MYLOTARG
PFIZER
gentuzumabe ozogamicina
Antineoplásico.
Apresentações.
Mylotarg® 4,5 mg em embalagens contendo 1 frasco-ampola com 4,5 mg de pó liofilizado para solução injetável.
VIA DE ADMINISTRAÇÃO: VIA INTRAVENOSA
USO ADULTO E PEDIÁTRICO ACIMA DE 15 ANOS
CUIDADO: AGENTE CITOTÓXICO
Composição.
Cada frasco-ampola de Mylotarg® contém o equivalente a 4,5 mg de gentuzumabe ozogamicina. Após reconstituição, a solução concentrada contém 1 mg/mL de gentuzumabe ozogamicina com volume extraível de 4,5 mL (4,5 mg) por frasco-ampola.
Excipientes: sacarose, dextrana, cloreto de sódio, fosfato de sódio monobásico monoidratado e fosfato de sódio dibásico anidro.
AVISO: HEPATOTOXICIDADE
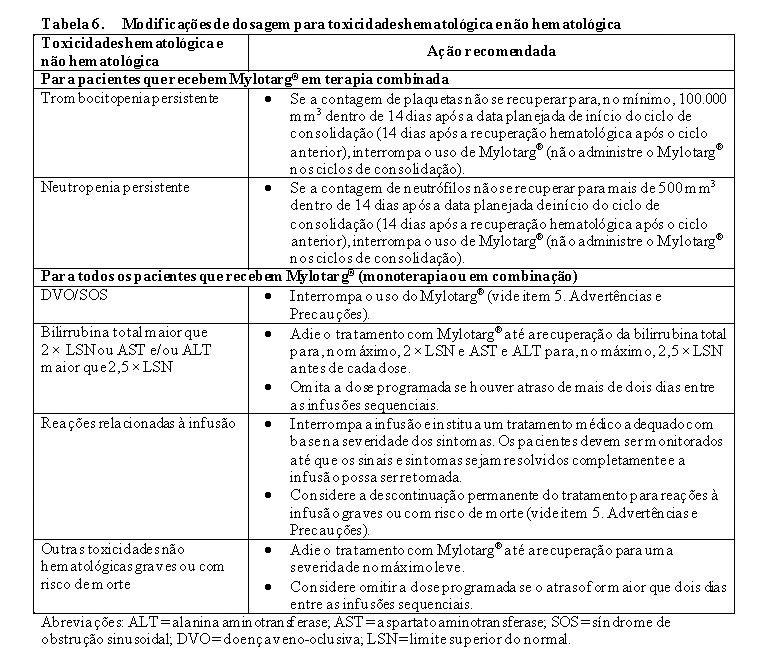
Foi notificada hepatotoxicidade, incluindo doença veno-oclusiva hepática (DVO) grave ou fatal, também conhecida como síndrome de obstrução sinusoidal (SOS), em associação com a utilização de Mylotarg® como agente único e como parte de um regime de quimioterapia combinada. Monitorar frequentemente os sinais e sintomas de DVO após o tratamento com Mylotarg® (vide item 5. Advertências e Precauções - Hepatotoxicidade, Incluindo Doença Veno-oclusiva Hepática/Síndrome de Obstrução Sinusoidal (DVO/SOS) e 2. RESULTADOS DE EFICÁCIA).
Informações técnicas.
1.INDICAÇÕES
Mylotarg® (gentuzumabe ozogamicina) é indicado para:
- Leucemia mieloide aguda (LMA) recém diagnosticada (terapia de combinação):
•Terapia de combinação com daunorrubicina (DNR) e citarabina (AraC) para o tratamento depacientes com 15 anos de idade ou mais, com LMA primária CD33-positivo não tratados previamente, com exceção de leucemia promielocítica aguda (LPA) (vide item 5. Advertências e Precauções e item 2. Resultados de Eficácia).
2.RESULTADOS DE EFICÁCIA
Estudos de pacientes com LMA primária não tratados previamente
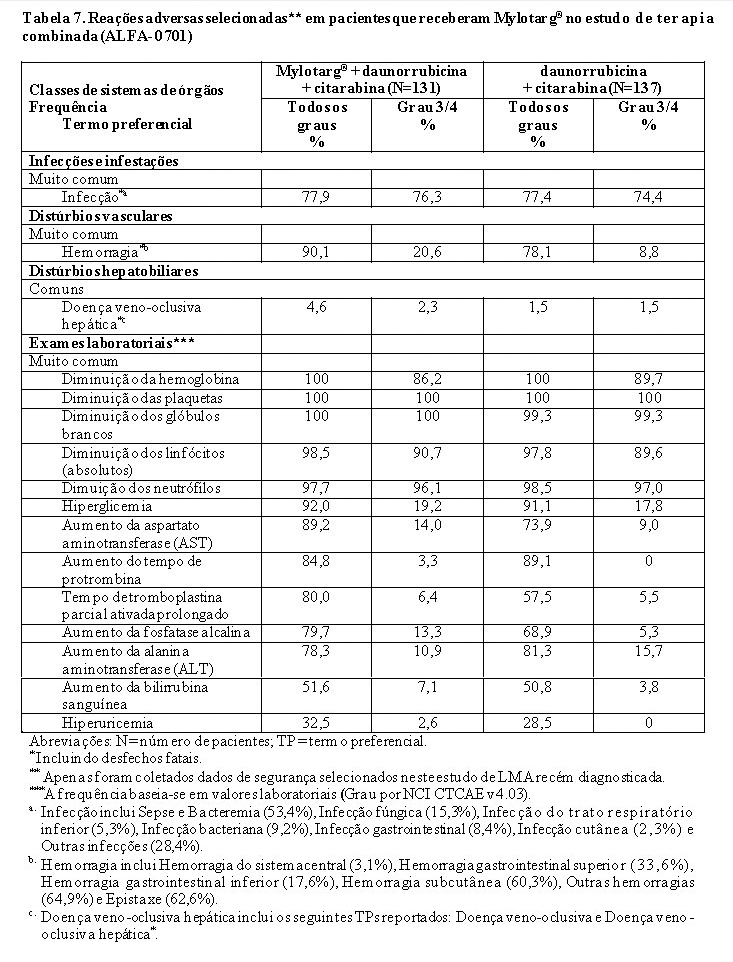
A eficácia e a segurança de Mylotarg® foram avaliadas em um estudo clínico de fase 3, multicêntrico, randomizado, aberto (ALFA-0701) comparando a adição de Mylotarg® ao regime de quimioterapia de indução padrão com daunorrubicina e citarabina (DA) versus DA isolada. Os pacientes elegíveis eram adultos entre 50 e 70 anos de idade com LMA primária não tratados previamente.
Pacientes que não atingiram uma resposta após a primeira indução puderam receber uma segunda indução com DNR e AraC isolada. Pacientes com resposta receberam terapia de consolidação com 2 ciclos de tratamento incluindo DNR (60 mg/m2 no Dia 1 do ciclo de consolidação 1; 60 mg/m2 nos Dias 1 e 2 do ciclo de consolidação 2) e AraC (1 g/m2 a cada 12 horas nos Dias 1 a 4) com ou sem Mylotarg® 3 mg/m2 (até o máximo de um frasco-ampola) no Dia 1 de acordo com sua randomização inicial. Os pacientes que apresentaram remissão também foram elegíveis ao transplante alogênico. Foi recomendado um intervalo de ao menos 2 meses entre a última dose de Mylotarg® e o transplante.
O desfecho primário foi a sobrevida livre de evento (SLE). O desfecho secundário incluiu taxas de RC e RCp, sobrevida geral livre de recidiva (SLR), sobrevida global (SG) e segurança da combinação de DA com ou sem Mylotarg®.
No total, 271 indivíduos foram randomizados nesse estudo com 135 para o tratamento de indução de 3+7 DA mais doses fracionadas de 3 mg/m2 × 3 de Mylotarg® e 136 para 3+7 DA isolada (vide item 8. Posologia e Modo de Usar). Um segundo ciclo de terapia de indução com DA mas sem Mylotarg® foi permitido, independentemente do braço de randomização. Pacientes em ambos os braços que não receberam o segundo ciclo da terapia de indução e não atingiram uma RC após indução puderam receber um ciclo de salvamento composto de idarrubicina, AraC e fator estimulante de colônia de granulócitos (G-CSF).
Pacientes com RC ou RCp receberam terapia de consolidação com dois ciclos de tratamento, incluindo DNR e AraC com ou sem Mylotarg® de acordo com sua randomização inicial. Os pacientes que apresentaram remissão também foram elegíveis ao transplante alogênico. Foi recomendado um intervalo de pelo menos 2 meses entre a última dose de Mylotarg® e o transplante.
Dados de segurança que consistem em TEAEs selecionados e considerados mais importantes para a compreensão do perfil de segurança de Mylotarg®, bem como todos os eventos adversos (EAs) que levaram à descontinuação permanente do tratamento foram coletados de modo retrospectivo. Os TEAEs selecionados consistiam em todos os graus de hemorragia, todos os graus de DVO/SOS e infecções graves.
No geral, a idade mediana dos pacientes era de 62 anos e muitos pacientes (87,8%) apresentaram um estado de desempenho do Eastern Cooperative Oncology Group (ECOG PS) de 0 a 1 na avaliação inicial. As características basais foram equilibradas entre os braços de tratamento, com exceção do gênero, visto que uma porcentagem maior de homens foi inscrita no braço de Mylotarg® (54,8%) do que no braço de DA isolada (44,1%). No geral, 59,0% e 65,3% dos pacientes tinham documentado doença de risco favorável/intermediário conforme a classificação de risco da National Comprehensive Cancer Network (NCCN) e da European LeukemiaNet (ELN) de 2010, respectivamente. A expressão de CD33 nos blastos de LMA por citometria de fluxo harmonizada a partir dos resultados laboratoriais locais foi determinada em 194/271 (71,6%) pacientes em geral. Poucos pacientes (13,7%) apesentaram baixa expressão de CD33 (menos de 30% dos blastos).
O estudo atingiu seu objetivo principal de demonstrar que o Mylotarg® adicionado em doses fracionadas (3 mg/m2 × 3) à quimioterapia de indução para pacientes com LMA primária não tratados previamente resultou em uma melhora estatística e clinicamente significativa na SLE. A SLE mediana foi de 17,3 meses (intervalo de confiança [IC] de 95%: 13,4; 30,0) no braço de Mylotarg® versus 9,5 meses (IC de 95%: 8,1; 12,0) no braço de DA isolada; razão de risco (HR) 0,562 (IC de 95%: 0,415; 0,762), valor p bilateral = 0,0002 pelo teste log-rank. Os resultados de SLE derivados da avaliação do investigador estão resumidos na Tabela 1 e o gráfico de Kaplan-Meier está demonstrado na Figura 1.
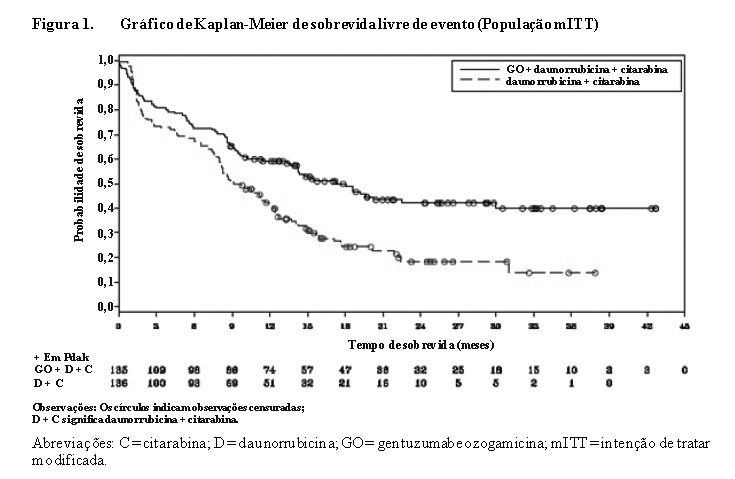
Uso em LMA com risco adverso citogenético
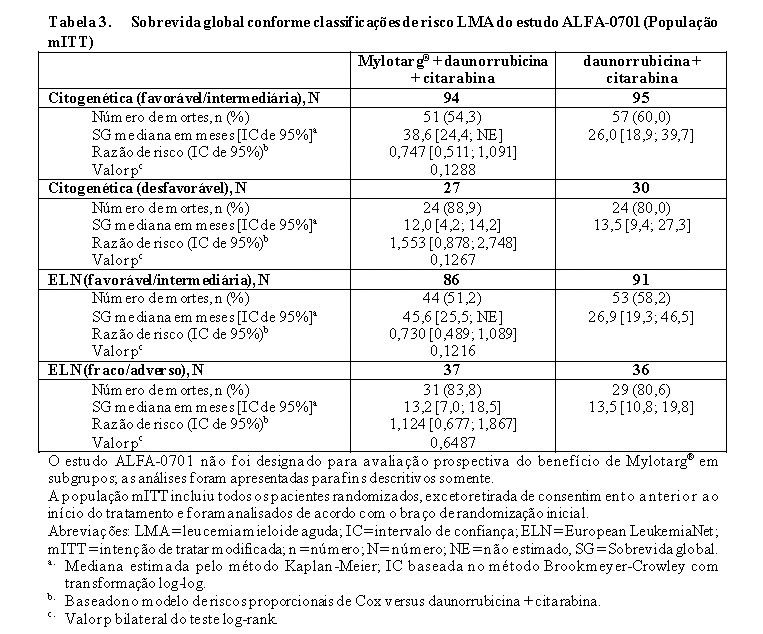
Em análises de subgrupos no ALFA-0701, a adição de Mylotarg® à quimioterapia de combinação padrão não melhorou o SLE no subgrupo de pacientes que apresentaram risco citogenético adverso (HR 1,11; IC 95%: 0,63, 1,95). SLE e SG analisados pela classificação de risco citogenético e classificação de risco citogenético/molecular estão apresentados na Tabela 2 e Tabela 3.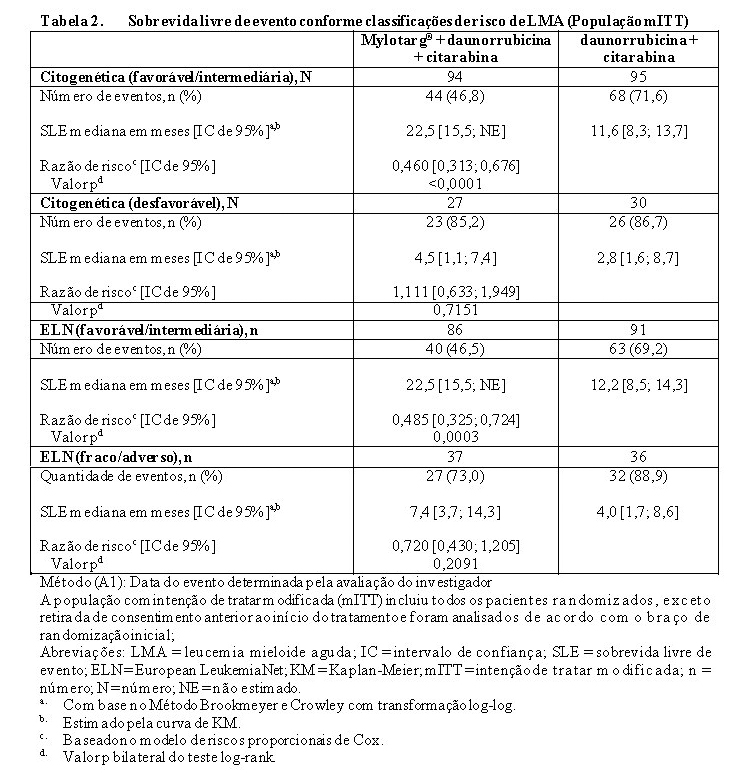
Referências
1. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patientswith de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet.Apr 21 2012;379(9825):1508-1516.
2. Castaigne S. Final Analysis of the ALFA 0701 Study ORAL PRESENTATION. 56th ASH AnnualMeeting and Exposition. 2014.
3.CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
O gentuzumabe ozogamicina é um ADC direcionado a CD33. O gentuzumabe é um anticorpo de imunoglobulina humanizada classe G subtipo 4 (IgG4) que reconhece especificamente o CD33 humano. A parte do anticorpo (hP67.6) é ligada especificamente ao antígeno CD33, uma proteína de adesão dependente de ácido siálico encontrada na superfície de blastos leucêmicos mieloides e células normais imaturas de linhagem mielomonocítica, mas não em células-tronco hematopoiéticas normais. A pequena molécula, N-acetil gama caliqueamicina, é um produto natural semissintético citotóxico. A N-acetil gama caliqueamicina é covalentemente ligada ao anticorpo via um ligante de ácido butanoico AcBut (4-(4'-acetilfenoxi). Dados não clínicos sugerem que a atividade anticancerígena do gentuzumabe ozogamicina deve-se à ligação do ADC às células cancerígenas que expressam CD33, seguida da internalização do complexo ADC-CD33 e à liberação intracelular de N-acetil gama caliqueamicina dimetil hidrazida via clivagem hidrolítica do ligante. A ativação da N-acetil gama dimetil hidrazida induz quebras na cadeia dupla do DNA, consequentemente levando a interrupção do ciclo celular e morte celular por apoptose.
Presume-se que a saturação de uma elevada porcentagem de sítios antigênicos CD33 é necessária para a entrega máxima de caliqueamicina em células blásticas leucêmicas. Observou-se uma saturação de CD33 periférico quase máxima nos estudos após administração de gentuzumabe ozogamicina a níveis de dose de 2 mg/m2 ou superiores.
Estudos in vitro também demostraram que, após uma dose de 3 mg/m2, a reexpressão de locais de CD33 disponíveis ocorreu a cada 72 horas para níveis próximos ao pré-tratamento antes da dose seguinte. Esta observação levou à hipótese de que administração repetida de doses menores de gentuzumabe ozogamicina pode ser capaz de melhorar o processo de internalização e, consequentemente, o acúmulo intracelular do medicamento, melhorando simultaneamente a segurança em comparação com o regime posológico mais elevado não fracionado.
Efeitos farmacodinâmicos
Os ensaios de citotoxicidade in vitro demonstraram que o gentuzumabe ozogamicina foi eficaz em matar seletivamente as células alvo na linhagem de células de leucemia humana (HL-60). Em modelos murinos não clínicos, o gentuzumabe ozogamicina demonstra efeitos antitumorais no tumor de xenoenxerto de leucemia promielocítica humana de HL-60 em camundongos atímicos. A combinação de quimioterapia AraC e DNR com gentuzumabe ozogamicina foi eficaz na eliminação da doença e no prolongamento de sobrevida em modelos LMA não clínicos.
Propriedades Farmacocinéticas
O gentuzumabe ozogamicina é um ADC composto de anticorpo (hP67.6) monoclonal direcionado a CD33 que é covalentemente ligado ao agente citotóxico N-acetil-gama caliqueamicina. A farmacocinética (PK) de gentuzumabe ozogamicina está descrita pela medição de características de PK do anticorpo (hP67.6), bem como pelos derivados de caliqueamicina total e não conjugada. Dado que a porção hP67.6 fornece seletividade alvo na molécula intacta, e que as dosagens de Mylotarg® são reportadas em termos de miligramas de proteína (hP67.6), os resultados de concentração de hP67.6 são reportados como medidas de PK primárias. Após o gentuzumabe ozogamicina se ligar ao alvo, ele é internalizado e a N-acetil caliqueamicina é liberada por clivagem hidrolítica. A determinação dos parâmetros de PK para caliqueamicina não conjugada foi limitada devido aos baixos níveis de concentração sistêmica.
Não foram coletados dados clínicos de PK usando o regime fracionado, no entanto a PK foi simulada usando o modelo de PK da população. Embora a dose total do regime posológico fracionado seja a metade da dose no regime posológico original (9 versus 18 mg/m2), a área total prevista sob a curva de concentração plasmática versus tempo (AUC) de hP67.6 no ciclo de tratamento é 25%, e a concentração máxima observada (Cmáx) é 24%, dos valores do regime posológico original de 9 mg/m2, uma vez que a PK não é linear. Quando o gentuzumabe ozogamicina é administrado a 3 mg/m2 nos Dias 1, 4 e 7, prevê-se que a Cmáx de hP67.6, que ocorreria no final da infusão, seja 0,38 mg/L após a primeira dose e aumente para 0,63 mg/L após a terceira dose.
Distribuição
In vitro, a ligação da N-acetil gama caliqueamicina dimetil hidrazida a proteínas plasmáticas humanas é aproximadamente 97%. In vitro, N-acetil gama caliqueamicina dimetil hidrazida é um substrato de glicoproteína-P (P-gp). Análises de PK da população demonstraram que o volume total de distribuição do anticorpo hP67.6 (soma de V1 [10 L] e V2 [15 L]) era de aproximadamente 25 L.
Biotransformação
A via metabólica primária de gentuzumabe ozogamicina é prevista que seja a liberação hidrolítica de N-acetil gama caliqueamicina dimetil hidrazida. Estudos in vitro demonstraram que N-acetil gama caliqueamicina dimetil hidrazida é extensamente metabolizada, principalmente através da redução não enzimática da fração de dissulfeto. Espera-se que a atividade (citotoxicidade) dos metabólitos resultantes seja significativamente atenuada. Em pacientes, os níveis plasmáticos de caliqueamicina não conjugada tipicamente são baixos, com uma média geométrica prevista de Cmáx de 1,5 ng/mL (IC de 95%: 1,4; 1,6) após a terceira dose.
Eliminação
Com base nas análises de PK populacional, o valor da depuração prevista (CL) de hP67.6 do plasma foi 3 L/h imediatamente após a primeira dose e depois 0,3 L/h. Prevê-se que a meia-vida plasmática terminal (t½) para hP67,6 seja de aproximadamente 160 horas para um paciente adulto típico do sexo masculino no nível de dose recomendado (3 mg/m2) de Mylotarg®.
Efeito de outros medicamentos sobre gentuzumabe ozogamicina
In vitro, N-acetil gama caliqueamicina dimetil hidrazida é metabolizada principalmente via redução não enzimática. Assim, é pouco provável que a coadministração de Mylotarg® com inibidores ou indutores do citocromo P450 (CYP) ou das enzimas metabolizadoras de fármaco difosfato de uridina glucuronosiltransferase (UGT) altere a exposição à N-acetil gama caliqueamicina dimetil hidrazida.
Com base nas análises de PK da população, não é esperado que a combinação de gentuzumabe ozogamicina com hidroxiureia, DNR, e AraC cause alterações clinicamente significativas na PK de hP67.6 ou caliqueamicina não conjugada.
Efeito de gentuzumabe ozogamicina sobre outros medicamentos
Efeito sobre os substratos da CYP
In vitro, N-acetil gama caliqueamicina dimetil hidrazida e gentuzumabe ozogamicina demonstraram um baixo potencial de inibir as atividades de CYP1A2, CYP2A6 (testado somente com gentuzumabe ozogamicina), CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4/5 nas concentrações clinicamente relevantes. In vitro, N-acetil gama caliqueamicina dimetil hidrazida e gentuzumabe ozogamicina apresentaram baixo potencial de induzir as atividades de CYP1A2, CYP2B6 e CYP3A4 em concentrações clinicamente relevantes.
Efeito sobre os substratos de UGT
In vitro, N-acetil gama caliqueamicina dimetil hidrazida demonstrou baixo potencial de inibir as atividades de UGT1A1, UGT1A4, UGT1A6, UGT1A9 e UGT2B7 em concentrações clinicamente relevantes.
Efeitos sobre os substratos transportadores de medicamentos
In vitro, N-acetil gama caliqueamicina dimetil hidrazida demonstrou baixo potencial para inibir as atividades de P-gp, proteína de resistência ao câncer da mama (BCRP), bomba de exportação de sais biliares (BSEP), proteína associada à resistência a vários medicamentos (MRP) 2, proteína de extrusão de vários medicamentos e toxinas (MATE)1 e MATE2K, transportador de ânion orgânico (OAT)1 e OAT3, transportador de cátion orgânico (OCT) 1 e OCT 2, e polipeptídio transportador de ânion orgânico (OATP)1B1 e OATP1B3 em concentrações clinicamente relevantes.
Efeito sobre AraC e DNR
Com base em uma análise da PK populacional, não está previsto que a combinação de gentuzumabe ozogamicina com DNR e AraC cause alterações clinicamente significativas na PK desses agentes.
Farmacocinética em grupos específicos de participantes de pesquisa ou pacientes
Idade, raça e gênero
Com base em uma análise da PK populacional, a idade, a raça e o gênero não afetaram a disposição do Mylotarg® de forma significativa.
Insuficiência hepática
Nenhum estudo formal da PK do Mylotarg® foi conduzido em pacientes com insuficiência hepática.
Com base em uma análise da PK populacional, não é esperado que a depuração de gentuzumabe ozogamicina (anticorpo hP67.6 e caliqueamicina não conjugada) seja afetada pela situação da insuficiência hepática leve ou moderada conforme definido pelo National Cancer Institute Organ Dysfunction Working Group (NCI ODWG). A análise incluiu 405 pacientes nas seguintes categorias de situação de insuficiência do NCI ODWG: leve (B1, n=58 e B2, n=19), moderada (C, n=6) e função hepática normal (n=322). Não se estudou a PK da gentuzumabe ozogamicina em pacientes com insuficiência hepática grave (vide item 8. Posologia e Modo de Usar).
Insuficiência renal
Nenhum estudo formal da PK de gentuzumabe ozogamicina foi conduzido em pacientes com insuficiência renal.
Com base em uma análise da PK populacional em 406 pacientes, a depuração de gentuzumabe ozogamicina em pacientes com insuficiência renal leve (CLcr 60-89 mL/min; n=149) ou insuficiência renal moderada (CLcr 30-59 mL/min; n=47) foi semelhante aos pacientes com função renal normal (CLcr ≥90 mL/min; n=209). O impacto da insuficiência renal grave sobre a PK de gentuzumabe ozogamicina não pode ser avaliado, já que estão disponíveis dados de apenas um paciente (CLcr 15-29 mL/min; n=1).
Uso geriátrico
O uso de Mylotarg® em combinação com DNR e AraC nos pacientes adultos recém-diagnosticados com LMA primária está apoiado em um estudo controlado, randomizado que incluiu 50 pacientes com 65 anos ou mais. Não foram observadas diferenças gerais de segurança ou efetividade entre esses indivíduos e indivíduos mais jovens.
Uso pediátrico
A segurança e a eficácia de Mylotarg® em combinação com a quimioterapia não foram estabelecidas nos pacientes pediátricos com idade < 15 anos com LMA primária recém-diagnosticada.
Dados de segurança pré-clínicos
Toxicidade de dose repetida
Em estudos de toxicidade de dose repetida em ratos e/ou macacos com até 12 semanas de duração, as toxicidades importantes ocorreram no fígado (elevações da enzima hepática, alterações hepatocelulares, células ovais/hiperplasia do ducto biliar e dilatação sinusoidal com atrofia hepatocitária), medula óssea e órgãos linfoides (hipocelularidade), parâmetros de hematologia (diminuição da massa de eritrócitos (RBC) e contagens de leucócitos, principalmente linfócitos), rins (alterações tubulares e/ou glomerulares, e proteinúria), olhos (degeneração e pigmentação do epitélio corneano, e inchaço peripapilar do nervo óptico) e órgãos reprodutores masculinos (atrofia de túbulos seminíferos, oligospermia e atrofia da glândula mamária) e femininos (atrofia do ovário, oviduto, útero e colo uterino). Efeitos no fígado, rins e órgãos reprodutores masculinos em ratos, e em tecidos linfoides em macacos não foram reversíveis nos estudos de 6 semanas seguidos de um período de 4 semanas sem dosagem (aproximadamente 18 vezes para ratos e 36 vezes para macacos a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168). Os efeitos sobre os órgãos reprodutores femininos e os olhos em macacos foram adversos no estudo de 12 semanas (aproximadamente 193 e 322 vezes, respectivamente, a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168).
Genotoxicidade
O gentuzumabe ozogamicina foi clastogênico in vivo na medula óssea de camundongos a ≥22,1 mg/m2. Isso é consistente com a indução conhecida de quebras de DNA por caliqueamicina e outros antibióticos antitumorais enediinos. A n-acetil gama caliqueamicina dimetil hidrazida (a citotoxina liberada) foi mutagênica no ensaio de mutação reversa bacteriana e clastogênica no ensaio de micronúcleo in vitro em células humanas TK6.
Carcinogenicidade
Estudos formais de carcinogenicidade não foram conduzidos com gentuzumabe ozogamicina. Após 6 semanas de administração de gentuzumabe ozogamicina em ratos, foram observadas lesões pré-neoplásicas (hiperplasia de célula oval mínima a leve) no fígado a 7,2 mg/m2/semana (aproximadamente 54 vezes a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168). Não houve lesões pré-neoplásicas ou neoplásicas observadas em macacos até 22 mg/m2/semana (aproximadamente 115 vezes a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168). Foram observadas lesões pré-neoplásicas e neoplásicas nos fígados de ratos com outros anticorpos conjugados à caliqueamicina.
Toxicidade reprodutiva
No estudo de fertilidade feminina, onde ratas tratadas foram acasaladas com ratos não tratados no final do período de dosagem, não foram observados efeitos relacionados com gentuzumabe ozogamicina na cópula ou fertilidade; no entanto, números ligeiramente menores de corpos lúteos a 1,08 mg/m2/dia e aumento da letalidade embrionária a ≥0,36 mg/m2/dia foram observados na presença de toxicidade maternal. Foram observados achados relacionados com gentuzumabe ozogamicina no trato reprodutivo de macacas após 12 semanas de dosagem a ≥2,2 mg/m2/semana (atrofia no ovário, oviduto, útero e colo uterino; aproximadamente 66 vezes a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168). Achados no trato reprodutivo feminino foram adversos a ≥6,6 mg/m2/semana (aproximadamente 193 vezes a exposição clínica humana após a terceira dose de 3 mg/m2 com base na AUC168) em razão do potencial previsto para interrupção ou perda de um ciclo menstrual normal e, assim, função reprodutiva normal.
No estudo de fertilidade masculina, onde ratos tratados foram acasalados com ratas não tratadas no final do período de dosagem, os efeitos relacionados com gentuzumabe ozogamicina na reprodução masculina incluíram espermatogônia e espermatócitos menores, diminuição das espermátides testiculares e do esperma do epidídimo, vacuolização do núcleo das espermátides e/ou aparecimento de células gigantes a ≥0,12 mg/ m2/dia. Achados adicionais incluíram efeitos nos testículos (≥0,12 mg/ m2/dia) e epidídimos (≥0,36 mg/ m2/dia); ambos os órgãos eram macroscopicamente pequenos e diminuíram em peso, bem como fertilidade (1,08 mg/ m2/dia). Quando ratos foram acasalados novamente depois de um período sem dosagem de 9 semanas, os efeitos sobre o esperma e a fertilidade foram piores, mas houve recuperação parcial da baixa espermatogônia e espermatócitos nos testículos. No estudo de toxicidade de 6 semanas com gentuzumabe ozogamicina, efeitos sobre os órgãos reprodutivos masculinos (testículos, epidídimos e glândula mamária) foram observados a ≥2,4 mg/ m2/semana (aproximadamente 18 vezes a exposição clínica humana após a terceira dose humana de 3 mg/ m2 com base na AUC). Os efeitos sobre os órgãos reprodutivos masculinos de ratos foram parcialmente reversíveis ou não reversíveis após um período sem dosagem de 4 semanas. Os efeitos nos órgãos reprodutivos de macacos machos em um estudo de toxicidade de 6 semanas incluíram achados nos testículos e epidídimos e redução do peso médio dos testículos em 18 mg/ m2/semana (aproximadamente 81 vezes a exposição clínica humana após a terceira dose humana de 3 mg/ m2 com base na AUC168). Durante o estudo de 12 semanas em macacos, os achados adversos no trato reprodutivo de machos sexualmente maduros foram observados a ≥2,2 mg/ m2/semana (aproximadamente 66 vezes a exposição clínica humana após a terceira dose de 3 mg/ m2 com base na AUC168) e consistiram de leve degeneração de túbulos seminíferos no testículo; detritos celulares no lúmen mínimos ou leves e oligospermia e degeneração epitelial mínima a moderada no epidídimo; e leve atrofia epitelial, leve ectasia do ducto, e estase mínima ou leve de esperma na vesícula seminal.
Toxicidade de desenvolvimento
Em um estudo de desenvolvimento embrionário-fetal em ratos, animais gestantes receberam doses diárias intravenosas até 1,2 mg/ m2/dia de gentuzumabe ozogamicina durante o período de organogênese. Menor peso corpóreo fetal, maior incidência de costelas onduladas fetais e menor incidência de ossificação esquelética fetal foram observados a ≥0,15 mg/ m2/dia. Aumento da letalidade embrionária e anomalias morfológicas fetais (malformações digitais, ausência do arco aórtico, anomalias nos ossos longos nos membros dianteiros, escápula disforme, ausência de centro vertebral e esterno fundido) foram observados a 0,36 mg/m2/dia. O aumento da letalidade embrionária também foi observado na presença de toxicidade materna a ≥0,36 mg/ m2/dia em estudos de fertilidade feminina e desenvolvimento embrionário precoce. Todas as doses com efeitos embrionário-fetais foram observadas na presença de toxicidade materna. A menor dose com efeitos embrionário-fetais em ratos (0,15 mg/ m2/dia) foram 9,7 vezes a exposição clínica humana após a terceira dose humana de 3 mg/ m2 com base na AUC168.
4.CONTRAINDICAÇÕES
Este medicamento é contraindicado a pacientes com hipersensibilidade ao gentuzumabe ozogamicina ou a qualquer outro componente da formulação do produto.
5.ADVERTÊNCIAS E PRECAUÇÕES
Hepatotoxicidade, Incluindo Doença Veno-oclusiva Hepática (DVO)
Foram notificados casos de hepatotoxicidade, incluindo eventos de DVO hepática potencialmente fatais e por vezes fatais, em pacientes recebendo Mylotarg® como agente único ou como parte de um regime de quimioterapia combinada (vide item 9. Reações Adversas).
No ALFA-0701, foram reportados eventos de DVO em 6/131 (5%) pacientes adultos durante ou após o tratamento com Mylotarg®, ou após transplante hematopoiético posterior de células tronco (HSCT). O tempo mediano desde a dose de Mylotarg® até o início da DVO foi de 9 dias (intervalo: 2-298 dias), ocorrendo 5 eventos no período de 28 dias após qualquer dose de Mylotarg® e 1 evento ocorrendo mais de 28 dias após a última dose de Mylotarg®. Três dos 6 eventos de DVO foram fatais. A DVO foi também reportada em 2 pacientes no braço controle do ALFA-0701 após receber Mylotarg® como terapêutica para a LMA recidivante.
Com base numa análise efetuada em ensaios clínicos, o risco de DVO foi mais elevado em pacientes adultos que receberam doses mais elevadas de Mylotarg® em monoterapia, em pacientes com insuficiência hepática moderada ou grave antes de receber Mylotarg®, em pacientes tratados com Mylotarg® após HSCT e em pacientes que foram submetidos a HSCT após tratamento com Mylotarg®. Os pacientes com insuficiência hepática moderada/grave antes do tratamento com Mylotarg® tinham uma probabilidade 8,7 vezes superior de desenvolver DVO em comparação com os pacientes sem insuficiência hepática moderada/grave no início do tratamento. Os pacientes tratados com Mylotarg® para recidiva após HSCT tinham uma probabilidade 2,6 vezes superior de desenvolver DVO em comparação com os pacientes sem HSCT prévia. Os pacientes que foram submetidos a HSCT após o tratamento com Mylotarg® apresentaram uma probabilidade 2,9 vezes superior de desenvolver DVO após HSCT em comparação com os pacientes sem HSCT após o tratamento com Mylotarg®. Apesar de não se ter encontrado qualquer relação entre a DVO e o tempo de HSCT relativo a doses mais elevadas de Mylotarg® em monoterapia, o estudo ALFA-0701 recomendou um intervalo de 2 meses entre a última dose de Mylotarg® e HSCT.
Avaliar os níveis de ALT, AST, bilirrubina total e fosfatase alcalina antes de cada dose de Mylotarg®. Após o tratamento com Mylotarg®, monitorar frequentemente os sinais e sintomas de DVO; estes podem incluir aumentos da ALT, AST, bilirrubina total, hepatomegalia (que pode ser dolorosa), rápido aumento de peso e ascite. A monitorização apenas da bilirrubina total pode não identificar todos os pacientes com risco de DVO. Em pacientes que desenvolvam testes hepáticos anormais, recomenda-se uma monitorização mais frequente dos testes hepáticos e dos sinais e sintomas clínicos de hepatotoxicidade. Para os pacientes que seguem para o HSCT, monitorar frequentemente os testes hepáticos durante o período pós-HSCT, conforme apropriado.
Gerenciar os sinais ou sintomas de toxicidade hepática através da interrupção ou descontinuação de Mylotarg® (vide item 8. Posologia e Modo de Usar). Em pacientes que apresentam DVO, descontinue o tratamento com Mylotarg® e trate de acordo com a prática clínica habitual.
Reações relacionadas à infusão (incluindo anafilaxia)
Podem ocorrer reações relacionadas à infusão fatais ou potencialmente fatais durante ou nas 24 horas seguintes à infusão de Mylotarg® (vide item 9. Reações Adversas). Os sinais e sintomas de reações relacionadas com a infusão podem incluir febre, calafrios, hipotensão, taquicardia, hipóxia e insuficiência respiratória.
Medicar antes da infusão de Mylotarg® (vide item 8. Posologia e administração). Monitorar frequentemente os sinais vitais durante a infusão. Interromper imediatamente a infusão em pacientes que desenvolvam evidência de reação à infusão, especialmente dispneia, broncoespasmo ou hipotensão. Monitorar os pacientes durante e pelo menos 1 hora após o final da infusão ou até que os sinais e sintomas desapareçam completamente. Descontinuar a utilização de Mylotarg® em pacientes que desenvolvam sinais ou sintomas de anafilaxia, incluindo sintomas respiratórios graves ou hipotensão clinicamente significativa (vide item 8. Posologia e Modo de Usar).
Hemorragia
Mylotarg® é mielossupressor e pode causar hemorragias fatais ou potencialmente fatais devido à trombocitopenia prolongada. No ALFA-0701 (Mylotarg® em associação com quimioterapia), foram reportados eventos hemorrágicos de todos os graus e de Grau 3-4 em 118/131 (90%) e 27/131 (21%) pacientes, respectivamente. Ocorreram eventos hemorrágicos fatais (incluindo hematoma cerebral, hematoma intracraniano e hematoma subdural) em 4/131pacientes (3%). Ocorreu trombocitopenia com contagens de plaquetas inferiores a 50×109/L, persistindo por mais de 42 dias em 19 pacientes (19%) na fase de indução (vide item 9. Reações Adversas). A proporção de pacientes com trombocitopenia persistente aumentou com fases de tratamento progressivas e foi mais elevada em pacientes tratados com Mylotarg® mais quimioterapia do que com quimioterapia isolada (vide item 9. Reações Adversas).
Avaliar as contagens sanguíneas antes de cada dose de Mylotarg® e monitorar frequentemente as contagens sanguíneas após o tratamento com Mylotarg® até à resolução das citopenias. Monitorar os pacientes quanto a sinais e sintomas de hemorragia durante o tratamento com Mylotarg® . Gerenciar sangramento grave, hemorragia ou trombocitopenia persistente usando atraso na dose ou descontinuação permanente de Mylotarg® (vide item 8. Posologia e Modo de Usar) e fornecer cuidados de suporte de acordo com a prática habitual.
Prolongamento do intervalo QT
Foi observado um prolongamento do intervalo QT em pacientes tratados com outros medicamentos contendo caliqueamicina. Ao administrar Mylotarg® a pacientes com história ou predisposição para prolongamento do intervalo QTc, que estejam tomando medicamentos que são conhecidos por prolongarem o intervalo QT, e a pacientes com distúrbios eletrolíticos, obtenham eletrocardiogramas (ECGs) e eletrólitos antes do início do tratamento e conforme necessário durante a administração.
Utilização em LMA com risco adverso citogenético
A eficácia de Mylotarg® foi demonstrada em pacientes com LMA com citogenética de risco intermediário e favorável, com incerteza quanto ao efeito em pacientes com citogenética adversa (vide item 2. Resultados de Eficácia). Para os pacientes que estão sendo tratados com Mylotarg® em combinação com DNR e AraC para LMA primária recém-diagnosticada, quando os resultados dos testes citogenéticos estiverem disponíveis, considere se o benefício potencial de continuar o tratamento com Mylotarg® supera os riscos para cada paciente.
Toxicidade embrio-fetal
Com base no seu mecanismo de ação e resultados de estudos em animais, Mylotarg® pode causar dano embrio-fetal quando administrado a uma mulher grávida. Em estudos em animais, o gentuzumabe ozogamicina causou toxicidade embrio-fetal, começando com uma dose aproximadamente 0,4 vezes a exposição em pacientes com a dose máxima recomendada, com base na área sob a curva de concentração-tempo (AUC).
Informe as mulheres grávidas sobre o potencial risco para o feto. Aconselhar as mulheres em idade reprodutiva a utilizarem métodos contraceptivos eficazes durante o tratamento com Mylotarg® e por pelo menos 6 meses após a dose final. Aconselhar os homens com parceiras com potencial reprodutivo para utilizar um método contraceptivo eficaz durante o tratamento com Mylotarg® e durante pelo menos 3 meses após a última dose (vide item Uso em populações específicas).
Eletrofisiologia cardíaca
Há dados limitados disponíveis para descrever os efeitos de gentuzumabe ozogamicina na eletrofisiologia cardíaca.
Uso em populações específicas
Gravidez
•Resumo do Risco
Com base no seu mecanismo de ação e resultados de estudos em animais (vide itens 3. Características Farmacológicas - Mecanismo de Ação e 3. Características Farmacológicas - Dados de Segurança Pré-Clínicos), Mylotarg® pode causar dano embrio-fetal quando administrado a uma mulher grávida. Não existem dados disponíveis sobre a utilização de Mylotarg® em mulheres grávidas para avaliar o risco associado ao fármaco. Em estudos de reprodução em animais, o gentuzumabe ozogamicina causou toxicidade embrio-fetal, incluindo anomalias estruturais e alterações no crescimento, a exposições sistêmicas maternas superiores ou iguais a 0,4 vezes a exposição em pacientes com a dose máxima recomendada com base na AUC (vide subitem Dados). Informe as mulheres grávidas sobre o potencial risco para o feto.
Desconhece-se o risco histórico estimado de anomalias congênitas graves e aborto espontâneo para a população indicada. Todas as gravidezes apresentam um risco de ocorrência de malformações congênitas, interrupção ou outros efeitos adversos. Na população geral dos EUA, o risco estimado de ocorrência de anomalias congênitas graves e aborto espontâneo em gravidezes clinicamente reconhecidas é de 2-4% e 15-20%, respectivamente.
•Dados
- Dados em Animais
Em um estudo de desenvolvimento embrio-fetal em ratos, as fêmeas grávidas receberam doses intravenosas diárias de até 1,2 mg/ m2 /dia de gentuzumabe ozogamicina durante o período de organogênese. Toxicidades embrio-fetais incluindo atraso do crescimento fetal, como evidenciado pela diminuição do peso dos fetos vivos, incidência de costelas onduladas fetais e ossificação óssea retardada foram observadas com doses superiores ou iguais a 0,15 mg/ m2/dia. Observou-se um aumento da letalidade embrio-fetal e anomalias morfológicas fetais (malformações digitais, ausência do arco aórtico, anomalias nos ossos longos nos membros anteriores, escápulas em forma errada, ausência de um centro vertebral e esterno fundido) com dose superior ou igual a 0,36 mg/m2/dia. Todas as doses com efeitos embrio-fetais foram observadas na presença de toxicidade materna que incluiu diminuições no aumento de peso corporal gestacional, no consumo de alimentos e no peso do útero gravídico. A dose mais baixa em que se observaram efeitos embrio-fetais em ratos (0,15 mg/m2/dia) foi 0,4 vezes a exposição em pacientes com a dose máxima recomendada em humanos, com base na AUC.
Lactação
•Resumo do Risco
Não existem dados sobre a presença de gentuzumabe ozogamicina ou dos seus metabólitos no leite humano, os efeitos no lactente ou os efeitos na produção de leite. Devido ao potencial de ocorrência de reações adversas graves no lactente, aconselhar as mulheres a não amamentarem durante o tratamento com Mylotarg® e durante pelo menos 1 mês após a dose final.
Mulheres e Homens com Potencial Reprodutivo
Mylotarg® pode causar dano embrio-fetal quando administrado a uma mulher grávida (vide item Uso em Populações Específicas).
•Teste de Gravidez
Verifique a condição de gravidez em mulheres com potencial reprodutivo antes de iniciar o tratamento com Mylotarg®.
•Contracepção
- Mulheres
Aconselhar as mulheres com potencial reprodutivo a utilizarem métodos contraceptivos eficazes durante o tratamento com Mylotarg® e durante pelo menos 6 meses após a última dose (vide item 3. Propriedades Farmacológicas - Dados de Segurança Pré-Clínica).
- Homens
Aconselhar os homens com parceiras com potencial reprodutivo a utilizar um método contraceptivo eficaz durante o tratamento com Mylotarg® e durante pelo menos 3 meses após a última dose (vide item 3. Propriedades Farmacológicas - Dados de Segurança Pré-Clínica).
•Infertilidade
- Mulheres
Com base nos resultados obtidos em animais, Mylotarg® pode prejudicar a fertilidade feminina (vide item 3. Propriedades Farmacológicas - Dados de Segurança Pré-Clínicos).
- Homens
Com base nos resultados obtidos em animais, Mylotarg® pode prejudicar a fertilidade masculina (vide item 3. Propriedades Farmacológicas - Dados de Segurança Pré-Clínicos).
Uso pediátrico
A segurança e eficácia de Mylotarg® em associação com daunorrubicina e citarabina não foram estabelecidas em pacientes pediátricos com LMA primária recém-diagnosticada.
Uso geriátrico
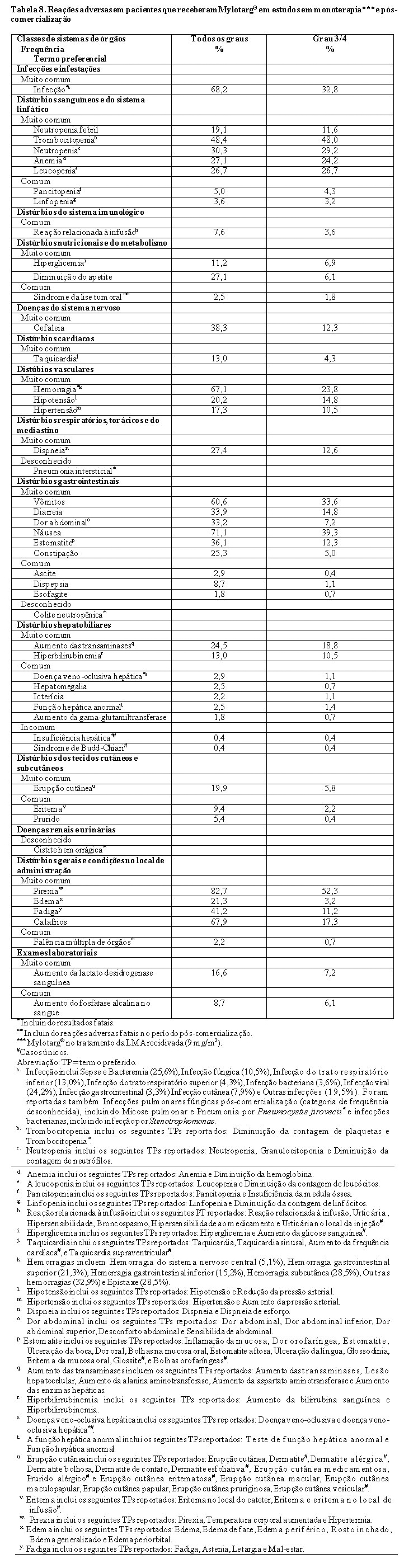
A utilização de Mylotarg® em associação com a daunorrubicina e citarabina em pacientes adultos com LMA primária recém-diagnosticada é suportada por um ensaio randomizado, controlado, que incluiu 50 pacientes com idade igual ou superior a 65 anos. Não se observaram diferenças gerais de segurança ou eficácia entre estes indivíduos e os indivíduos mais jovens. A utilização de Mylotarg® em monoterapia em pacientes adultos com LMA recém-diagnosticada é suportada por um ensaio randomizado controlado com 118 pacientes tratados com Mylotarg®. Todos os pacientes tinham mais de 60 anos e 65% dos pacientes tinham mais de 75 anos. No geral, não se observaram diferenças na eficácia por idade.
Mylotarg® é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
Efeitos na capacidade de dirigir veículos e operar máquinas
Não foram realizados estudos sobre o efeito de Mylotarg® na capacidade de dirigir e usar máquinas. Foi relatada fadiga durante o tratamento com Mylotarg® (vide item 9. Reações Adversas). Porta