LUMAKRAS
AMGEN
sotorasibe
Antineoplásico.
Apresentações.
Comprimidos de 120 mg revestidos em embalagens com 2 frascos com 120 comprimidos cada.
USO ORAL
USO ADULTO
Composição.
Cada comprimido contém:
Informações técnicas.
1. INDICAÇÕES
LUMAKRAS é indicado para o tratamento de pacientes com câncer de pulmão de células não pequenas (CPCNP) localmente avançado ou metastático com mutação de KRAS G12C que receberam pelo menos uma linha de tratamento sistêmico anterior.
2. RESULTADOS DE EFICÁCIA
A eficácia do LUMAKRAS foi demonstrada em um estudo multicêntrico de braço único aberto (CodeBreaK 100) que incluiu pacientes com CPCNP com mutação no gene KRAS G12C localmente avançado ou metastático que tiveram progressão da doença depois do recebimento de tratamento anterior. Os principais critérios de elegibilidade incluíram progressão em um inibidor imune do ponto de verificação e/ou quimioterapia à base de platina, a escala do desempenho Eastern Cooperative Oncology Group (ECOG PS) de 0 ou 1 e pelo menos uma lesão mensurável, conforme definido pelos Critérios de avaliação de resposta em tumores sólidos (RECIST v1.1).
Todos os pacientes deveriam ter o CPCNP com mutação de KRAS G12C identificado prospectivamente em amostras de tecido tumoral utilizando um teste validado realizado em um laboratório central. Dos pacientes com mutações de KRAS G12C confirmadas no tecido tumoral, as amostras de plasma de 112 pacientes foram testadas retrospectivamente usando um teste validado separado. 78 pacientes (70%) apresentaram mutação de KRAS G12C identificada na amostra de plasma e 31 pacientes (28%) não tiveram mutação de KRAS G12C identificada na amostra de plasma.
Um total de 126 pacientes foram inscritos e tratados com LUMAKRAS 960 mg uma vez ao dia até a progressão da doença ou toxicidade inaceitável; 123 pacientes tiveram pelo menos uma lesão mensurável na avaliação inicial, conforme avaliado pela Blinded Independent Central Review (BICR), de acordo com RECIST v1.1, e foram incluídos na análise de resultados de eficácia relacionados à resposta. A duração média do tratamento foi de 5,5 meses (faixa de 0 a 12) com 48% dos pacientes tratados por ≥ 6 meses e 29% dos pacientes tratados por ≥ 9 meses. A medida dos resultados de maior eficácia foi a taxa de resposta objetiva (TRO) e a duração da resposta (DOR), conforme avaliado pela BICR, de acordo com RECIST v1.1. As medidas adicionais dos resultados de eficácia incluem a taxa de controle da doença (TCDs), o tempo para resposta (TR), a sobrevida livre de progressão (SLP) e a sobrevida global (SG).
As características demográficas e da doença da população do estudo na avaliação inicial foram: idade média de 64 anos (faixa de 37 a 80) com 47% ≥ 65 anos e 8% ≥ 75 anos; 50% do sexo feminino; 82% brancos, 15% asiáticos, 2% negros; 70% ED ECOG 1; 96% tinham doença em estágio IV; 99% com histologia não escamosa; 81% ex-fumantes, 12% fumantes, 5% nunca fumaram. Todos os pacientes receberam pelo menos 1 linha anterior de terapia sistêmica para CPCNP metastático; 43% receberam apenas 1 linha de terapia anterior, 35% receberam 2 linhas de terapia anteriores, 22% receberam 3 linhas de terapia anteriores; 91% receberam imunoterapia anterior anti-PD-1/PD-L1, 90% receberam quimioterapia anterior à base de platina, 81% receberam quimioterapia à base de platina e anti-PD-1/PD-L1. Os locais de metástase extratorácica conhecidos incluíram 48% no osso, 21% no cérebro e 21% no fígado.
Os resultados da eficácia estão resumidos na Tabela 1. A TRO foi de 37% (IC de 95%: 29, 47). Os pacientes com respostas objetivas tiveram DOR variando de 1,3 a 8,4 meses e 52% ainda estavam em terapia com resposta contínua, depois de um tempo médio de acompanhamento de 6,9 meses. A TR mediana foi de 1,4 mês (intervalo de 1,2 a 6,1), com 70% das respostas ocorrendo nas primeiras 7 semanas. Os resultados consistentes de eficácia foram observados em pacientes com mutação de KRAS G12C identificada em amostras de tecido ou de plasma.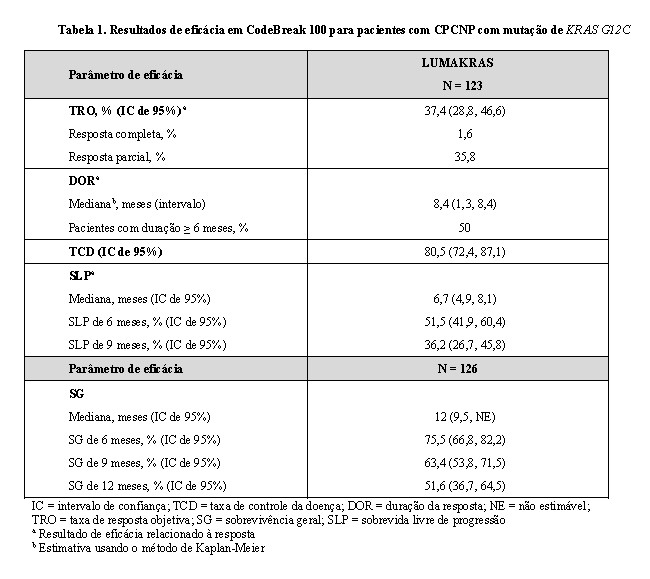
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O sotorasibe é um inibidor potente e altamente seletivo de KRASG12C, que se liga, de forma covalente e irreversível, à cisteína exclusiva de KRASG12C. A inativação do KRASG12C pelo sotorasibe bloqueia a sinalização e a sobrevida das células tumorais, inibe o crescimento das células e promove a apoptose seletivamente em tumores que apresentam KRASG12C, um impulsionador oncogênico de tumorigênese em vários tipos de câncer. A potência e seletividade do sotorasibe são melhoradas pela ligação exclusiva ao compartimento P2 e à ranhura de superfície His95, bloqueando a proteína em um estado inativo que impede a sinalização a jusante sem afetar o KRAS tipo selvagem.
O sotorasibe demonstrou inibição in vitro e in vivo do KRASG12C com atividade mínima detectável fora do alvo contra outras proteínas e processos celulares. O sotorasibe prejudicou a sinalização oncogênica e a sobrevida das células tumorais em exposições clinicamente relevantes em diversos modelos pré-clínicos que expressavam KRASG12C. O sotorasibe também melhorou a apresentação de antígenos e a produção de citocinas inflamatórias somente em células tumorais com KRASG12C. O sotorasibe induziu respostas inflamatórias antitumorais e imunidade, gerando regressões tumorais permanentes e completas em camundongos imunocompetentes implantados com KRASG12C que expressavam tumores.
Eletrofisiologia cardíaca
O efeito do sotorasibe sobre o intervalo QT foi avaliado em 156 pacientes que receberam LUMAKRAS 960 mg uma vez ao dia em estudos clínicos. O LUMAKRAS não prolongou o intervalo QT para nenhuma extensão clinicamente relevante. Em concentrações de pico, a alteração média da avaliação inicial foi inferior a 5 ms. Nenhum paciente teve um grande aumento médio no QTc ( > 20 ms) no estudo.
Propriedades farmacocinéticas
A farmacocinética do sotorasibe foi caracterizada em pacientes com tumores sólidos com mutação de KRAS G12C, incluindo CPCNP, e indivíduos saudáveis.
Absorção
Depois da administração oral de dose única, o sotorasibe foi absorvido com tempo médio para atingir a concentração de pico de 1 hora.
Efeito dos alimentos
Depois da administração de sotorasibe com uma refeição altamente calórica e com alto teor de gordura, não houve efeito sobre a Cmáx, e a AUC aumentou 38% em comparação com a administração em condições de jejum. O sotorasibe pode ser administrado com ou sem alimentos.
Distribuição
O volume médio de distribuição em estado estável do sotorasibe foi de 211 l. A ligação da proteína no plasma in vitro do sotorasibe foi de 89%.
Metabolismo
As principais vias metabólicas do sotorasibe foram conjugação e metabolismo oxidativo.
Eliminação
A 960 mg uma vez ao dia, a depuração aparente em estado estável é de 26,2 l/h. A meia-vida média é de 5 horas. O estado estável foi alcançado em 22 dias e permaneceu estável. Nenhum acúmulo com várias dosagens foi observado. O sotorasibe é eliminado principalmente nas fezes, com aproximadamente 74% da dose recuperada nas fezes e 6% (1% inalterada) recuperada na urina.
Populações especiais
Não foram observadas diferenças clinicamente significativas na farmacocinética do sotorasibe com base na idade, sexo, raça, peso corporal, linha de terapia, ED ECOG, comprometimento renal leve (depuração da creatinina: ≥ 60 ml/min) ou comprometimento hepático leve (AST ou ALT < 2,5 x LSN ou bilirrubina total < 1,5 x LSN). O efeito do comprometimento renal ou hepático moderado a grave na farmacocinética do sotorasibe não foi estudado.
Estudos de interação medicamentosa
Estudos clínicos
Efeito de outros medicamentos no sotorasibe
Agente redutor de acidez: Em condições alimentadas (refeições de caloria padrão com quantidade moderada de gordura), a coadministração de várias doses de omeprazol (IBP) com uma dose única de 960 mg de LUMAKRAS diminuiu a Cmáx do sotorasibe em 65% e a AUC em 57%.
A coadministração de uma única dose de famotidina (antagonista do receptor H2) administrada 10 horas antes e 2 horas depois de uma única dose de 960 mg de LUMAKRAS diminuiu a Cmáx do sotorasibe em 35% e a AUC em 38%.
Em condições de jejum, a coadministração de várias doses de omeprazol com uma única dose de 960 mg de LUMAKRAS diminuiu a Cmáx do sotorasibe em 57% e a AUC em 42% [consulte INTERAÇÕES MEDICAMENTOSAS].
Indutores fortes de CYP3A4: A coadministração de LUMAKRAS com várias doses de rifampicina (um indutor forte de CYP3A4) reduziu a Cmáx do sotorasibe em 35% e a AUC em 51% [consulte INTERAÇÕES MEDICAMENTOSAS].
Inibidores fortes de CYP3A4 e sistemas de transporte: Nenhum efeito clinicamente significativo sobre a exposição do sotorasibe foi observado depois da coadministração de LUMAKRAS com itraconazol (um forte inibidor de CYP3A4 e de glicoproteína P [gp-P]), dose única de rifampicina (um inibidor de OATP1B1/1B3) ou metformina (um substrato de MATE1/MATE2-K).
Efeito do sotorasibe sobre outros medicamentos
Substratos de CYP3A4: A coadministração de LUMAKRAS com midazolam (um substrato sensível de CYP3A4) reduziu a Cmáx do midazolam em 48% e a AUC em 53% [consulte INTERAÇÕES MEDICAMENTOSAS].
Substratos da BCRP
A coadministração de LUMAKRAS com rosuvastatina (um substrato da BCRP) aumentou a Cmáx de rosuvastatina em 70% e a AUC em 34% [vide INTERAÇÕES MEDICAMENTOSAS].
Estudos in vitro
Enzimas CYP: Os dados in vitro indicados para o sotorasibe não inibem as CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ou CYP2D6 em concentrações clinicamente relevantes.
Sistemas de transporte: Os dados in vitro indicaram que o sotorasibe pode ter potencial para inibir a BCRP. A relevância clínica desses achados é desconhecida.
Dados de segurança pré-clínicos/Toxicologia não clínica
Mutagenicidade
O sotorasibe não foi mutagênico em um ensaio de mutagenicidade bacteriana (Ames). O sotorasibe não foi genotóxico nos ensaios do micronúcleo e cometa in vivo em ratos.
Carcinogenicidade
Não foram realizados estudos de carcinogenicidade com o sotorasibe.
Comprometimento da fertilidade
Estudos de fertilidade/desenvolvimento embrionário precoce não foram realizados com o sotorasibe. Não houve efeitos adversos nos órgãos reprodutivos masculinos ou femininos nos estudos toxicológicos gerais realizados em cães e ratos.
4. CONTRAINDICAÇÕES
Hipersensibilidade
O LUMAKRAS é contraindicado em pacientes com hipersensibilidade clinicamente significativa conhecida ao sotorasibe ou a qualquer componente da formulação do produto.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hepatotoxicidade
O sotorasibe pode causar hepatotoxicidade, que pode causar lesão hepática induzida por medicamento (DILI) e hepatite. O sotorasibe foi associado a elevações transitórias das transaminases séricas (ALT e AST). Estas elevações melhoraram ou se resolveram com modificação da dose ou descontinuação permanente do tratamento e não resultaram em quaisquer casos de insuficiência hepática ou casos fatais em estudos clínicos. Entre os pacientes que apresentaram hepatotoxicidade, 38% tiveram hepatotoxicidade levando à interrupção ou redução da dose. No geral, 26% dos pacientes com hepatotoxicidade receberam corticosteroides concomitantes. Os casos de aumento das enzimas hepáticas podem ser assintomáticos. Os pacientes devem ser monitorados quanto à função hepática (ALT, AST, bilirrubina total) antes do início de LUMAKRAS, a cada 3 semanas durante os primeiros 3 meses de tratamento, depois uma vez por mês ou conforme clinicamente indicado, com testes mais frequentes em pacientes que desenvolveram elevações das transaminases e / ou bilirrubina. Com base na gravidade das anormalidades laboratoriais, o tratamento com LUMAKRAS deve ser interrompido até recuperação para grau ≤ 1 ou grau basal, e a dose deve ser modificada ou permanentemente descontinuada conforme recomendado (vide POSOLOGIA E USO).
Doença pulmonar intersticial (DPI) / pneumonite
A DPI / pneumonite ocorreu em pacientes tratados com LUMAKRAS com exposição anterior a imunoterapia ou radioterapia (vide EVENTOS ADVERSOS). Monitore os pacientes quanto a novos ou agravamento de sintomas pulmonares indicativos de DPI/ pneumonite (por exemplo, dispneia, tosse, febre). Suspenda imediatamente o uso LUMAKRAS em pacientes com suspeita de DPI/ pneumonite e descontinue o LUMAKRAS permanentemente se nenhuma outra causa potencial de DPI/ pneumonite for identificada (vide POSOLOGIA E USO)
Populações especiais
Gravidez
Não há estudos clínicos com o uso de LUMAKRAS em gestantes.
Em estudos de desenvolvimento embrionário-fetal em ratos e coelhos, o sotorasibe oral não foi teratogênico.
Em rato, não houve efeitos sobre o desenvolvimento embrionário-fetal até a dose mais alta testada (3,9 vezes mais alta do que a exposição na dose humana máxima recomendada [DHMR] de 960 mg baseada na área sob a curva [AUC]).
Em coelho, os pesos corporais fetais mais baixos e uma redução do número de metacarpos ossificados nos fetos foram observados somente no nível de dose mais alto testado (2,2 vezes mais alto que a exposição na DHMR de 960 mg com base na AUC), que estava associada a efeitos maternos como diminuição do ganho de peso corporal e diminuição do consumo de alimentos durante a fase de dosagem. A ossificação reduzida, como evidência de supressão do crescimento associada à redução do peso corporal fetal, foi interpretada como efeito não específico na presença de toxicidade materna significativa.
Informe a paciente sobre os possíveis riscos para o feto se o LUMAKRAS for usado durante a gravidez ou se a paciente ficar grávida enquanto estiver tomando o LUMAKRAS.
Categoria B para gravidez: Este medicamento não deve ser usado por gestantes sem orientação médica.
Lactação
Devido ao possível risco de o tratamento com LUMAKRAS causar efeitos adversos em crianças amamentadas, é necessário tomar a decisão de interromper a amamentação durante o tratamento e por uma semana após a dose, ou de interromper o tratamento com LUMAKRAS durante a amamentação.
Fertilidade
Não há estudos clínicos para avaliar o efeito do LUMAKRAS na fertilidade.
Pediatria
A segurança e a eficácia do LUMAKRAS em pacientes pediátricos não foram estabelecidas.
Geriatria
Em estudos clínicos, não foram observadas diferenças gerais de segurança ou eficácia entre pacientes geriátricos (≥ 65 anos) e pacientes mais jovens. Nenhum ajuste de dose é necessário para pacientes geriátricos.
Comprometimento hepático
Nenhum ajuste de dose é recomendado para pacientes com comprometimento hepático leve (AST ou ALT < 2,5 x LSN ou bilirrubina total < 1,5 x LSN). O LUMAKRAS não foi estudado em pacientes com comprometimento hepático moderado ou grave.
Insuficiência renal
Com base na análise farmacocinética da população, nenhum ajuste de dose é recomendado para pacientes com insuficiência renal leve (depuração de creatinina: ≥ 60 ml/min). O LUMAKRAS não foi estudado em pacientes com grau moderado ou grave (depuração da creatinina: < 60 ml/min) de insuficiência renal.
Efeitos sobre a capacidade de dirigir e de usar máquinas
O LUMAKRAS não tem influência ou é insignificante na capacidade de dirigir e usar máquinas.
Atenção diabéticos: contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
Efeitos de outros medicamentos no LUMAKRAS
Agentes redutores de acidez
A coadministração de LUMAKRAS com IBP (omeprazol) ou antagonista do receptor H2 (famotidina) levou a uma diminuição das concentrações de sotorasibe. A coadministração de IBPs e de antagonistas do receptor H2 com LUMAKRAS não é recomendada porque o impacto na eficácia é desconhecido. Se o tratamento com um agente redutor de acidez for necessário, tome o LUMAKRAS 4 horas antes ou 10 horas depois da administração de um antiácido local [consulte Propriedades farmacocinéticas].
Indutores fortes de CYP3A4
A coadministração do LUMAKRAS com um indutor forte de CYP3A4 (rifampina) levou a uma diminuição das concentrações de sotorasibe. A coadministração de indutores fortes de CYP3A4 com LUMAKRAS não é recomendada porque o impacto na eficácia é desconhecido [consulte Propriedades farmacocinéticas].
Efeitos do LUMAKRAS sobre outros medicamentos
Substratos de CYP3A4
O LUMAKRAS é um indutor de CYP3A4 moderado. A coadministração de LUMAKRAS com substratos de CYP3A4 levou a uma diminuição das suas concentrações plasmáticas, o que pode reduzir a eficácia desses substratos [consulte Propriedades farmacocinéticas]. Evite a coadministração de LUMAKRAS com substratos de CYP3A4 com índices terapêuticos restritos. Se a coadministração não puder ser evitada, ajuste a dosagem do substrato de CYP3A4 de acordo com a rotulagem aprovada do produto.
Substratos da BCRP
LUMAKRAS é um inibidor fraco da BCRP. A coadministração de LUMAKRAS com um substrato da BCRP levou a um aumento nas concentrações plasmáticas do substrato da BCRP o que pode aumentar os efeitos desses substratos [vide CARACTERÍSTICAS FARMACOLÓGICAS]. Quando coadministrado com LUMAKRAS, monitore as reações adversas do substrato da BCRP e reduza a dosagem do substrato da BCRP, de acordo com a bula do produto.
Efeito do sotorasibe em substratos de glicoproteína P
A co-administração de sotorasibe com digoxina (um substrato da glicoproteína P) aumentou a Cmax em 1,9 vezes e a AUCinf em 1,2 vezes da digoxina administrada isoladamente. A coadministração de LUMAKRAS com substratos da gp-P com índices terapêuticos estreitos não é recomendada. Se a co-administração não puder ser evitada, ajuste a dosagem do substrato gp-P de acordo com a bula do produto.
7. CUIDADOS COM O ARMAZENAMENTO DO MEDICAMENTO
Armazene o LUMAKRAS em temperatura ambiente entre 15 °C e 30 °C.
Incompatibilidades: não há incompatibilidades conhecidas.
Validade: 24 meses
Número do lote e datas de fabricação e validade: consulte a embalagem.
Não use o medicamento da data de validade. Mantenha o medicamento na embalagem original.
Antes de usar, observe a aparência do medicamento.
Depois de aberto, é válido por 30 dias.
Todos os medicamentos devem ser mantidos fora do alcance das crianças.
8. POSOLOGIA E USO
Confirme a presença da mutação KRAS G12C usando um teste validado antes de iniciar o uso do LUMAKRAS.
Posologia
A dose recomendada de LUMAKRAS é de 960 mg (oito comprimidos de 120 mg) via oral uma vez por dia até a progressão da doença ou toxicidade inaceitável.
Se uma dose de LUMAKRAS for perdida, não tome a dose se tiverem passado 6 horas do tempo de dosagem programado. Retome o tratamento conforme prescrito no dia seguinte.
Não tome uma dose adicional se vomitar depois de tomar LUMAKRAS. Retome o tratamento conforme prescrito no dia seguinte.
Modificações da dose
Os níveis de redução de dose de LUMAKRAS estão resumidos na Tabela 2. As modificações na dose para reações adversas são fornecidas na Tabela 3.
Se ocorrerem eventos de toxicidade, são permitidas no máximo duas reduções de dose. Interrompa o LUMAKRAS se os pacientes não conseguirem tolerar a dose mínima de 240 mg uma vez por dia.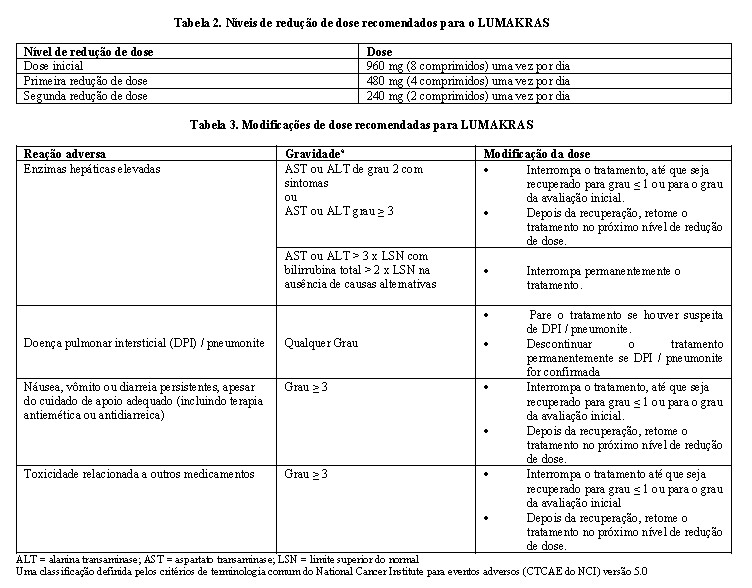
Coadministração de LUMAKRAS com agentes redutores de acidez
A coadministração de IBPs e de antagonistas do receptor H2 com LUMAKRAS não é recomendada. Se o tratamento com um agente redutor de acidez for necessário, tome LUMAKRAS 4 horas antes ou 10 horas depois da administração de um antiácido local [consulte INTERAÇÕES MEDICAMENTOSAS e Propriedades farmacocinéticas].
Método de administração
Tome LUMAKRAS no mesmo horário todos os dias, com ou sem ingestão de alimentos. Engula os comprimidos inteiros. Não mastigue, amasse nem divida os comprimidos.
Administração em pacientes com dificuldade de engolir sólidos
Disperse os comprimidos em 120 ml de água sem gás em temperatura ambiente e sem esmagar. Não use outros líquidos. Agite até que os comprimidos sejam dispersos em pequenos pedaços (os comprimidos não se dissolverão completamente) e beba imediatamente. A aparência da mistura pode variar de amarelo pálido a amarelo brilhante. Enxágue o recipiente com mais 120 ml (4 onças) de água e beba imediatamente. Se a mistura não for consumida imediatamente, agite a mistura novamente para garantir que os comprimidos sejam dispersos. Consuma até duas horas depois da preparação.
Insuficiência hepática
Nenhum ajuste de dose é recomendado para pacientes com insuficiência hepática leve (AST ou ALT < 2,5 x LSN ou bilirrubina total < 1,5 x LSN). A administração de sotorasibe em indivíduos com insuficiência hepática moderada e grave não é recomendada.
Insuficiência renal
Nenhum ajuste de dose é recomendado para pacientes com insuficiência renal leve (depuração da creatina, CrCL, ≥ 60 mL/min). LUMAKRAS não foi estudado em pacientes com insuficiência renal moderada ou grave (CrCL < 60 mL/min). Portanto, deve-se ter cuidado ao tratar pacientes com insuficiência renal moderada, grave e terminal (vide CARACTERÍSTICAS FARMACOLÓGICAS)
Este medicamento não deve ser dividido, aberto nem mastigado.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
As reações adversas mais comuns foram diarreia (34%), náuseas (25%) e fadiga (21%). As reações adversas graves mais comuns (grau ≥ 3) foram aumento da ALT (5%), aumento da AST (4%) e diarreia (4%). As reações adversas mais comuns que conduzem à descontinuação permanente do tratamento foram o aumento da ALT (1%) e o aumento da AST (1%) e DILI (1%). As reações adversas mais comuns que levam à modificação da dose foram aumento de ALT (6%), diarreia (6%), aumento de AST (6%), náuseas (3%), aumento da fosfatase alcalina no sangue (3%) e vômitos (2%).
Lista tabelada de reações adversas
As reações adversas notificadas em estudos clínicos com LUMAKRAS são apresentadas na tabela 3 abaixo. As categorias de frequência são definidas como segue: muito comuns (≥ 1/10), comuns (≥ 1/100 a < 1/10), incomuns (≥ 1/1.000 a < 1/100), raros (≥ 1/10.000 a < 1/1.000) muito raro ( < 1/10.000) e desconhecido (não pode ser calculado a partir dos dados disponíveis). Dentro de cada classe de sistemas de órgãos, as reações adversas são apresentadas por ordem decrescente de gravidade.
A segurança de LUMAKRAS foi avaliada em 359 pacientes com tumores sólidos com mutação KRAS G12C que receberam 960 mg por via oral uma vez ao dia como monoterapia. A duração mediana da exposição ao LUMAKRAS foi de 4,1 meses (intervalo: 0,02 a 21).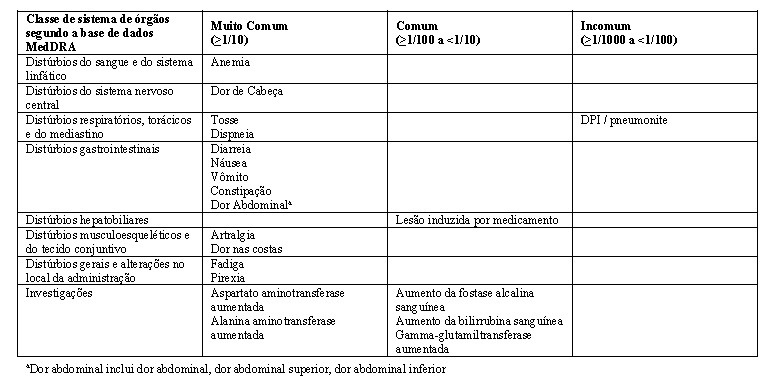
Descrição das reações adversas selecionadas
Enzimas hepáticas elevadas
Em estudos clínicos, foram observadas elevações transitórias das transaminases séricas (vide ADVERTÊNCIAS E PRECAUÇÕES). Elevações de ALT ocorreram em 14% dos indivíduos e elevações de AST em 16% dos indivíduos, com um tempo médio de início de 8 semanas (intervalo: 1 a 42) e 8 semanas (intervalo: 0 a 42), respectivamente. As elevações da ALT resultaram na interrupção e / ou redução da dose em 6,1% dos indivíduos, e as elevações da AST resultaram na interrupção e / ou redução da dose em 6,1% dos indivíduos.
DPI / pneumonite
Em estudos clínicos, entre 359 pacientes que receberam LUMAKRAS, DPI / pneumonite ocorreu em 0,8% dos pacientes, todos os casos foram de grau 3 ou 4 no início. O tempo médio para o primeiro aparecimento de DPI / pneumonite foi de 2 semanas (intervalo: 2 a 18 semanas). LUMAKRAS foi descontinuado devido a DPI / pneumonite em 0,6% dos pacientes (ver POSOLOGIA E USO e AVISOs E PRECAUÇÕES).
Idosos
Em estudos clínicos, não foram observadas diferenças gerais na segurança ou eficácia entre pacientes idosos (≥ 65 anos de idade) e pacientes mais jovens (vide CARACTERÍSTICAS FARMACOLÓGICAS e POSOLOGIA E USO).
Aviso: trata-se de um novo medicamento e, embora a pesquisa tenha indicado eficácia e segurança aceitáveis, mesmo se indicado e usado corretamente, pode haver eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo sistema VigiMed, disponível no portal da Anvisa.
10. SUPERDOSE
Não há experiência clínica com sobredosagem de LUMAKRAS. Em caso de sobredosagem, trate o paciente sintomaticamente e institua medidas de suporte, conforme necessário.
Em caso de envenenamento, ligue para 0800 722 6001, se precisar de mais orientação.
Dizeres legais.
VENDA SOB PRESCRIÇÃO MÉDICA.
MS 1.0244.0021.001-3