MEKINIST
NOVARTIS
trametinibe
Antineoplásico.
Apresentações.
Mekinist® 0,5 mg ou 2,0 mg - embalagens contendo 30 comprimidos revestidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 0,5635 mg de dimetilsulfóxido de trametinibe equivalente a 0,5 mg de trametinibe
Cada comprimido revestido contém 2,254 mg de dimetilsulfóxido de trametinibe equivalente a 2,0 mg de trametinibe
Excipientes: manitol, celulose microcristalina, hipromelose, croscarmelose sódica, lauril sulfato de sódio, dióxido de silício coloidal, estearato de magnésio, opadry® amarelo, opadry® rosa.
Excipientes do revestimento: opadry® amarelo: hipromelose, dióxido de titânio, macrogol, óxido de ferro amarelo e opadry® rosa: hipromelose, dióxido de titânio, macrogol, polissorbato 80, óxido de ferro vermelho.
Informações técnicas.
1. INDICAÇÕES
Mekinist® (dimetilsulfóxido de trametinibe) em combinação com dabrafenibe é indicado para o tratamento de pacientes com melanoma não ressecável ou metastático com mutação BRAF V600 (veja resultados de eficácia).
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança da dose recomendada de dimetilsulfóxido de trametinibe (2 mg uma vez ao dia) em combinação com dabrafenibe (150 mg duas vezes ao dia) para o tratamento de pacientes adultos com melanoma não ressecável ou metastático com mutação BRAF V600 foram estudadas em dois estudos pivotais de fase III.
MEK115306 (COMBI-d)
O MEK115306 (COMBI-d) foi um estudo de fase III, randomizado, duplo-cego, comparando a combinação de dimetilsulfóxido de trametinibe e dabrafenibe versus dabrafenibe e placebo como terapia de primeira linha em indivíduos com melanoma cutâneo não ressecável (estágio IIIC) ou metastático (estágio IV) com mutação BRAF V600E/K positiva. O desfecho primário do estudo foi a sobrevida livre de progressão (PFS) avaliada pelo investigador com um desfecho secundário de sobrevida global (SG). Os participantes foram estratificados por nível de lactato desidrogenase (DHL) ( > o limite superior da normalidade (ULN) versus ≤ ULN) e mutação BRAF (V600E versus V600K).
Um total de 423 participantes foi randomizado 1:1 para o braço de terapia combinada (dimetilsulfóxido de trametinibe 2 mg uma vez ao dia e dabrafenibe 150 mg duas vezes ao dia) (N = 211) ou braço de monoterapia com dabrafenibe (150 mg duas vezes ao dia) (N = 212). As características basais estavam equilibradas entre os grupos de tratamento. O sexo masculino constituiu 53% dos pacientes e a idade média foi de 56 anos. A maioria dos pacientes obteve uma pontuação de desempenho do ECOG [Eastern Cooperative Oncology Group (Grupo Oncológico Cooperativo do Leste)] de 0 (72%) e tinha doença estágio IVM1c (66%). A maioria dos pacientes teve mutação BRAF V600E (85%). Os 15% de pacientes restantes tiveram mutação BRAF V600K. Indivíduos com metástase cerebral não foram incluídos neste estudo.
No momento da análise de SG final, foi relatado um total de 222 óbitos (52,5%) [grupo combinado 99 óbitos (47%) e grupo do dabrafenibe 123 óbitos (58%)] fora da população randomizada (ou ITT [Intent-to-treat (intenção de tratamento)]). O tempo mediano de acompanhamento do tratamento no estudo foi de 20 meses para o braço de terapia combinada e 16 meses para o braço de monoterapia com dabrafenibe. O estudo MEK115306 demonstrou uma redução estatisticamente significativa de 29% no risco de morte para o braço de terapia combinada comparado ao braço de monoterapia com dabrafenibe (PR [proporção de risco] = 0,71, IC [intervalo de confiança] de 95%: 0,55, 0,92; p = 0,011). A SG mediana foi de 25,1 meses para o braço de terapia combinada e de 18,7 meses para o braço de monoterapia com dabrafenibe. As estimativas de SG de 12 meses (74%) e 24 meses (51,4%) para a combinação também foram maiores em relação às da monoterapia com dabrafenibe (67,6 e 42,1%, respectivamente).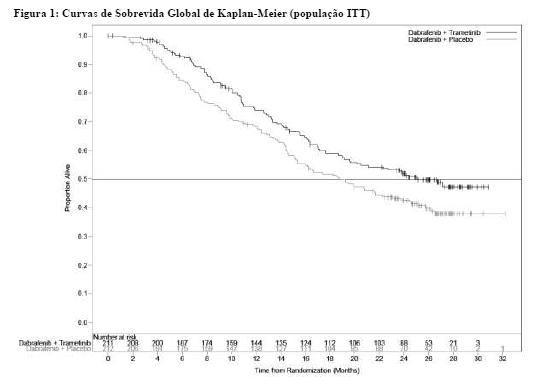

Os resultados de eficácia de PFS, ORR [taxa de resposta geral] e duração da resposta estão resumidos na tabela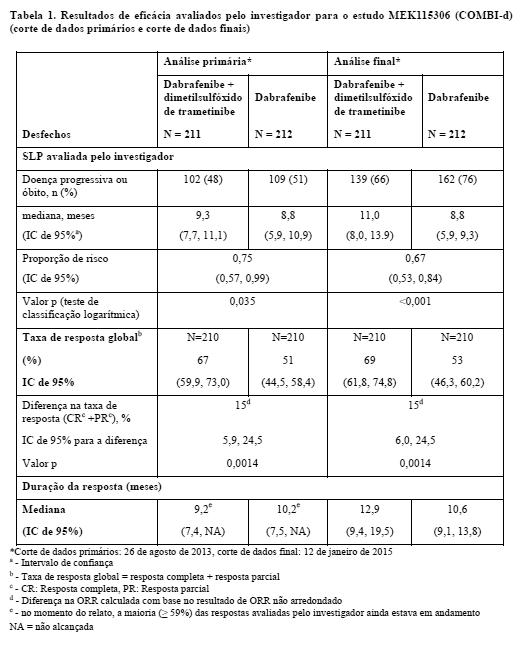
MEK116513 (COMBI-v):
O estudo MEK116513 foi um estudo de fase III de dois braços, aberto, randomizado, que comparou a terapia combinada de dabrafenibe e dimetilsulfóxido de trametinibe à monoterapia com vemurafenibe em melanoma metastático com mutação BRAF V600 positiva. O desfecho primário do estudo foi sobrevida global. Os indivíduos foram estratificados por nível de lactato desidrogenase (DHL) ( > o limite superior da normalidade (ULN) versus ≤ ULN) e mutação BRAF (V600E versus V600K).
Um total de 704 indivíduos foi randomizado 1:1 para o braço de terapia combinada (dimetilsulfóxido de trametinibe 2 mg uma vez ao dia e dabrafenibe 150 mg duas vezes ao dia) ou braço de monoterapia com vemurafenibe (960 mg duas vezes ao dia). A maioria dos indivíduos era branca ( > 96%) e do sexo masculino (55%), com uma idade média de 55 anos (24% tinham ≥ 65 anos). A maioria dos indivíduos tinha doença M1c estágio IV (61%). A maioria dos indivíduos tinha DHL ≤ ULN (67%), situação de desempenho ECOG de 0 (70%), e doença visceral (78%) na avaliação inicial. No geral, 54% dos indivíduos tinham < 3 locais da doença na avaliação inicial. A maioria dos indivíduos tinha mutação BRAF V600E (89%). Indivíduos com metástase cerebral não foram incluídos neste estudo.
A análise de SG foi conduzida quando ocorreram 222 óbitos totais (77% dos eventos necessários para a análise final). O Comitê de Monitoramento de Dados Independente (Independent Data Monitoring Committee, IDMC) recomendou a interrupção do estudo, pois os resultados de SG ultrapassaram o limite de eficácia pré-especificado. Como consequência, o resumo parcial de SG foi considerado como a análise de SG comparativa final.
A análise de SG do estudo MEK116513 foi baseada em 222 óbitos (32%) [combinação; 100 óbitos (28%) e vemurafenibe 122 óbitos (35%)]. O tempo mediano de acompanhamento do tratamento no estudo foi de 11 meses para o braço de terapia combinada e 9 meses para o braço de vemurafenibe. O estudo MEK116513 demonstrou uma redução estatisticamente significativa de 31% no risco de morte com a terapia combinada em comparação ao vemurafenibe (HR = 0,69; IC de 95%: 0,53, 0,89; p = 0,005). A SG mediana para o braço de terapia combinada não foi atingida ainda e, para a monoterapia com vemurafenibe, foi de 17,2 meses.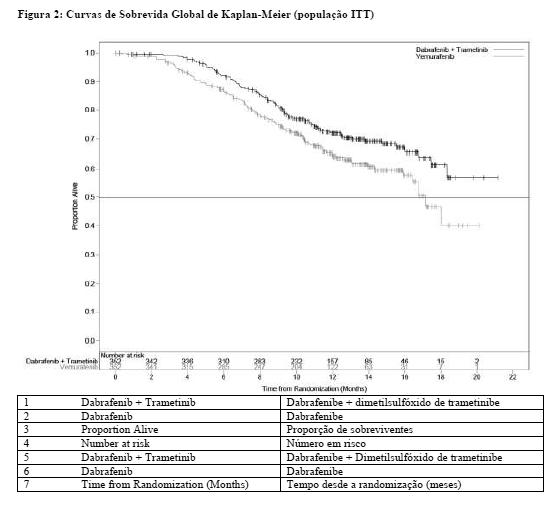
Os resultados dos desfechos de PFS, ORR e duração da resposta estão resumidos na tabela 2.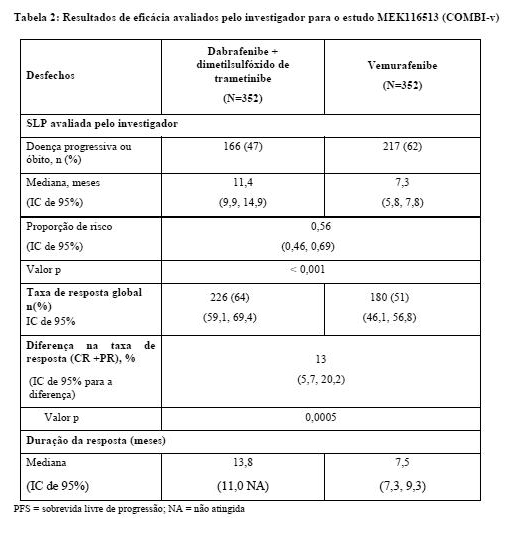
Pacientes com metástase cerebral
A segurança e eficácia da combinação de dimetilsulfóxido de trametinibe e dabrafenibe não foi avaliada em pacientes com melanoma com metástase cerebral com mutação BRAF V600 positiva.
Pacientes que progrediram de um inibidor BRAF
Existem dados limitados em pacientes utilizando a combinação de dimetilsulfóxido de trametinibe com dabrafenibe que progrediram previamente de um inibidor BRAF. Esses dados demonstram que a eficácia da combinação será menor nesses pacientes. Portanto, outras opções de tratamento devem ser consideradas antes do tratamento com a combinação dimetilsulfóxido de trametinibe e dabrafenibe na população tratada anteriormente com outro inibidor de BRAF.
Referências
1. [COMBI-d]: LONG G.V., Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N ENGL J MED. November 11, 2014.
2. [COMBI-d]: SCHADENDORF, D. Health-related quality of life impact in a randomised phase III study of the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600 metastatic melanoma. Eur J Cancer (2015), http://dx.doi.org/10.1016/j.ejca.2015.03.004.
3. [COMBI-v]: ROBERT, C. Improved overall survival in melanoma with combined dabrafenib and trametinib. N ENGL J MED. November 17, 2014.
4. [COMBI-v]: GROB, J.J. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol 2015; 16: 1389-98. October, 2015.
5. Module 2.5, Clinical Overview Rationale for labelling change to Core Data Sheet (CDS) - - supporting the addition of colitis and GI perforation, deletion of Bradycardia and Rhabdomyolysis from Post Marketing Data and moving enzyme-related disorders to Investigations section. Novartis. Jun-2016.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Leia a bula de dabrafenibe para informações específicas sobre as Características Farmacológicas.
Grupo farmacoterapêutico
Agentes antineoplásicos, inibidor da proteína quinase, código ATC [Anatomical Therapeutic Chemical (Anatômico Terapêutico Químico)]: L01XE25
Propriedades farmacodinâmicas
O dimetilsulfóxido de trametinibe suprimiu os níveis de ERK fosforilada em linhagens de células tumorais de melanoma mutante BRAF e em modelos de xenoenxerto de melanoma.
Em indivíduos com melanoma mutante BRAF e NRAS, a administração de dimetilsulfóxido de trametinibe resultou em alterações dependentes da dose nos biomarcadores tumorais, incluindo inibição de ERK fosforilada, inibição de Ki67 (um marcador de proliferação celular) e aumentos da p27 (um marcador de apoptose). As concentrações médias de dimetilsulfóxido de trametinibe observadas após administração de doses repetidas de 2 mg uma vez ao dia excedem a concentração alvo pré-clínica durante o intervalo de administração de 24 horas proporcionando, assim, inibição sustentada da via MEK.
Eletrofisiologia cardíaca
Estudo MEK111054
Inicialmente, o potencial de prolongamento QT pelo dimetilsulfóxido de trametinibe foi avaliado como parte do primeiro estudo em humanos para determinar a relação entre o intervalo QTc lido manualmente de forma independente e as concentrações plasmáticas de dimetilsulfóxido de trametinibe usando um modelo não linear de efeitos mistos. Os dados estavam disponíveis em 50 indivíduos, com um total de 498 valores de QTc equiparados. Com base na análise de concentração-QTc, o dimetilsulfóxido de trametinibe não demonstrou potencial aparente para alterar o intervalo QTc. No valor médio de Cmáx observado na dose recomendada de 2 mg uma vez ao dia, o aumento mediano no QTc é de 2,2 msec (IC de 90%: 0,2, 4,0).
Para confirmar a ausência de efeito na QTc, o potencial de prolongamento QT do dimetilsulfóxido de trametinibe foi avaliado adicionalmente em um estudo de fase I independente, dedicado em 35 indivíduos (30 indivíduos concluíram o estudo) com tumores sólidos. Os indivíduos receberam 3 mg de placebo correspondente no dia 1 do estudo seguidos de uma dose de 2 mg uma vez ao dia de dimetilsulfóxido de trametinibe e 2 comprimidos de 0,5 placebo correspondente nos dias 2 a 14 do estudo. No dia 15 do estudo, todos os participantes receberam uma dose única de 3 mg de dimetilsulfóxido de trametinibe (dose supraterapêutica). O estudo não demonstrou nenhum potencial do dimetilsulfóxido de trametinibe para alterar o intervalo QTcF após administração de doses repetidas de 2 mg de dimetilsulfóxido de trametinibe, incluindo a dose supraterapêutica de 3 mg no dia 15.
Em uma dose 1,5 vezes da dose máxima recomendada, Mekinist® não prolonga o intervalo QT a nenhuma extensão clinicamente relevante.
Mecanismo de ação
O dimetilsulfóxido de trametinibe é um inibidor reversível, altamente seletivo, alostérico da ativação da quinase regulada de sinal extracelular mitógeno-ativado 1 (MEK1) e inibidor da atividade da quinase MEK 2. Proteínas MEK são componentes críticos da via de sinalização da quinase relacionada ao sinal extracelular (ERK). No melanoma e em outros cânceres, esta via é frequentemente ativada por formas mutadas de BRAF a qual ativa MEK e estimula o crescimento de células tumorais. O dimetilsulfóxido de trametinibe inibe a ativação de MEK pelo BRAF e inibe a atividade da quinase MEK. O dimetilsulfóxido de trametinibe inibe o crescimento das linhagens de célula do melanoma com mutação BRAF V600 e demonstra efeitos antineoplásicos em modelos animais com melanoma BRAF V600 mutado.
O dabrafenibe é um inibidor potente, seletivo, ATP-competitivo, das quinases mutantes do BRAF V600 e das quinases dos genes BRAF e CRAF do tipo selvagem. As mutações oncogênicas do BRAF resultam em ativação constitutiva da via RAS/RAF/MEK/ERK e na estimulação do crescimento de células tumorais. O dabrafenibe e o dimetilsulfóxido de trametinibe inibem duas quinases nesta via, BRAF e MEK, e a combinação proporciona inibição concomitante da via. A combinação de dabrafenibe com dimetilsulfóxido de trametinibe é sinérgica nas linhagens de células do melanoma com mutação BRAF V600 positivo in vitro e retarda o surgimento de resistência in vivo em xenoenxertos de melanoma com mutação BRAF V600 positivo.
Propriedades farmacocinéticas
- Absorção
O dimetilsulfóxido de trametinibe é absorvido por via oral com tempo mediano para atingir pico de concentrações de 1,5 horas pós-dose. A biodisponibilidade absoluta média de uma única dose de comprimido de 2 mg é de 72% em relação a uma microdose por via intravenosa (IV). O aumento na exposição (Cmáx e AUC [área sob a curva]) foi proporcional à dose após a administração repetida. Após a administração de 2 mg diários, a média geométrica Cmáx, AUC(0-
- Distribuição
A ligação de dimetilsulfóxido de trametinibe às proteínas plasmáticas humanas é de 97,4%. O dimetilsulfóxido de trametinibe tem um volume de distribuição de 1060 L, determinado após a administração de uma microdose de 5 mg IV.
- Biotransformação/metabolismo
Estudos in vitro e in vivo demonstraram que o dimetilsulfóxido de trametinibe é metabolizado predominantemente através de desacetilação isolada ou em combinação ou com mono-oxigenação. O metabólito desacetilado foi adicionalmente metabolizado por glicuronidação. A desacetilação é mediada por esterases carboxílicas 1b, 1c e 2, e também pode ser mediada por outras enzimas hidrolíticas.
- Eliminação
O dimetilsulfóxido de trametinibe se acumula com administração diária repetida com razão média de acumulação de 6,0 após uma dose de 2 mg uma vez ao dia. A meia-vida terminal média é de 127 horas (5,3 dias) após a administração de uma dose única. O nível do estado estacionário foi alcançado ao dia 15. A depuração plasmática IV de dimetilsulfóxido de trametinibe é de 3,21 L/h.
A recuperação total da dose é baixa após um período de coleta de 10 dias ( < 50%) após administração de uma dose oral única de dimetilsulfóxido de trametinibe radiomarcado como solução, em função da meia-vida longa. O material relacionado ao medicamento foi excretado predominantemente nas fezes (≥81% da radioatividade recuperada) e em uma pequena parcela na urina ( < 19%). Menos do que 0,1% da dose excretada foi recuperada como progenitor na urina.
Farmacocinética da combinação
A coadministração de doses repetidas de 150 mg de dabrafenibe duas vezes ao dia e de 2 mg de dimetilsulfóxido de trametinibe uma vez ao dia resultou em um aumento de 16% na Cmáx do dabrafenibe e em um aumento de 23% na área sobre a curva do dabrafenibe. Estimou-se uma pequena diminuição na biodisponibilidade do dimetilsulfóxido de trametinibe, correspondente a uma diminuição na área sobre a curva de 12%, quando o dimetilsulfóxido de trametinibe é administrado em combinação com dabrafenibe usando uma análise farmacocinética populacional. As mudanças na Cmáx e área sob a curva de dabrafenibe ou dimetilsulfóxido de trametinibe não são consideradas clinicamente relevantes.
Populações especiais
Comprometimento hepático
A farmacocinética do dimetilsulfóxido de trametinibe foi caracterizada em 64 pacientes incluídos em estudos clínicos com dimetilsulfóxido de trametinibe que apresentavam comprometimento hepático leve (definido pela classificação do Instituto Nacional do Câncer dos EUA) usando uma análise farmacocinética da população. A depuração oral do dimetilsulfóxido de trametinibe e, portanto, a exposição ao dimetilsulfóxido de trametinibe não foram significativamente diferentes nesses pacientes em relação a pacientes com função hepática normal. Não há dados disponíveis em pacientes com comprometimento hepático moderado ou grave (veja posologia e administração).
Comprometimento renal
É improvável que o comprometimento renal exerça efeito clinicamente relevante na farmacocinética do dimetilsulfóxido de trametinibe em função da baixa excreção renal do dimetilsulfóxido de trametinibe. A farmacocinética do dimetilsulfóxido de trametinibe foi caracterizada em 223 pacientes incluídos em estudos clínicos com dimetilsulfóxido de trametinibe que apresentavam comprometimento renal leve e 35 pacientes com comprometimento renal moderado usando uma análise farmacocinética da população. O comprometimento renal leve e moderado não exerceu nenhum efeito na exposição ao dimetilsulfóxido de trametinibe ( < 6% em cada grupo). Não há dados disponíveis em pacientes com comprometimento renal grave (veja posologia e administração).
População geriátrica (65 anos de idade ou mais)
Com base na análise farmacocinética da população, a idade não exerceu efeito clínico relevante na farmacocinética do dimetilsulfóxido de trametinibe.
População pediátrica (menor que 18 anos)
Não foram conduzidos estudos para investigar a farmacocinética do dimetilsulfóxido de trametinibe em pacientes pediátricos.
Gênero/Peso
Baseado na análise farmacocinética da população, foi demonstrado que gênero e peso influenciam na liberação oral de dimetilsulfóxido de trametinibe. Apesar de indivíduos do sexo feminino serem predispostos a uma maior exposição do que os indivíduos do sexo masculino, essas diferenças são improváveis de serem clinicamente relevante e nenhum ajuste de dose é garantido.
Raça/Etnicidade
Existem dados insuficientes para avaliar o potencial efeito da raça na farmacocinética de dimetilsulfóxido de trametinibe.
Dados de segurança pré-clínicos
Carcinogênese/mutagênese
Não foram realizados estudos de carcinogenicidade com o dimetilsulfóxido de trametinibe. O dimetilsulfóxido de trametinibe não foi genotóxico em estudos para avaliar mutações reversas em bactérias, aberrações cromossômicas em células de mamíferos e micronúcleos na medula óssea de ratos.
Toxicologia reprodutiva
Desenvolvimento embriofetal e fertilidade
O dimetilsulfóxido de trametinibe pode comprometer a fertilidade em humanos. Em estudos de doses repetidas em ratas adultas e jovens com dimetilsulfóxido de trametinibe, foram observadas alterações na maturação folicular, consistindo em aumentos nos folículos císticos e diminuições nos corpos lúteos císticos a ≥ 0,016 mg/kg/dia (aproximadamente 0,3 vez a exposição clínica humana com base na área sobre a curva).
Além disso, em ratas jovens que receberam dimetilsulfóxido de trametinibe, foram observadas diminuições nos pesos dos ovários, discretos atrasos nos marcos de maturação sexual das fêmeas (abertura vaginal e aumento da incidência de brotos finais terminais proeminentes na glândula mamária) e discreta hipertrofia do epitélio superficial do útero. Todos esses efeitos foram reversíveis após um período sem tratamento e atribuíveis à farmacologia. No entanto, em estudos de toxicidade de ratos e cães de até 13 semanas de duração, não foram observados efeitos do tratamento nos tecidos reprodutivos de machos.
Estudos em animais jovens
Em um estudo de toxicidade em ratos jovens, as principais toxicidades foram no crescimento (peso corpóreo e comprimento de ossos longos), achados adversos microscópicos incluíram mudanças no osso, mineralização e/ou degeneração em vários órgãos, primariamente no estômago em todas as doses. Achados adversos em doses mais altas incluídas no olho, rim, artéria aorta e/ou cavidade/seio nasal, coração, fígado e na pele, e pesos cardíacos maiores e o atraso em um ponto de referência físico da maturidade sexual em fêmeas (abertura da vagina).
A maioria dos achados são reversíveis com exceção do osso, fósforo sérico e mineralização de tecido mole o qual progrediu/piorou durante o período sem medicamento. Também, basofilia tubular renal e pesos cardíacos maiores foram presentes ainda no final do período de recuperação.
Com exceção da distrofia/mineralização da córnea e aumento do peso cardíaco, efeitos similares foram observados em animais adultos que receberam dimetilsulfóxido de trametinibe. No menor nível de dose da combinação avaliado, a exposição sistêmica é aproximadamente 0,3 vezes a exposição humana na dose clínica de 2mg/dia baseada na área sob a curva.
Gravidez
Em estudos de toxicidade reprodutiva em ratas, foi observada toxicidade materna e no desenvolvimento (diminuição dos pesos fetais) a ≥ 0,031 mg/kg/dia (aproximadamente 0,3 vezes a exposição clínica humana com base na área sobre a curva). Em coelhas prenhas, foram observados aumentos nos abortos e toxicidade materna e no desenvolvimento (diminuição do peso corporal fetal e aumento da incidência de defeitos de ossificação) a ≥ 0,039 mg/kg/dia (aproximadamente 0,1 vez a exposição clínica humana com base na área sobre a curva).
Segurança farmacológica toxicidade de doses repetidas
Em estudos de doses repetidas em ratos, foram observadas necrose hepatocelular e elevações de transaminase após 8 semanas a ≥ 0,062 mg/kg/dia (aproximadamente 0,8 vez a exposição clínica humana com base na área sobre a curva).
Em camundongos, foi observada diminuição da frequência cardíaca, do peso do coração e da função ventricular esquerda, sem histopatologia cardíaca, após 3 semanas a ≥ 0,25 mg/kg/dia de dimetilsulfóxido de trametinibe (aproximadamente 3 vezes a exposição clínica humana com base na área sobre a curva) por até 3 semanas. Em ratos adultos, foi observada mineralização e necrose do miocárdio associadas a aumento do fósforo sérico com doses ≥ 1 mg/kg/dia (aproximadamente 12 vezes a exposição clínica humana com base na área sobre a curva). Em ratos jovens, foi observado aumento do peso cardíaco sem histopatologia a 0,35 mg/kg/dia (aproximadamente 2 vezes a exposição clínica de humanos adultos com base na área sobre a curva).
O dimetilsulfóxido de trametinibe foi fototóxico em um ensaio in vitro de captação de vermelho neutro 3T3 (Neutral Red Uptake, NRU) em fibroblastos de camundongos em concentrações significativamente maiores do que as exposições clínicas (CI50 [concentração inibitória de 50%] a 2,92 micrograma/ml, ≥ 130 vezes a exposição clínica com base na Cmáx), indicando haver risco reduzido de fototoxicidade para pacientes que usam dimetilsulfóxido de trametinibe.
Terapia em combinação
Os cães que receberam dimetilsulfóxido de trametinibe e dabrafenibe em combinação por 4 semanas demonstraram toxicidades semelhantes às observadas em estudos de monoterapia comparáveis. Consulte a bula para informações de prescrição completas do dabrafenibe.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer excipiente.
5. ADVERTÊNCIAS E PRECAUÇÕES
Leia a bula de dabrafenibe para informações específicas sobre as advertências e precauções.
Redução da FEVE/disfunção do ventrículo esquerdo: foi relatado que o dimetilsulfóxido de trametinibe reduz a FEVE [fração de ejeção do ventrículo esquerdo] (veja reações adversas). Em estudos clínicos, o tempo mediano até o início da primeira ocorrência de disfunção ventricular esquerda, insuficiência cardíaca e diminuição da FEVE em pacientes tratados com dimetilsulfóxido de trametinibe em combinação com dabrafenibe foi entre dois e cinco meses. O dimetilsulfóxido de trametinibe deve ser usado com cautela em pacientes com condições que possam prejudicar a função ventricular esquerda. A FEVE deve ser avaliada em todos os pacientes antes do início do tratamento com dimetilsulfóxido de trametinibe com uma recomendação de acompanhamento periódico oito semanas após o início da terapia, conforme clinicamente adequado. A FEVE deve continuar a ser avaliada durante o tratamento com dimetilsulfóxido de trametinibe conforme clinicamente adequado (veja posologia e administração).
Hemorragia: eventos hemorrágicos, incluindo eventos hemorrágicos importantes, ocorreram em pacientes recebendo dimetilsulfóxido de trametinibe em combinação com dabrafenibe (veja reações adversas). Dos 559 pacientes com melanoma metastático tratados com Mekinist® em combinação com dabrafenibe, houve seis casos fatais de hemorragia intracraniana (1%). Três casos eram do estudo MEK115306 (COMBI-d) e três casos eram do estudo MEK116513 (COMBI-v). Se os pacientes desenvolverem sintomas de hemorragia, eles devem procurar ajuda médica imediatamente.
Comprometimento visual: distúrbios associados a disfunções visuais, incluindo coriorretinopatia ou descolamento do epitélio pigmentado da retina (DEPR) e oclusão da veia retiniana (OVR) foram observados com o dimetilsulfóxido de trametinibe. Foram relatados sintomas como visão embaçada, diminuição da acuidade e outros fenômenos visuais nos estudos clínicos com dimetilsulfóxido de trametinibe (veja reações adversas). O dimetilsulfóxido de trametinibe não é recomendado em pacientes com histórico de OVR.
Deve-se realizar uma avaliação oftalmológica minuciosa na visita inicial e durante o tratamento com dimetilsulfóxido de trametinibe, se clinicamente justificado. Se os pacientes relatarem distúrbios visuais a qualquer momento durante a terapia com dimetilsulfóxido de trametinibe, deve-se realizar uma avaliação oftalmológica adicional. Se for observada uma anormalidade retiniana, deve-se interromper o tratamento com dimetilsulfóxido de trametinibe imediatamente e considerar o encaminhamento a um especialista em retina. Se for diagnosticado DEPR, seguir o cronograma de modificação da dose (intolerável) descrito na tabela 2 (veja posologia e administração). Em pacientes que apresentarem OVR, o tratamento com dimetilsulfóxido de trametinibe devem ser descontinuado permanentemente.
Erupção cutânea: em estudos clínicos com dimetilsulfóxido de trametinibe, foi observada erupção cutânea em cerca de 20 - 30% na combinação com dabrafenibe (veja reações adversas). A maioria dos casos foi de grau 1 ou 2 e não exigiu interrupção ou redução da dose.
Trombose venosa profunda (TVP)/embolia pulmonar (EP): TVP e EP podem ocorrer quando o dimetilsulfóxido de trametinibe é usado em combinação com o dabrafenibe. Se os pacientes desenvolverem sintomas de embolia pulmonar ou trombose venosa profunda, eles deverão procurar atendimento médico imediatamente.
Pirexia: a pirexia foi relatada em estudos clínicos com dimetilsulfóxido de trametinibe. A incidência e a gravidade da pirexia aumentam quando o dimetilsulfóxido de trametinibe é usado em combinação com dabrafenibe (veja reações adversas). Em pacientes com melanoma que receberam a dose combinada de 150 mg de dabrafenibe duas vezes ao dia e 2 mg de dimetilsulfóxido de trametinibe uma vez ao dia e desenvolveram pirexia, aproximadamente metade das primeiras ocorrências de pirexia ocorreu no primeiro mês de terapia. Cerca de um terço dos pacientes recebendo terapia de combinação que apresentaram pirexia teve 3 ou mais eventos. A pirexia pode vir acompanhada por calafrios, desidratação e hipotensão graves que, em alguns casos, podem resultar em insuficiência renal aguda. Monitorar a creatinina sérica e outras evidências de função renal durante e após eventos graves de pirexia.
Foram observados eventos febris sérios não infecciosos. Esses eventos responderam bem à interrupção e/ou redução da dose e cuidados de apoio em estudos clínicos.
Para o tratamento de pirexia, veja a bula de dabrafenibe, seções posologia e administração, modificações da dose.
Colite e perfuração gastrointestinal: colite e perfuração gastrointestinal, incluindo desfecho fatal foram reportadas em pacientes usando Mekinist® em combinação com dabrafenibe (veja reações adversas). Tratamento com Mekinist® em combinação com dabrafenibe deve ser realizado com cautela em pacientes com fatores de risco para perfuração gastrointestinal, incluindo histórico de diverticulite, metástase para o trato gastrointestinal e uso concomitante de medicamentos com um risco reconhecido de perfuração gastrointestinal.
Se os pacientes desenvolverem sintomas de colite e perfuração gastrointestinal, eles devem procurar ajuda médica imediatamente.
Populações especiais
População pediátrica (abaixo de 18 anos)
Não foi estabelecida a segurança e a eficácia do dimetilsulfóxido de trametinibe em crianças e adolescentes ( < 18 anos).
População geriátrica (65 anos de idade ou mais)
Não são necessários ajustes de dose em pacientes acima de 65 anos (ver propriedades farmacocinéticas).
Comprometimento renal
Não é necessário ajustar a dose em pacientes com comprometimento renal leve ou moderado. O comprometimento renal leve ou moderado não teve efeito significativo na farmacocinética populacional do dimetilsulfóxido de trametinibe (veja propriedades farmacocinéticas). Não há dados clínicos com dimetilsulfóxido de trametinibe em pacientes com comprometimento renal grave. Portanto, não é possível determinar a necessidade potencial de ajuste da dose inicial. O dimetilsulfóxido de trametinibe deve ser usado com cautela em pacientes com comprometimento renal grave.
Comprometimento hepático
Não há necessidade de ajuste de dose em pacientes com comprometimento hepático leve. Em uma análise farmacocinética da população, a depuração oral do dimetilsulfóxido de trametinibe e, portanto, a exposição, não foram significativamente diferentes em pacientes com comprometimento hepático leve comparadas a pacientes com função hepática normal (veja propriedades farmacocinéticas).
Não há dados clínicos de pacientes com comprometimento hepático moderado ou grave. Portanto, não é possível determinar a necessidade potencial de ajuste da dose inicial. O dimetilsulfóxido de trametinibe deve ser usado com cautela em pacientes com comprometimento hepático moderado ou grave.
Gravidez e lactação
Infertilidade
Não há informações sobre o efeito do dimetilsulfóxido de trametinibe na fertilidade humana. Em animais, não foram realizados estudos de fertilidade, mas foram observados efeitos adversos nos órgãos reprodutores femininos (veja informações pré-clínicas). O dimetilsulfóxido de trametinibe pode comprometer a fertilidade em humanos.
Gravidez
Não há estudos adequados e bem controlados do dimetilsulfóxido de trametinibe em gestantes. Estudos com dimetilsulfóxido de trametinibe em animais mostraram toxicidade reprodutiva (veja informações pré- clínicas). O dimetilsulfóxido de trametinibe não deve ser administrado a gestantes ou lactantes. Mulheres em idade fértil devem usar métodos eficazes de contracepção durante a terapia e por 4 meses após a descontinuação do dimetilsulfóxido de trametinibe. Se o dimetilsulfóxido de trametinibe, com ou sem dabrafenibe, for usado durante a gestação ou se a paciente engravidar enquanto estiver tomando dimetilsulfóxido de trametinibe em combinação com dabrafenibe, ela deve ser informada do perigo potencial para o feto.
Potencial reprodutivo de indivíduos do sexo feminino e masculino Contracepção
Indivíduos do sexo feminino com potencial reprodutivo que estiverem recebendo dimetilsulfóxido de trametinibe em combinação com dabrafenibe devem ser avisadas de que o dabrafenibe pode diminuir a eficácia de contraceptivos hormonais e que deve ser usado um método alternativo de contracepção, como métodos de barreira.
Lactação
Não se sabe se o dimetilsulfóxido de trametinibe é excretado no leite materno humano. Como vários produtos medicinais são excretados no leite materno humano, o risco ao lactente não pode ser descartado. Deve-se tomar uma decisão sobre descontinuar a amamentação ou descontinuar o dimetilsulfóxido de trametinibe, levando em consideração a importância do dimetilsulfóxido de trametinibe para a mãe.
Mekinist® pertence à categoria D de risco na gestação.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Efeitos sobre a capacidade de dirigir e/ou operar máquinas
Não houve estudos para investigar o efeito do dimetilsulfóxido de trametinibe no desempenho ao dirigir ou na capacidade de operar máquinas. Um efeito negativo em tais atividades não seria previsto pela farmacologia do dimetilsulfóxido de trametinibe. Deve-se ter em mente o estado clínico do paciente e o perfil de eventos adversos do dimetilsulfóxido de trametinibe ao considerar a capacidade do paciente realizar tarefas que requeiram habilidades de discernimento, motoras e cognitivas.
6. INTERAÇÕES MEDICAMENTOSAS
Leia a bula de dabrafenibe para informações específicas sobre as Interações Medicamentosas.
Uma vez que o dimetilsulfóxido de trametinibe é metabolizado predominantemente por desacetilação mediada por enzimas hidrolíticas (p. ex., carboxilesterases), é improvável que sua farmacocinética seja afetada por outros agentes por meio de interações metabólicas. A exposição a doses repetidas de dimetilsulfóxido de trametinibe não foi afetada pela coadministração com um indutor do citocromo CYP3A4.
Efeitos do dimetilsulfóxido de trametinibe em enzimas metabolizantes e transportadores de fármacos: dados in vitro e in vivo sugerem que é improvável que o dimetilsulfóxido de trametinibe afete a farmacocinética de outros fármacos. Com base em estudos in vitro, o dimetilsulfóxido de trametinibe não é um inibidor do CYP1A2, CYP2A6, CYP2B6, CYP2D6 e CYP3A4. Foi verificado que o dimetilsulfóxido de trametinibe é um inibidor in vitro do CYP2C8, CYP2C9 e CYP2C19, um indutor do CYP3A4 e um inibidor dos transportadores OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp e BCRP [Breast Cancer Resistance Protein (proteína de resistência do câncer de mama)]. No entanto, com base na dose e exposição clínica sistêmica reduzidas em relação à potência de inibição ou indução in vitro, o dimetilsulfóxido de trametinibe não é considerado um inibidor ou indutor in vivo dessas enzimas ou transportadores. A administração da dose repetida de 2 mg de dimetilsulfóxido de trametinibe uma vez ao dia não teve efeito sobre a Cmáx e área sobre a curva de dose única de dabrafenibe, um substrato de CYP2C8/CYP3A4.
Efeitos de outros fármacos sobre o dimetilsulfóxido de trametinibe: Dados in vivo e in vitro sugerem que a farmacocinética do dimetilsulfóxido de trametinibe provavelmente não é afetada por outros fármacos. O dimetilsulfóxido de trametinibe é desacetilado por carboxilesterases e possivelmente por outras enzimas hidrolíticas. Existe uma pequena evidência de ensaios clínicos para interação medicamentosa mediada por carboxilesterases. Enzimas CYP desempenham um papel menor na eliminação do dimetilsulfóxido de trametinibe e o componente não é um substrato dos seguintes transportadores: proteína de resistência do câncer de mama (BCRP), polipeptídeo de transporte de ânion orgânico (OATP) 1B1, OATP1B3, OATP2B1, transportador de cátion orgânico (OCT) 1, proteína associada à resistência multi-medicamentosa (MRP) 2, e proteína de extrusão de toxina e multimedicamento (MATE) 1. O dimetilsulfóxido de trametinibe é um substrato in vitro do transportador de efluxo de glicoproteína-P (Pgp), mas provavelmente não é afetado de maneira significativa pela inibição desse transportador, considerando sua alta permeabilidade passiva e alta biodisponibilidade.
A coadministração de doses repetidas de dabrafenibe 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez ao dia não resultou em alterações clinicamente significativas na Cmáx e área sobre a curva do dabrafenibe ou dimetilsulfóxido de trametinibe (veja características farmacológicas). Veja as informações de prescrição do dabrafenibe para diretrizes sobre interações medicamentosas na monoterapia com dabrafenibe.
Interação com alimentos
O dimetilsulfóxido de trametinibe deve ser administrado sem alimentos pelo menos 1 hora antes ou 2 horas após uma refeição.
Veja as informações de prescrição do dabrafenibe para diretrizes sobre interações com alimentos na monoterapia com dabrafenibe.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTOS
Armazenar sob refrigeração de 2 °C a 8 °C. Não congelar. Proteger contra umidade e luz. Conservar o produto em seu frasco original. Manter o frasco hermeticamente fechado. Não remover o dessecante.
Prazo de validade: 24 meses a partir da data de fabricação.
Número de lote, datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características do medicamento
Mekinist® 0,5 mg - comprimidos revestidos amarelos, ovais modificados, biconvexos, com "GS" gravado em uma face e "TFC" na face oposta.
Mekinist® 2 mg - comprimidos revestidos rosas, redondos, biconvexos, com "GS" gravado em uma face e "HMJ" na face oposta.
Antes de usar, observe o aspecto do medicamento.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
8. POSOLOGIA E MODO DE USAR
É necessária a confirmação da mutação BRAF V600 usando um teste aprovado/validado para a seleção de pacientes adequados para o dimetilsulfóxido de trametinibe em combinação com dabrafenibe (veja resultados de eficácia).
Considerando que o dimetilsulfóxido de trametinibe é usado em combinação com dabrafenibe, consulte a bula para informações de prescrição completas do dabrafenibe, posologia e modo de usar, para instruções de dosagem.
Adultos
A dose recomendada de dimetilsulfóxido de trametinibe em combinação com dabrafenibe é de 2 mg administrados por via oral uma vez ao dia com um copo cheio de água.
O dimetilsulfóxido de trametinibe deve ser administrado sem alimentos, pelo menos1 hora antes ou 2 horas após uma refeição (veja características farmacológicas).
Quando o dimetilsulfóxido de trametinibe e dabrafenibe forem tomados em combinação, tome a dose diária única de dimetilsulfóxido de trametinibe no mesmo horário todos os dias junto da dose matinal ou da dose noturna de dabrafenibe.
Se uma dose de dimetilsulfóxido de trametinibe for perdida, tome a dose somente se estiverem faltando mais de 12 horas até a próxima dose agendada.
Ajustes da dose
O controle de eventos adversos/reações adversas pode exigir interrupção do tratamento, redução da dose ou descontinuação do tratamento (veja a tabela 3 e a tabela