepte uma vez por semana, por mais 24 semanas. As respostas observadas durante o estudo aberto foram semelhantes às respostas observadas durante período duplo-cego.
Durante um período de retirada randomizado, significativamente mais pacientes re-randomizados para receberem placebo experimentaram recidiva da doença (perda de resposta PASI 75) em comparação com os pacientes re-randomizados para receberem etanercepte. Com a continuação da terapia, as respostas foram mantidas até 48 semanas. A segurança e eficácia a longo prazo de etanercepte 0,8 mg/kg (até 50 mg), uma vez por semana, foram avaliadas em uma extensão de estudo aberto com 181 pacientes pediátricos com psoríase em placas por 2 anos, além do estudo de 48 semanas exposto acima. A experiência de longo prazo com etanercepte foi, em geral, comparável ao estudo original de 48 semanas e não revelou novos dados de segurança.
-Resultados de eficácia do produto biossimilar (Nepexto®)
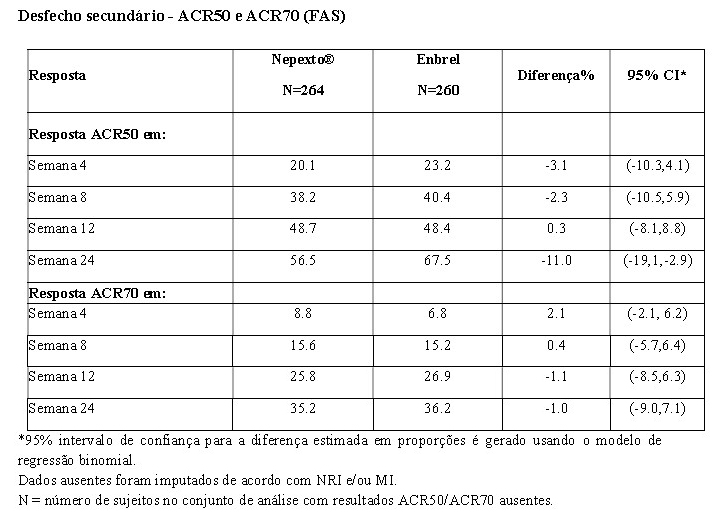
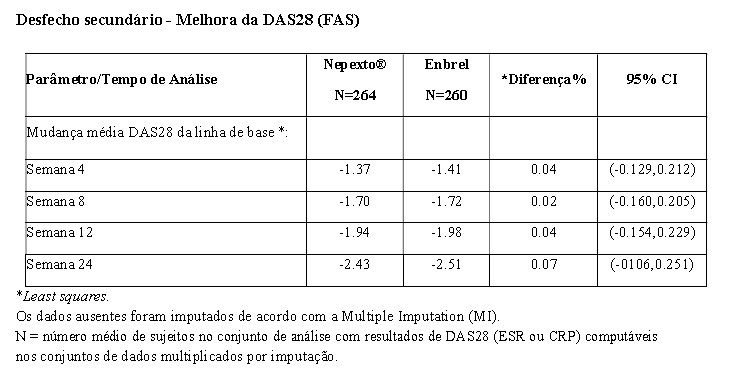
Em ensaio clínico fase 3, YLB113-002, foram avaliados 524 pacientes portadores de artrite reumatoide moderada a grave, onde Nepexto® demonstrou equivalência ao produto biológico comparador Enbrel no desfecho primário, ACR 20, em 24 semanas de tratamento. Nepexto® foi semelhante ao Enbrel nos desfechos secundários, como ACR 50 e ACR 70 e escore DAS 28 (Disease Activity Score 28) nas 24 semanas de tratamento. A tabela a seguir mostra os resultados de eficácia no desfecho primário do estudo YLB113-002: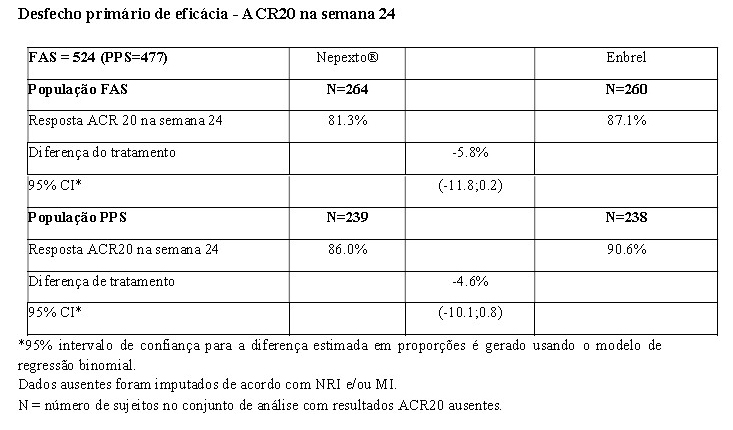
Os desfechos secundários foram avaliados em nível descritivo, conforme ilustração nas tabelas abaixo: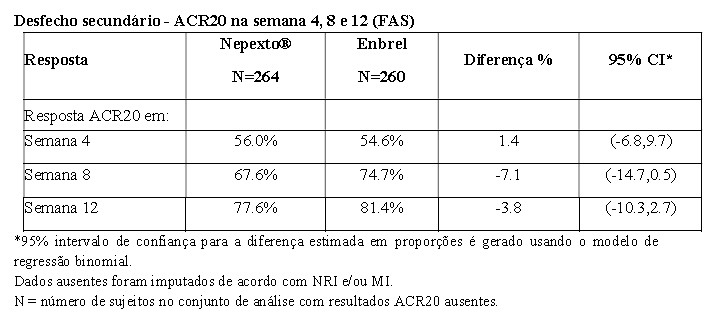
Referências
• Heijde D.V.D., Landewe R., Einstein S., at al. Radiographic Progression of Ankylosing Spondylitis After Up to Two Years of Treatment With Etanercept. Arthritis & Rheumatism. v.58 (5), p. 1324- 1331. May 2008.
• Barthon JM, Martin RW, Fleischmann RM, at al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. The New England Journal of Medicine. v.343 (22).
p. 1586-1593. November 30, 2000.
• Calin A, Dijkmans BAC, Emery P, at al. Outcomes of a multicentre randomised clinical trial of etanercept to treat ankylosing spondylitis. Ann Rheum Dis. 63: p.1594-1600. 2004.
• Dougados M, van der Heijde D, Sieper J, et al. Symptomatic efficacy of etanercept and its effects on objective signs of inflammation in early nonradiographic axial spondyloarthritis: a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2014 Aug;66(8):2091-102.
• Sterry W, Ortonne JP, Kirkham B, at al. Comparison of two etanercept regimens for treatment of psoriasis and psoriatic arthritis: PRESTA randomised double blind multicentre trial. BMJ. 340:c147. 2010.
• Gordon KB, Gottlieb AB, Leonardi CL, at al. Clinical response in psoriasis patients discontinued from and then reinitiated on etanercept therapy. Journal of Dermatological Treatment. 17: p. 9-
17. 2006.
• Gorman JDG, Sack KE, Davis JR JC. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor a. N Engl J Med, v. 346, No. 18. p.1349-1356. May 2, 2002.
• Gottlieb AB, Matheson RT, Lowe N, at al. A Randomized Trial of Etanercept as Monotherapy for Psoriasis. Arch Dermatol. v. 139, p. 1627-1632. Dec 2003.
• Keystone EC, Schiff MH, Kremer JM, at al. Once-Weekly Administration of 50 mg Etanercept in Patients With Active Rheumatoid Arthritis. Results of a Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis & Rheumatism. Vol. 50 (2), p. 353-363. February 2004.
• Kietz DA, Pepmueller PH, Moore PH. Therapeutic use of etanercept in polyarticular course juvenile idiopathic arthritis over a two years period. Ann Rheum Dis.61:171-173. 2002.
• Klareskog L, Jager JP, Gough A, at al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. The Lancet. v. 363. February 28, 2004.
• Mease PJ, Goffe BS, Metz J, at al. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised Trial. The Lancet. v. 356. July 29, 2000.
• Moreland LW, Schiff MH, Baumgartner SW, at al. Etanercept Therapy in Rheumatoid Arthritis. A Randomized, Controlled Trial. Ann Intern Med. 130: p.478-486. 1999.
• Paller AS, Siegfried EC, Langley RG, at al. Etanercept Treatment for Children and Adolescents with Plaque Psoriasis. N Engl J Med. 358: p. 241-51. 2008.
• Papp KA, Tyring S, Lahfa M, at al. A global phase III randomized controlled trial of etanercept in psoriasis: safety, efficacy, and effect of dose reduction. British Journal of Dermatology. 152, p.1304- 1312. 2005.
• Tyring S, Gordon KB, Poulin Y, at al. Long-term Safety and Efficacy of 50 mg of Etanercept Twice Weekly in Patients With Psoriasis. Arch Dermatol. 143:p.719-726. 2007.
• Heijde DVD, Silva JC, Dougados M, at al. Etanercept 50 mg once weekly is as effective as 25 mg twice weekly in patients with ankylosing Spondylitis. Ann Rheum Dis. 65: p. 1572-1577. 2006.
• Van den Bosch F, Wei JCC, Nash P, et al. OP0107 Etanercept withdrawal and re-treatment in patients with inactive non-radiographic axial spondyloarthritis at 24 weeks: results of re-embark, an open-label, phase iv trial Annals of the Rheumatic Diseases 2020;79:70.
• YLB113-002. A comparative study phase 3 to assess the Efficacy, Safety and Immunogenicity of Nepexto® (Etanercept) and Enbrel for the Treatment of Rheumatoid Arthritis.
3. CARACTERÍSTICAS FARMACOLÓGICAS
FARMACODINÂMICA
Nepexto® é uma proteína de fusão do receptor p75 do TNF humano com o fragmento Fc, produzida por tecnologia de DNA recombinante em um sistema mamífero de expressão em células de ovário de hamster chinês.
Trata-se de um dímero de uma proteína quimérica, obtido por engenharia genética pela fusão do domínio de ligação extracelular do receptor 2 do fator de necrose tumoral humano (TNFR2/p75) com o domínio Fc da IgG1 humana. Este componente Fc contém as regiões CH2 e CH3, mas não possui a região CH1 da IgG1.
Nepexto® é solúvel em água e seu peso molecular aparente é de 125 quilodaltons.
Uso geriátrico: Não se recomenda ajuste posológico específico de etanercepte de acordo com a idade do
paciente.
Mecanismo de ação
Nepexto® é a forma dimérica solúvel do receptor p75 do TNF que pode ligar-se a duas moléculas diferentes: TNFa e linfotoxina-alfa [LTa](TNFb).
Nepexto® inibe a ligação do TNF (TNFa) e da linfotoxina-alfa [LTa] (TNFb) aos receptores de TNF na superfície celular, tornando o TNF biologicamente inativo e impedindo as respostas celulares mediadas pelo mesmo. O TNF é uma citocina dominante no processo inflamatório da artrite reumatoide. Os níveis do TNF no fluido sinovial de pacientes com artrite reumatoide e artrite idiopática juvenil estão elevados. Na psoríase em placas, a infiltração por células inflamatórias, incluindo as células T, resultou em níveis aumentados do TNF nas lesões psoriásicas em comparação aos níveis na pele não envolvida.
Existem dois receptores naturais diferentes para o TNF (TNFRs), uma proteína de 55 quilodaltons (p55) e outra de 75 quilodaltons (p75), que existem naturalmente como moléculas monoméricas na superfície celular e sob a forma solúvel. A atividade biológica do TNF depende da ligação a um ou ambos os receptores da superfície celular. Nepexto® também pode modular respostas biológicas, controladas por outras moléculas de etapas posteriores da cadeia (por ex.: citocinas, moléculas de adesão ou proteinases), que são induzidas ou reguladas pelo TNF.
Etanercepte inibe a atividade do TNF in vitro e tem demonstrado alterar vários modelos animais de inflamação, entre eles, o de artrite induzida por colágeno em camundongos.
A atividade biológica do Nepexto® foi comprovada em múltiplos testes funcionais ortogonais in vitro, conduzidos em comparação com o produto biológico Enbrel, os quais demonstraram similaridade.
FARMACOCINÉTICA
Absorção: etanercepte é absorvido lentamente do local da administração subcutânea, atingindo concentração máxima aproximadamente 48 horas após uma dose única. A biodisponibilidade absoluta é de 76%.
Distribuição: Após uma dose única subcutânea de 25 mg de etanercepte, a média da concentração sérica máxima em voluntários saudáveis foi de 1,65 ± 0,66 m/mL e a área sob a curva (AUC) foi de 235 ± 96,6 m.h/mL. A proporcionalidade à dose ainda não foi avaliada formalmente, mas não há saturação aparente do processo de depuração ao longo do intervalo de doses.
O volume de distribuição no estado de equilíbrio após a administração subcutânea é de 13,9 ± 9,4 litros.
Após a administração contínua de etanercepte em pacientes com artrite reumatoide (n=25) por 6 meses, na dose de 25 mg duas vezes por semana, o nível mediano observado foi de 3,0 m/mL (variação entre 1,7 e 5,6 m/mL). Com base nos dados disponíveis, alguns pacientes podem apresentar aumento de duas a cinco vezes nos níveis séricos com a administração repetida.
Eliminação: etanercepte é depurado lentamente do organismo. A meia-vida é de aproximadamente 80 horas.
A depuração é de cerca de 175 ± 116 mL/h em pacientes com artrite reumatoide e de 131 ± 81 mL/h em voluntários saudáveis.
Após a administração de etanercepte radiomarcado a pacientes e voluntários o composto radioativo é eliminado na urina.
Disfunção renal ou hepática: Embora haja eliminação de radioatividade na urina após a administração de etanercepte radiomarcado a pacientes e voluntários, não foi observado aumento nas concentrações de etanercepte em pacientes com insuficiência renal aguda ou falência hepática. A presença de insuficiência renal ou hepática não deve requerer modificação de dose.
Sexo: Não há diferença farmacocinética aparente entre homens e mulheres.
Relação Concentração-Efeito: As concentrações séricas no estado de equilíbrio de 1 a 2 mg/L de etanercepte estão associadas a efeito ideal e são obtidas com as doses de 25 mg, duas vezes por semana. Em um estudo cruzado, aberto, de dose única e de dois tratamentos em 28 voluntários saudáveis, foi observado que etanercepte administrado em injeção única de 50 mg/mL é bioequivalente a duas injeções simultâneas de 25 mg/mL.
O tempo médio estimado para início de ação de etanercepte é de 2 semanas, podendo se modificar a depender da gravidade dos sintomas.
DADOS DE SEGURANÇA PRÉ-CLÍNICOS
Carcinogenicidade: Não foram conduzidos estudos de longo prazo em animais para avaliar o potencial carcinogênico de etanercepte. Esses estudos não são viáveis, pois animais podem desenvolver anticorpos para etanercepte, que é uma proteína humana.
Mutagenicidade: Foram conduzidos estudos de mutagênese in vitro e in vivo e não foi observada nenhuma evidência de atividade mutagênica.
Prejuízo à fertili