MINJUVI
UNITED MEDICAL
tafasitamabe
Agente antineoplásico, tafasitamabe é um anticorpo monoclonal humanizado específico para CD19 da subclasse de imunoglobulina G (IgG) produzido em células de mamífero (ovário de hamster chinês) por tecnologia de DNA recombinante
Apresentações.
TAMANHO DE EMBALAGEM COMERCIAL
Pó para Solução para Infusão 200 mg (pó concentrado)
Caixa contendo um frasco de 200 mg de pó concentrado por frasco (40 mg/mL após reconstituição)
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada frasco-ampola contém tafasitamabe 200mg. Excipientes: citrato de sódio dihidratado, ácido cítrico monohidratado, trealose dihidratada e polissorbato 20.
Após a reconstituição, cada ml de solução contém 40 mg tafasitamabe.
Informações técnicas.
1. INDICAÇÕES
MINJUVI® é indicado em combinação com lenalidomida seguida de monoterapia com MINJUVI® para o tratamento de pacientes adultos com linfoma difuso de grandes células B (LDGCB) recidivante ou refratário, incluindo LDGCB decorrente de linfoma de baixo grau, e que não são elegíveis para transplante autólogo de células-tronco (ASCT).
2. RESULTADOS DE EFICÁCIA
Eficácia clínica
Tafasitamabe mais lenalidomida, seguido de monoterapia com tafasitamabe estudado no estudo L-MIND, um estudo multicêntrico, de braço único, em regime aberto. Este estudo foi realizado em pacientes adultos com LDGCB recidivante ou refratário após 1 a 3 terapias sistêmicas para LDGCB anteriores, que no momento do estudo não eram candidatos para quimioterapia de alta dose seguida de (ASCT) ou que recusaram (ASCT). Uma das terapias sistêmicas anteriores tinha de incluir uma terapia direcionada para CD20. O estudo excluiu pacientes com comprometimento hepático grave (bilirrubina sérica total > 3 mg/dl) e pacientes com comprometimento renal (CrCL < 60 ml/min), bem como pacientes com histórico ou evidências de doença cardiovascular, do SNC e/ou outras doenças sistêmicas clinicamente significativas. Pacientes com histórico conhecido de LDGCB double/triple-hit também foram excluídos na entrada do estudo.
Para os primeiros três ciclos, os pacientes receberam 12 mg/kg de tafasitamabe através de infusão nos dias 1, 8, 15 e 22 de cada ciclo de 28 dias, mais uma dose de carga no dia 4 do ciclo 1. Depois disso, tafasitamabe foi administrado nos dias 1 e 15 de cada ciclo até progressão da doença. Foi administrada pré-medicação incluindo antipiréticos, bloqueadores dos receptores H1 e H2 da histamina e glicocorticoides 30 a 120 minutos antes das primeiras três infusões de tafasitamabe. Os pacientes autoadministraram diariamente 25 mg de lenalidomida nos dias 1 a 21 de cada ciclo de 28 dias, até 12 ciclos.
Foi incluído no estudo L-MIND um total de 81 pacientes. A idade mediana foi de 72 anos (intervalo dos 41 aos 86 anos), 89% eram brancos e 54% eram do sexo masculino. Dos 81 pacientes, 74 (91,4%) tiveram uma pontuação de desempenho ECOG de 0 ou 1, e 7 pacientes (8,6%) tiveram uma pontuação ECOG de 2. O número médio de terapêuticas anteriores foi de dois (intervalo: 1 a 4), com 40 pacientes (49,4%) terem recebido uma terapia anterior e 35 pacientes (43,2%) terem recebido 2 linhas anteriores de tratamento. Cinco pacientes (6,2%) tinham 3 linhas anteriores de terapias, 1 (1,2%) tinha 4 linhas anteriores de tratamento. Todos os pacientes tinham recebido uma terapia anterior que continha CD20. Oito pacientes tiveram um diagnóstico de LDGCB transformado de linfoma de baixo grau. Quinze pacientes (18,5%) tinham doença refratária primária, 36 (44,4%) eram refratários à sua última terapia anterior, e 34 (42,0%) eram refratários ao rituximabe. Nove pacientes (11,1%) tinham recebido um (ASCT) anterior. Os principais motivos para que os pacientes não fossem candidatos para (ASCT) incluíram idade (45,7%), serem refratários a quimioterapia de resgate (23,5%), comorbilidades (13,6%) e recusa de (ASCT)/quimioterapia de alta dose (16,0%).
Um paciente recebeu tafasitamabe, mas não lenalidomida. Os 80 pacientes restantes receberam pelo menos uma dose de tafasitamabe e lenalidomida. Todos os pacientes incluídos no estudo L-MIND tinham um diagnóstico de LDGCB baseado na patologia local. Contudo, segundo a revisão centralizada da patologia, 10% dos pacientes não puderam ser classificados como LDGCB.
A duração mediana de exposição ao tratamento foi de 9,2 meses (intervalo: 0,23; 54,67 meses). Trinta e dois (39,5%) pacientes concluíram 12 ciclos de tafasitamabe. Trinta (37,0%) pacientes concluíram 12 ciclos de lenalidomida.
O parâmetro de avaliação primária de eficácia foi a melhor taxa de resposta objetiva (ORR), definida como a proporção de pacientes com resposta parcial e completa, conforme avaliado por uma comissão de revisão independente (IRC). Outros parâmetros de avaliação de eficácia incluíam duração da resposta (DoR), sobrevida livre de progressão (PFS) e a sobrevida global (OS). Os resultados de eficácia estão resumidos na Tabela 1.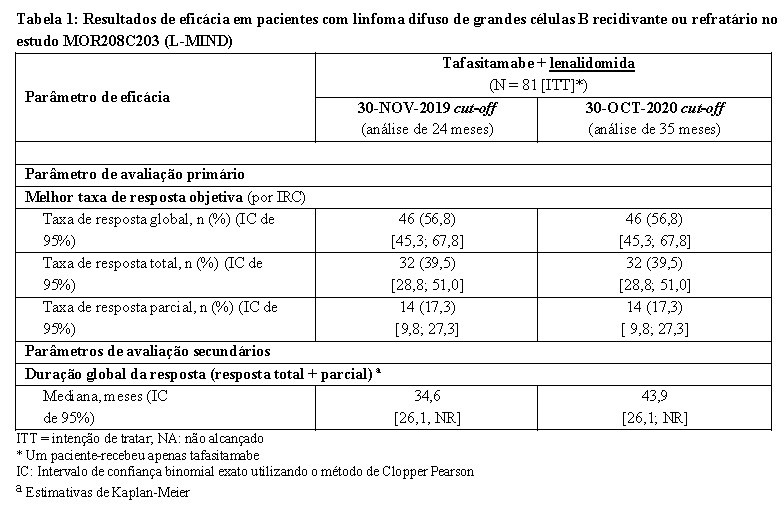
A sobrevida global (OS) foi um parâmetro de avaliação secundário no estudo. Depois de um tempo de seguimento mediano de 42,7 meses (IC de 95%: 38,0; 47,2), a OS mediana foi de 31,6 meses (IC de 95%: 18,3; não alcançado). Entre os oito pacientes que tinham LDGCB transformado a partir de um linfoma indolente anterior, sete pacientes tiveram uma resposta objetiva (três pacientes uma resposta completa, quatro pacientes uma resposta parcial) e um paciente teve uma doença estável como a melhor resposta ao tratamento com tafasitamabe + lenalidomida.
Idosos
No conjunto ITT, 36 de 81 pacientes tinham idade igual ou inferior a 70 anos e 45 de 81 pacientes tinham idade superior a 70 anos. Não foram observadas diferenças globais na eficácia para os pacientes com idade igual ou inferior a 70 anos versus pacientes com idade superior a 70 anos.
Referências bibliográficas
• Cordoba R, Prawitz T, Westley T, et al. Tafasitamab Plus Lenalidomide Versus 3 Rituximab-Based Treatments for Non-Transplant Eligible Relapsed/Refractory Diffuse Large B-Cell Lymphoma: A Matching-Adjusted Indirect Comparison. Adv Ther. 2022;39(6):2668-2687.
• Duell J, Maddocks KJ, Gonzalez Barca E, et al. Long-term outcomes from the Phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. 2021;106(9):2417-2426.
• Duell J, Obr A, Augustin M, et al. CD19 expression is maintained in DLBCL patients after treatment with tafasitamab plus lenalidomide in the L-MIND study. Leuk Lymphoma. 2022;63(2):468-472.
• Nowakowski GS, Yoon DH, Peters A, et al. Improved Efficacy of Tafasitamab plus Lenalidomide versus Systemic Therapies for Relapsed/Refractory DLBCL: RE-MIND2, an Observational Retrospective Matched Cohort Study. Clin Cancer Res. 2022 Jun 8;OF1-OF15. doi: 10.1158/1078-0432.CCR-21-3648. Online ahead of print.
• Salles G, Duell J, Gonzalez Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre,
• Zinzani PL, Rodgers T, Marino D, et al. RE-MIND: Comparing tafasitamab + lenalidomide (L-MIND) with a real-world lenalidomide monotherapy cohort in relapsed or refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2021;27(22):6124-6134.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Grupo farmacoterapêutico: agentes antineoplásicos, anticorpos monoclonais, código ATC: L01FX12.
Mecanismo de ação
Tafasitamabe é um anticorpo monoclonal com Fc aprimorado cujo alvo é o antígeno CD19 expresso na superfície de linfócitos pré-B e B maduros.
Ao ligar-se ao CD19, o tafasitamabe medeia a lise das células B através de:
• envolvimento de células efetoras imunitárias como células NK (natural killer), células T cd e fagócitos
• indução direta de morte celular (apoptose)
A modificação do Fc resulta na acentuação da citotoxicidade celular dependente de anticorpos da fagocitose celular dependente de anticorpos.
Efeitos farmacodinâmicos
Em pacientes com LDGCB recidivante ou refratário, o tafasitamabe levou à redução das contagens de células B no sangue periférico. A redução relativa à contagem de células B basal atingiu 97% após oito dias de tratamento no estudo L-MIND. A redução máxima de células B em cerca de 100% (mediana) foi atingida no prazo de 16 semanas de tratamento.
Embora a depleção de células B no sangue periférico seja um efeito farmacodinâmico mensurável, não está diretamente correlacionada com a depleção de células B em órgãos sólidos ou depósitos malignos.
Farmacocinética
A absorção, distribuição, biotransformação e eliminação foram documentadas com base numa análise farmacocinética da população.
Absorção
Com base na análise de tafasitamabe em associação com lenalidomida, as concentrações séricas médias de tafasitamabe no vale (± desvio padrão) foram de 179 (± 53) mg/ml durante as administrações intravenosas semanais de 12 mg/kg (mais uma dose adicional no dia 4 do ciclo 1). Durante a administração, a cada 14 dias a partir do ciclo 4 em diante, as concentrações séricas médias no vale foram de 153 (± 68) mg/ml. As concentrações séricas máximas globais de tafasitamabe foram de 483 (± 109) mg/ml.
Distribuição
O volume total de distribuição de tafasitamabe foi 9,3 l.
Biotransformação
Não foi caracterizada a via metabólica exata através da qual tafasitamabe é metabolizado. Sendo um anticorpo monoclonal humano do tipo IgG, prevê-se que o tafasitamabe seja degradado em pequenos peptídeos e aminoácidos através de vias catabólicas do mesmo modo que a IgG endógena.
Eliminação
A depuração de tafasitamabe foi de 0,41 l/dia e a meia vida de eliminação terminal foi de 16,9 dias. Após observações a longo prazo, descobriu-se que a depuração do tafasitamabe diminuiu ao longo do tempo para 0,19 l/dia após dois anos.
Populações especiais
A idade, peso corporal, sexo, tamanho do tumor, tipo de doença, contagem de células B ou contagem absoluta de linfócitos, anticorpos antifármaco, níveis de desidrogenase lática e níveis de albumina sérica não tiveram qualquer efeito relevante na farmacocinética do tafasitamabe. Desconhece-se a influência da raça e etnia na farmacocinética do tafasitamabe.
Disfunção renal
O efeito no comprometimento renal não foi formalmente testado em ensaios clínicos dedicados; no entanto, não foram observadas diferenças clinicamente significativas na farmacocinética do tafasitamabe para disfunção renal leve a moderada (depuração da creatinina [CrCL] ≥ 30 e < 90 ml/min estimada pela equação de Cockcroft-Gault). O efeito do comprometimento renal grave na doença renal terminal (CrCL < 30 mL/min) é desconhecido.
Disfunção hepática
O efeito do comprometimento hepático não foi formalmente testado em ensaios clínicos dedicados; no entanto não foram observadas diferenças clinicamente significativas na farmacocinética do tafasitamabe para comprometimento hepático leve (bilirrubina total ≤ limite superior do normal [LSN] e aspartato aminotransferase [AST] > LSN, ou bilirrubina total de 1 a 1,5 vezes o LNS e qualquer AST). O efeito do comprometimento hepático moderado a grave (bilirrubina total > 1,5 vezes LSN e qualquer AST) é desconhecido.
Dados de segurança pré-clínica
Os dados pré-clínicos não revelam riscos especiais para os seres humanos
Repetir estudos de toxicologia de dose
Tafasitamabe mostrou ser altamente específico para o antígeno CD19 em células B. Estudos de toxicidade após administração intravenosa em macacos cynomolgus não mostraram outro efeito além da depleção farmacológica esperada de células B no sangue periférico e nos tecidos linfoides. Essas alterações reverteram após a interrupção do tratamento.
Mutagenicidade/carcinogenicidade
Como o tafasitamabe é um anticorpo monoclonal, não foram realizados estudos de genotoxicidade e carcinogenicidade, uma vez que tais testes não são relevantes para esta molécula na indicação proposta.
Toxidade reprodutiva
Não foram realizados estudos de toxicidade reprodutiva e de desenvolvimento, bem como estudos específicos para avaliar os efeitos na fertilidade com tafasitamabe. No entanto, nenhum efeito adverso nos órgãos reprodutivos em machos e fêmeas e nenhum efeito na duração do ciclo menstrual em fêmeas foram observados no estudo de toxicidade de dose repetida de 13 semanas em macacos cynomolgus.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes mencionados
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações relacionadas com a infusão
Podem ocorrer reações relacionadas com a infusão, estas foram notificadas mais frequentemente durante a primeira infusão. Os pacientes devem ser cuidadosamente monitorados ao longo da infusão. Os pacientes devem ser aconselhados a contatar o seu médico, farmacêutico ou enfermeiro se tiverem sinais e sintomas de reações relacionadas com a infusão, incluindo febre, arrepios, erupção na pele ou problemas de respiração no prazo de 24 horas após a infusão. Deve ser administrada uma pré-medicação aos pacientes antes de iniciar a infusão de tafasitamabe. Com base na gravidade da reação relacionada com a infusão, deve ser interrompida ou descontinuada a infusão de tafasitamabe e deve ser instituído manejo médico apropriado.
Mielossupressão
O tratamento com tafasitamabe pode causar mielossupressão grave e/ou severa, incluindo neutropenia, trombocitopenia e anemia. Os hemogramas completos devem ser monitorizados ao longo de todo o tratamento e antes da administração de cada ciclo de tratamento. Com base na gravidade da reação adversa, a infusão de tafasitamabe deve ser interrompida (ver Tabela 2).
Consultar a bula da lenalidomida para modificações de dosagem.
Neutropenia
Foi notificada neutropenia, incluindo neutropenia febril, durante o tratamento com tafasitamabe. Deve ser considerada a administração de fatores estimulantes de granulócitos (G-CSF), em particular em pacientes com neutropenia de Grau 3 ou 4. Quaisquer sintomas ou sinais de desenvolvimento de infeção devem ser antecipados, avaliados e tratados.
Trombocitopenia
Foi notificada trombocitopenia durante o tratamento com tafasitamabe. Deve ser considerada a interrupção do tratamento com medicamentos concomitantes que podem aumentar o risco de hemorragia (por exemplo, antiagregantes plaquetários, anticoagulantes). Os pacientes devem ser aconselhados a comunicar imediatamente sinais ou sintomas de hematomas ou hemorragia.
Leucoencefalopatia Multifocal Progressiva
Foi notificada leucoencefalopatia multifocal progressiva (LMP) durante a tratamento combinado com tafasitamabe. Os pacientes devem ser monitorados quanto aos sintomas ou sinais neurológicos novos ou agravados que possam ser sugestivos de PML. Se houver suspeita de PML, outras doses de tafasitamabe devem ser imediatamente suspensas. O encaminhamento para um neurologista deve ser considerado. Medidas diagnósticas apropriadas podem incluir ressonância magnética, teste de líquido cefalorraquidiano para DNA viral JC e avaliações neurológicas repetidas. Se a LMP for confirmada, o tafasitamabe deve ser descontinuado permanentemente.
Infeções
Ocorreram infeções graves e fatais, incluindo infeções oportunistas, em pacientes durante o tratamento com tafasitamabe. O tafasitamabe apenas deve ser administrado a pacientes com uma infeção ativa, se a infeção for tratada de forma adequada e estiver bem controlada. Os pacientes com história de infeções recorrentes ou crônicas podem estar em maior risco de infeção e devem ser monitorizados adequadamente.
Os pacientes devem ser aconselhados a contatar o seu médico, farmacêutico ou enfermeiro, se desenvolverem febre ou outros sinais de potencial infeção, tais como arrepios, tosse ou dor ao urinar.
Síndrome de lise tumoral
Os pacientes com elevada carga tumoral e tumores altamente proliferativos podem estar em maior risco de síndrome de lise tumoral. Em pacientes com LDGCB, foi observada síndrome de lise tumoral durante o tratamento com tafasitamabe. Devem ser tomadas medidas/profilaxia adequadas em conformidade com as orientações locais antes do tratamento com tafasitamabe. Os pacientes devem ser monitorados atentamente quanto à síndrome de lise tumoral durante o tratamento com tafasitamabe.
Imunizações
Não foi investigada a segurança da imunização com vacinas vivas após a terapia com tafasitamabe e não é recomendada a vacinação com vacinas vivas simultaneamente terapia de tafasitamabe.
Excipiente
Este medicamento contém 37,0 mg de sódio por 5 frascos para injetáveis (a dose para um paciente com 83 kg de peso), equivalente a 1,85% da ingestão diária máxima recomendada pela OMS de 2 g de sódio para um adulto.
Fertilidade, gravidez e aleitamento
Não deve ser iniciado o tratamento com tafasitamabe em associação com lenalidomida em pacientes do sexo feminino, a menos que a gravidez tenha sido excluída. Consultar também a bula da lenalidomida.
Mulheres com potencial para engravidar/Contracepção em mulheres
As mulheres com potencial para engravidar devem ser aconselhadas a utilizar contracepção eficaz durante o tratamento com tafasitamabe e, pelo menos, 3 meses após a conclusão do tratamento.
Gravidez
Não foram realizados estudos de toxicidade reprodutiva e do desenvolvimento com tafasitamabe.
A quantidade de dados sobre a utilização de tafasitamabe em mulheres grávidas, é limitada ou inexistente. Contudo, sabe-se que a IgG atravessa a placenta e o tafasitamabe poderá provocar depleção de células B fetais com base nas propriedades farmacológicas. Em caso de exposição durante a gravidez, os recém-nascidos devem ser monitorizados quanto a depleção de células B e a vacinação com vacinas de vírus vivos deve ser adiada até que a contagem de células B da criança tenha recuperado.
Tafasitamabe não é recomendado durante a gravidez e em mulheres com potencial para engravidar que não utilizem métodos contraceptivos.
A lenalidomida pode causar lesões embriofetais e é contraindicada para utilização na gravidez e em mulheres com potencial para engravidar, a menos que sejam cumpridas todas as condições do programa de prevenção de gravidez da lenalidomida.
Amamentação
Desconhece-se se tafasitamabe é excretado no leite humano. Contudo, sabe-se que a IgG materna é excretada no leite humano. Não existem dados sobre a utilização de tafasitamabe em mulheres a amamentar e não pode ser excluído qualquer risco para os lactentes. As mulheres devem ser aconselhadas a não amamentar durante o tratamento com tafasitamabe e, pelo menos, 3 meses após a última dose.
Fertilidade
Não foram realizados estudos específicos para avaliar os potenciais efeitos de tafasitamabe sobre a fertilidade. Não foram observados efeitos adversos nos órgãos reprodutivos masculinos e femininos num estudo de toxicidade de dose repetida em animais.
Efeitos sobre a capacidade de conduzir e utilizar máquinas
Os efeitos de MINJUVI® sobre a capacidade de conduzir e utilizar máquinas são nulos ou desprezíveis. No entanto, a fadiga tem sido relatada em pacientes que tomam tafasitamabe e isso deve ser levado em conta ao dirigir ou usar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar no refrigerador (2°C - 8°C).
Manter o frasco na embalagem externa para proteger da luz.
Prazo de validade
Frasco: 48 meses
Número do lote e datas de fabricação e validade: ver embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Condições de conservação do medicamento após reconstituição e diluição
Solução reconstituída (antes da diluição)
Foi demonstrada estabilidade química e física de utilização até 24 horas a 2°C - 25°C.
Do ponto de vista microbiológico, a menos que a reconstituição da solução exclua o risco de contaminação microbiana, a solução reconstituída deve ser utilizada imediatamente. Se não for utilizada imediatamente, os tempos e condições de conservação em utilização são da responsabilidade do utilizador. Não congelar ou agitar.
Solução diluída (para infusão)
Foi demonstrada estabilidade química e física de utilização até um máximo de 36 horas a 2°C - 8°C seguida de até 24 horas até 25°C.
Do ponto de vista microbiológico, a solução diluída deve ser utilizada imediatamente. Se não for utilizada imediatamente, os tempos e condições de conservação antes da utilização são da responsabilidade do utilizador e normalmente não serão superiores a 24 horas entre 2 °C e 8 °C, a menos que a diluição tenha sido efetuada em condições assépticas controladas e validadas. Não congelar ou agitar.
MINJUVI® é um pó liofilizado branco a levemente amarelado, uma vez reconstituído é uma solução incolor e levemente amarelada.
Antes de usar, observe a aparência do medicamento.
Todos os medicamentos devem ser mantidos fora do alcance das crianças.
Descarte medicamentos não utilizados e/ou vencidos.
8. POSOLOGIA E MODO DE ADMINISTRAÇÃO
MINJUVI® tem de ser administrado por um profissional de saúde com experiência no tratamento de pacientes com câncer.
Pré-medicação recomendada
Deve ser administrada uma pré-medicação para reduzir o risco de reações relacionadas com a infusão 30 minutos a 2 horas antes da infusão de tafasitamabe. Para pacientes que não tenham reações relacionadas com a infusão durante as primeiras 3 infusões, a pré-medicação é opcional para as infusões subsequentes.
A pré-medicação pode incluir antipiréticos (por exemplo, paracetamol), bloqueadores do receptor H1 da histamina (por exemplo, difenidramina), bloqueadores do receptor H2 da histamina (por exemplo, cimetidina), ou glicocorticoides (por exemplo, metilprednisolona).
Tratamento de reações relacionadas com a infusão
Se ocorrer uma reação relacionada com a infusão (Grau 2 e superior), a infusão deve ser interrompida. Além disso, o tratamento médico adequado de sintomas deve ser iniciado. Depois dos sinais e sintomas estarem resolvidos ou reduzidos para Grau 1, pode ser retomada a infusão de MINJUVI® a uma velocidade de infusão reduzida (ver Tabela 2).
Se um paciente teve uma reação relacionada com a infusão de Grau 1 a 3, deve ser administrada pré- medicação antes das infusões subsequentes de tafasitamabe.
Posologia
A dose recomendada de MINJUVI® é de 12 mg por kg de peso corporal administrada como uma infusão intravenosa de acordo com o seguinte esquema:
• Ciclo 1: infusão nos dias 1, 4, 8, 15 e 22 do ciclo.
• Ciclos 2 e 3: infusão nos dias 1, 8, 15 e 22 de cada ciclo.
• Ciclo 4 até progressão da doença: infusão nos dias 1 e 15 de cada ciclo. Cada ciclo tem 28 dias.
Além disso, os pacientes devem autoadministrar as cápsulas de lenalidomida na dose inicial recomendada de 25 mg diariamente nos dias 1 a 21 de cada ciclo. A dose inicial e a dosagem subsequente podem ser ajustadas de acordo com a bula da lenalidomida.
A associação de MINJUVI® mais lenalidomida é administrada até doze ciclos.
O tratamento com lenalidomida deve ser interrompido após um máximo de doze ciclos de associação terapia. Os pacientes devem continuar a receber infusões de MINJUVI® como agente único nos dias 1 e 15 de cada ciclo de 28 dias, até progressão de doença ou toxicidade inaceitável.
Modificações da dose
A Tabela 2 fornece modificações de dose no caso de reações adversas. Para modificações de dose relativamente à lenalidomida, consulte também a bula da lenalidomida.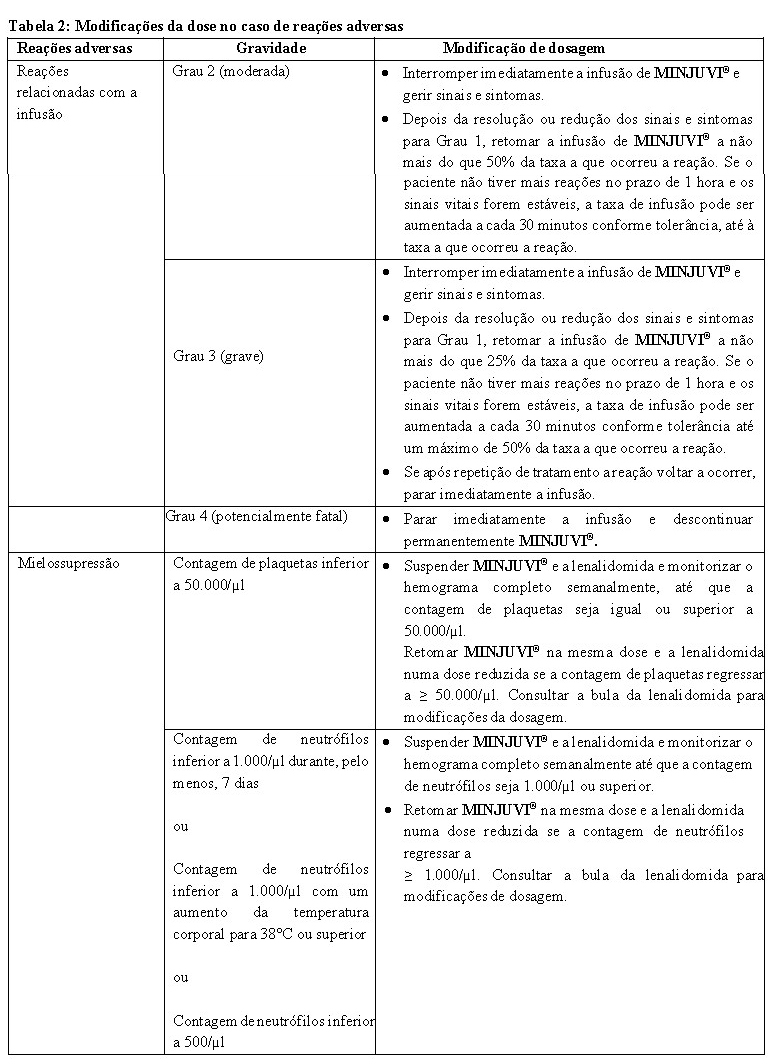
Populações especiais
População pediátrica
A segurança e eficácia de MINJUVI® em crianças com menos de 18 anos não foram estabelecidas. Não existem dados disponíveis.
Idosos
Não é necessário ajuste da dose em pacientes idosos (≥ 65 anos).
Disfunção renal
Não é necessário ajuste da dose em pacientes com disfunção renal leve a moderada. Não existem dados para se efetuarem recomendações de dosagem em pacientes com renal grave.
Disfunção Hepática
Não é necessário qualquer ajuste da dose para pacientes com comprometimento hepático leve. Não existem dados para se efetuarem recomendações de dosagem em pacientes com comprometimento hepático moderado ou grave.
Modo de administração
MINJUVI® destina-se a utilização por via intravenosa após reconstituição e diluição.
• Para a primeira infusão do ciclo 1, a taxa de infusão intravenosa deve ser de 70 ml/h para os primeiros 30 minutos. Depois disso, a taxa deve ser aumentada de modo a concluir a primeira infusão dentro de um período de 2,5 horas.
• Todas as infusões subsequentes devem ser administradas num período de 1,5 a 2 horas.
• No caso de reações adversas, considerar as modificações de dose recomendadas fornecidas na Tabela 2.
• MINJUVI® não pode ser coadministrado com outros medicamentos através da mesma linha de infusão.
• MINJUVI® não pode ser administrado por injeção intravenosa rápida ou bolus.
Precauções especiais de eliminação e manuseio
MINJUVI® é fornecido em frascos para injetáveis estéreis, de utilização única, isentos de conservantes. MINJUVI® deve ser reconstituído e diluído antes da infusão intravenosa.
Utilize uma técnica asséptica adequada para reconstituição e diluição.
Instruções de reconstituição
• Determine a dose de tafasitamabe com base no peso do paciente através da multiplicação de 12 mg pelo peso do paciente (kg). Em seguida, calcule o número de frascos de tafasitamabe necessários (cada frasco contém 200 mg de tafasitamabe).
• Utilizando uma seringa estéril, adicione com cuidado 5,0 ml de água para preparações injetáveis estéril a cada frasco de MINJUVI®. Direcione o jato para as paredes de cada frasco e não diretamente para o pó liofilizado.
• Gire suavemente o(s) frasco(s) reconstituído(s) para ajudar na dissolução do pó liofilizado. Não agitar ou girar vigorosamente. Não retire o conteúdo até que todos os sólidos tenham sido completamente dissolvidos. O pó liofilizado deve dissolver-se em 5 minutos.
• A solução reconstituída deve apresentar-se como uma solução incolor a ligeiramente amarela. Antes de prosseguir, certifique-se de que não há partículas ou descoloração através de inspeção visual. Se a solução estiver turva, descolorada ou contiver partículas visíveis, elimine o(s) frasco(s).
Instruções de diluição
• Deve ser utilizado uma bolsa de infusão que contenha 250 ml de solução injetável de cloreto de sódio a 9 mg/ml (0,9%).
• Calcule o volume total necessário da solução de 40 mg/ml de tafasitamabe reconstituída. Retire um volume igual a este da bolsa de infusão e elimine o volume retirado.
• Retire o volume total calculado (ml) da solução reconstituída de tafasitamabe do(s) frasco(s) e adicione lentamente ao saco de infusão com cloreto de sódio a 9 mg/ml (0,9%). Elimine qualquer porção não utilizada de tafasitamabe restante no frasco. A concentração final da solução diluída deve estar entre 2 mg/ml e 8 mg/ml de tafasitamabe.
• Misture suavemente o saco intravenoso invertendo lentamente o saco. Não agitar.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais.
Incompatibilidades
Este medicamento não pode ser misturado com outros medicamentos, exceto os mencionados na Precauções especiais de eliminação e manuseamento
Não foram observadas incompatibilidades com os materiais de infusão padrão.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
As reações adversas mais frequentes são:
• infeções (73%),
• neutropenia (51%),
• astenia (40%),
• anemia (36%),
• diarreia (36%),
• trombocitopenia (31%),
• tosse (26%),
• edema periférico (24%),
• febre (24%),
• diminuição do apetite (22%).
As reações adversas graves mais frequentes foram:
infeção (26%) incluindo pneumonia (7%), e neutropenia febril (6%). Ocorreu descontinuação permanente do tafasitamabe devido a reações adversas em 15% dos pacientes.
As reações adversas mais frequentes que levaram a descontinuação permanente do tafasitamabe foram:
• infeções e infestações (5%),
• doenças do sistema nervoso (2,5%),
• doenças respiratórias, torácicas e do mediastino (2,5%)
A frequência da modificação ou interrupção da dose devido a reações adversas foi de 65%. As reações adversas mais frequentes que levaram a interrupção do tratamento com tafasitamabe foram doenças do sangue e do sistema linfático (41%).
Lista tabelada das reações adversas
As reações adversas comunicadas em ensaios clínicos são enumeradas segundo as classes de sistemas de órgãos da base de dados MedDRA e por frequência. A frequência de reações adversas baseia-se no ensaio principal de Fase 2 MOR208C203 (L-MIND) com 81 pacientes. Os pacientes foram expostos ao tafasitamabe durante uma mediana de 7,7 meses. As frequências das reações adversas de ensaios clínicos são baseadas em frequências de acontecimentos adversos por todas as causas, onde uma proporção dos acontecimentos para uma reação adversa pode ter outras causas para além do medicamento, tais como a doença, outros medicamentos ou causas não relacionadas.
Dentro de cada grupo de frequência, as reações adversas são apresentadas por ordem decrescente de gravidade.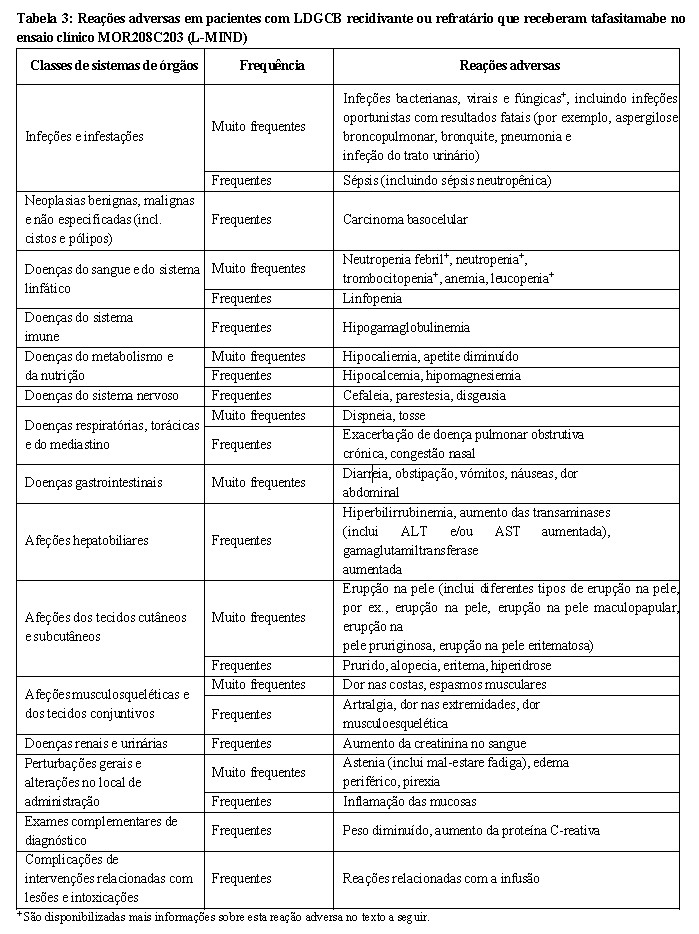
Em comparação com as incidências na associação terapia com lenalidomida, as incidências de reações adversas não hematológicas na monoterapia com tafasitamabe diminuíram, pelo menos, 10% para diminuição de apetite, astenia, hipocaliemia, obstipação, náuseas, espasmos musculares, dispneia e aumento da proteína C-reativa.
Descrição de reações adversas selecionadas
Mielossupressão
O tratamento com tafasitamabe pode causar mielossupressão grave ou severa, incluindo neutropenia, trombocitopenia e anemia (ver seções 5 e 8).
No estudo L-MIND, ocorreu mielossupressão (ou seja, neutropenia, neutropenia febril, trombocitopenia, leucopenia, linfopenia ou anemia) em 65,4% dos pacientes tratados com tafasitamabe. A mielosupressão foi tratada através da redução ou interrupção da lenalidomida, interrupção do tafasitamabe e/ou administração de G-CSF (ver seções 5 e 8). A mielosupressão levou à interrupção do tafasitamabe em 41% dos casos e a descontinuação do tafasitamabe em 1,2% dos casos.
Neutropenia/neutropenia febril
A incidência de neutropenia foi de 51%. A incidência de neutropenia de Grau 3 ou 4 foi de 49% e de neutropenia febril de Grau 3 ou 4 foi de 12%. A mediana da duração de qualquer reação adversa de neutropenia foi de 8 dias (intervalo 1 - 222 dias); a mediana do tempo até início da primeira ocorrência de neutropenia foi de 49 dias (intervalo 1 - 994 dias).
Trombocitopenia
A incidência de trombocitopenia foi de 31%. A incidência de trombocitopenia de Grau 3 ou 4 foi de 17%. A mediana da duração de qualquer reação adversa de neutropenia foi de 11 dias
(intervalo 1 - 470 dias); a mediana do tempo até início da primeira ocorrência de trombocitopenia foi de 71 dias (intervalo 1 - 358 dias).
Anemia
A incidência de anemia foi de 36%. A incidência de anemia de Grau 3 ou 4 foi de 7%. A mediana da duração de qualquer reação adversa de anemia foi de 15 dias (intervalo 1 - 535 dias); a mediana do tempo até início da primeira ocorrência de anemia foi de 49 dias (intervalo 1 - 1.129 dias).
Quando pacientes no estudo L-MIND mudaram de tafasitamabe e lenalidomida na fase de associação terapia para tafasitamabe isolado na fase de monoterapia prolongada, as incidências de acontecimentos hematológicos diminuíram, pelo menos, 20% para neutropenia, trombocitopenia e anemia; não foram notificadas quaisquer incidências de neutropenia febril com a monoterapia com tafasitamabe (ver seções 5 e 8).
Infeções
No estudo L-MIND, ocorreram infeções em 73% dos pacientes. A incidência de infeções de
Grau 3 ou 4 foi de 28%. As infeções de Grau 3 ou superior mais frequentemente notificadas foram pneumonia (7%), infeções do trato respiratório (4,9%), infeções do trato urinário (4,9%) e septicemia (4,9%). A infeção foi fatal em < 1% dos pacientes (pneumonia) no prazo de 30 dias do último tratamento.
O tempo mediano até ao início da infeção de Grau 3 ou 4 foi 62,5 dias (4 - 1,014 dias). A mediana da duração de qualquer infeção foi de 11 dias (1 - 392 dias).
São fornecidas recomendações para a gestão de infeções.
A infeção levou à interrupção da dose de tafasitamabe em 27% dos casos e a descontinuação do tafasitamabe em 4,9% dos casos.
Reações relacionadas com a infusão
No estudo L-MIND, ocorreram reações relacionadas com a infusão em 6% dos pacientes. Todas as reações relacionadas com a infusão foram de Grau 1 e resolveram-se no dia da ocorrência. Oitenta por cento (80%) destas reações ocorreram durante o ciclo 1 ou 2. Os sintomas incluíram arrepios, rubor, dispneia e hipertensão.
Imunogenicidade
Em 245 pacientes tratados com tafasitamabe, não foram observados anticorpos anti-tafasitamabe potenciados pelo tratamento ou emergentes do tratamento. Foram detectados anticorpos
anti-tafasitamabe preexistentes em 17/245 pacientes (6,9%) sem qualquer impacto na farmacocinética, eficácia ou segurança do tafasitamabe.
Populações especiais
Idosos
Entre 81 pacientes tratados no estudo L-MIND, 56 (69%) pacientes tinham mais de 65 anos de idade. Os pacientes com mais de 65 anos de idade tiveram uma incidência numericamente superior de acontecimentos adversos emergentes do tratamento (AAET) graves (55%) comparativamente aos pacientes com idade inferior a 65 anos (44%).
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que corretamente indicado e utilizado, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos por meio do Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPER DOSAGEM
No caso de uma sobredosagem, os pacientes devem ser cuidadosamente observados quanto a sinais ou sintomas de reações adversas e devem ser administrados cuidados de suporte, conforme adequado.
Em caso de envenenamento ligue para 0800 722 6001 se precisar de mais orientações sobre como proceder
Dizeres legais.
USO RESTRITO A ESTABELECIMENTOS DE SAÚDE
Venda sob pescrição
Registro no 1.2576.0039
Esta bula foi aprovada pela ANVISA em 10/07/2023.