LORBRENA
PFIZER
lorlatinibe
Inibidor da tirosina-quinase.
Apresentações.
Lorbrena® 25 mg em frascos contendo 90 comprimidos revestidos e 100 mg em frascos contendo 30 comprimidos revestidos.
VIA DE ADMINISTRAÇÃO: USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de Lorbrena® 25 mg contém 25 mg de lorlatinibe.
Cada comprimido revestido de Lorbrena®100 mg contém 100 mg de lorlatinibe.
Excipientes: celulose microcristalina, fosfato de cálcio dibásico, amidoglicolato de sódio, estearato de magnésio, hipromelose, lactose monoidratada, macrogol 400, triacetina, dióxido de titânio, óxido ferroso, óxido de ferro vermelho.
Informações técnicas.
1. INDICAÇÕES
Lorbrena® é indicado para o tratamento de pacientes adultos com câncer de pulmão de não pequenas células (CPNPC) avançado, positivo para quinase do linfoma anaplásico (ALK), previamente tratados com um ou mais inibidores da tirosina quinase (TKIs) ALK.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
O uso de Lorbrena® no tratamento de CPNPC avançado com ALK positivo, previamente tratado com 1 ou mais ALK TKIs, foi investigado no Estudo B7461001, um estudo multicêntrico de fase 1/2 de braço único. Um total de 197 pacientes com CPNPC avançado com ALK, previamente tratados com 1 ou mais ALK TKIs, foram incluídos na fase 2 do estudo. Os pacientes receberam Lorbrena® por via oral na dose recomendada de 100 mg uma vez ao dia, continuamente.
O parâmetro de avaliação de eficácia primário na parte da Fase 2 do estudo foi a taxa de resposta objetiva (TRO), incluindo TRO intracraniana, conforme a Revisão Central Independente (ICR) de acordo com os Critérios de Avaliação de Resposta modificados em Tumores Sólidos (RECIST versão 1.1 modificada). Os parâmetros de avaliação secundários incluíram duração da resposta (DDR), DDR intracraniana, tempo para resposta (TTR) e sobrevida livre de progressão (PFS).
Os dados demográficos dos 197 pacientes com CPNPC avançado de ALK positivo previamente tratados com 1 ou mais ALK TKIs foram 59% mulheres, 49% caucasianos, 36% asiáticos e a média de idade era de 53 anos (faixa de variação: 29 a 85 anos) com 19% ≥65 anos. O status de desempenho do Eastern Cooperative Oncology Group (ECOG) [Grupo Oncológico Cooperativo Oriental] no início do estudo era de 0 ou 1 em 97% dos pacientes e 2 em 4% dos pacientes. Metástases cerebrais estavam presentes no início do estudo em 62% dos pacientes. Todos os 197 pacientes receberam terapia sistêmica prévia, 20% receberam 1, 28% receberam 2, 19% receberam 3 e 34% receberam 4 ou mais terapias sistêmicas prévias. Dos 197 pacientes, 44% receberam previamente 1 tratamento com ALK TKI, 33% receberam 2 tratamentos com ALK TKIs e 23% receberam 3 ou mais ALK TKIs.
Os principais resultados de eficácia do Estudo B7461001 estão resumidos nas Tabelas 1 e 2.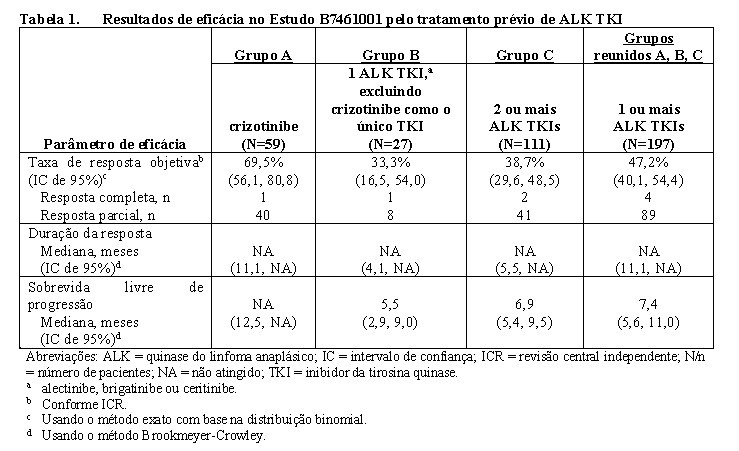

Entre os 93 pacientes com uma resposta objetiva confirmada pelo ICR, o TTR médio foi de 1,4 meses (faixa de variação: 1,1 a 11,0 meses). Entre os 70 pacientes com uma resposta objetiva do tumor confirmada pelo ICR, a TTR intracraniana média foi de 1,4 meses (faixa de variação: 1,1 a 6,2 meses).
Referências
1. Benjamin JS, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol 2018; 19: 1654-67
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
O lorlatinibe é um inibidor de pequenas moléculas inibidoras da tirosina quinase ALK e ROS1, com penetração em sistema nervoso central, adenosina trifosfato (ATP) competitiva, seletivo, que atua em mecanismos de resistência desenvolvidos após tratamento prévio com inibidores de ALK.
Em estudos não clínicos, o lorlatinibe inibiu potencialmente as atividades catalíticas de ALK não mutantes e uma ampla gama de quinases mutantes de ALK clinicamente relevantes em enzimas recombinantes e ensaios com base em células. As mutações de ALK analisadas incluíram aquelas que conferiram resistência a outros inibidores de ALK, incluindo alectinibe, brigatinibe, ceritinibe e crizotinibe.
O lorlatinibe demonstrou atividade antitumoral marcada em baixas concentrações plasmáticas livres nanomolares em camundongos portadores de xenoenxertos tumorais que expressam fusões equinodermes de proteínas semelhantes a microtúbulos 4 (EML4) com ALK variante 1 (v1), incluindo mutações ALK L1196M, G1269A, G1202R, e I1171T. Sabe-se que dois desses mutantes ALK, G1202R e I1171T, conferem resistência aos inibidores de ALK de primeira e segunda geração. O lorlatinibe também é capaz de penetrar na barreira hematoencefálica e alcançar exposição cerebral eficaz em camundongos e ratos. Em camundongos com implantes de tumor cerebral EML4ALK ou EML4ALKL1196M ortotópicos, o lorlatinibe causou redução tumoral e proporcionou sobrevida prolongada. A eficácia antitumoral global do lorlatinibe foi dependente da dose e fortemente correlacionada com a inibição da fosforilação de ALK.
Propriedades Farmacocinéticas
Absorção
As concentrações máximas de lorlatinibe no plasma são rapidamente atingidas com a Tmax média de 1,2 horas após uma dose única de 100 mg e de 2,0 horas após uma dose múltipla de 100 mg uma vez por dia.
Após a administração oral de comprimidos de lorlatinibe, a biodisponibilidade absoluta média é de 80,8% (IC de 90%: 75,7%, 86,2%) em comparação com a administração intravenosa.
A administração de lorlatinibe com alimentação com alto teor de gordura e alto teor calórico resultou em exposição 5% maior em comparação ao jejum noturno (razão AUCinf de 104,7%; IC de 90% para a razão: 101,3%, 108,3%). O lorlatinibe pode ser administrado com ou sem alimento. O rabeprazol, inibidor da bomba de prótons, teve um efeito mínimo sobre a exposição plasmática do lorlatinibe (razão AUCinf de 100,9%; IC de 90% para a razão: 97,6%, 104,3%). Nenhum ajuste de dose é necessário quando lorlatinibe é tomado com inibidores da bomba de prótons, antagonistas dos H2-receptores ou antiácidos de ação local.
Após múltiplas administrações de dose uma vez ao dia, o Cmax de lorlatinibe aumentou proporcionalmente à dose e o AUCtau aumentou ligeiramente menos do que proporcionalmente em uma dose de lorlatinibe de 10 a 200 mg uma vez ao dia. Na dose de 100 mg uma vez ao dia de lorlatinibe, a média geométrica de concentração plasmática máxima foi 577 ng/mL e a AUC24 5.650 ng h/mL em pacientes com câncer. A depuração oral média geométrica foi 17,7 L/h. A depuração oral de lorlatinibe aumentou no estado de equilíbrio em comparação com a dose única, indicando autoindução.
Distribuição
A ligação in vitro do lorlatinibe às proteínas plasmáticas humanas é de 66% com ligação moderada à albumina à glicoproteína ácida a1.
Metabolismo
Em humanos, o lorlatinibe sofre oxidação e glucuronidação como as principais vias metabólicas. Os dados in vitro indicam que o lorlatinibe é principalmente metabolizado pelo CYP3A4 e UGT1A4 com contribuições menores do CYP2C8, CYP2C19, CYP3A5 e UGT1A3.
No plasma, um ácido benzoico metabólito de lorlatinibe resultante da clivagem oxidativa das ligações amida e éter aromático do lorlatinibe foi observado como metabólito principal, responsável por 21% da radioatividade circulante. O metabólito de clivagem oxidativa é farmacologicamente inativo.
Eliminação
A meia-vida plasmática do lorlatinibe após uma dose única de 100 mg foi 23,6 horas. Após a administração oral de uma dose de 100 mg radiomarcada de lorlatinibe, uma média de 47,7% da radioatividade foi recuperada na urina e 40,9% da radioatividade foi recuperada nas fezes, com recuperação total média global de 88,6%.
O lorlatinibe inalterado era o principal componente do plasma e das fezes humanas, representando 44% e 9,1% da radioatividade total no plasma e nas fezes, respectivamente. Menos de 1% de lorlatinibe inalterado foi detectado na urina.
Eletrofisiologia cardíaca
• Intervalo QT
No Estudo B7461001, 2 pacientes (0,7%) apresentaram valores de QTc (QTcF) de correção de Fridericia absolutos > 500 ms e 5 pacientes (1,8%) tiveram uma mudança no QTcF do valor basal > 60 ms.
Além disso, o efeito de uma dose oral única de lorlatinibe (50 mg, 75 mg e 100 mg) com e sem 200 mg de itraconazol uma vez ao dia foi avaliado em um estudo cruzado de 2 vias em 16 voluntários saudáveis. Não foram observados aumentos no intervalo QTc médio para as concentrações médias observadas de lorlatinibe neste estudo.
Em 295 pacientes que receberam lorlatinibe na dose recomendada de 100 mg uma vez ao dia no Estudo B7461001, não foram detectados aumentos médios significativos do valor basal no intervalo QTcF (ou seja, > 20 ms).
• Intervalo PR
Em 295 pacientes que receberam lorlatinibe na dose recomendada de 100 mg uma vez ao dia e tinham uma medição de ECG no Estudo B7461001, a alteração média máxima do valor basal para o intervalo PR foi de 16,4 ms (IC de 90% superior de 2 lados: 19,4 ms). Entre os 284 pacientes com intervalo PR < 200 ms, 14% tiveram prolongamento do intervalo PR ≥200 ms após o início de lorlatinibe. O prolongamento do intervalo PR ocorreu de forma dependente da concentração. O bloqueio atrioventricular ocorreu em 1,0% dos pacientes.
Para aqueles pacientes que desenvolvem prolongamento PR, pode ser necessário (vide item 8. Posologia e Modo de usar).
Populações especiais
• Insuficiência hepática
Uma vez que o lorlatinibe é metabolizado no fígado, é provável que o comprometimento hepático aumente as concentrações plasmáticas do lorlatinibe. Os estudos clínicos que foram conduzidos excluíram pacientes com AST ou ALT > 2,5 x LSN, ou devido a malignidade subjacente, > 5,0 x LSN ou com bilirrubina total > 1,5 x LSN. Análises populacionais farmacocinéticas mostraram que a exposição ao lorlatinibe não era clinicamente significativa em pacientes com insuficiência hepática leve (n = 50). Não são recomendados ajustes da dose para pacientes com insuficiência hepática leve (vide item 8. Posologia e Modo de usar). O lorlatinibe não foi estudado em pacientes portadores de insuficiência hepática grave.
• Disfunção renal
Menos de 1% da dose administrada é detectada como lorlatinibe inalterado na urina. Estudos clínicos excluíram pacientes com creatinina sérica > 1,5 x LSN ou CLcr estimado < 60 mL/min. Análises farmacocinéticas populacionais mostraram que a exposição ao lorlatinibe não foi clinicamente significativa em pacientes com insuficiência renal leve (n = 103) ou moderada (n = 41) (CLcr ≥ 30 mL/min). Nenhum ajuste de dose é necessário para pacientes com insuficiência renal leve ou moderada (vide item 8. Posologia e Modo de usar). As informações para uso de lorlatinibe em pacientes com insuficiência renal grave (CLcr < 30 mL/min) é limitada (n = 1).
• Idosos (≥65 anos)
Dos 295 pacientes na população de segurança do Estudo B7461001, 18,3% dos pacientes tinham 65 anos ou mais. Dos 215 pacientes na população de eficácia do Estudo B7461001, 17,7% dos pacientes tinham 65 anos ou mais. Embora os dados sejam limitados, não foram observadas diferenças clinicamente relevantes na segurança ou eficácia entre pacientes com idade maior ou igual a 65 anos e pacientes mais jovens; não são recomendados ajustes de dose em pacientes idosos (vide item 8. Posologia e Modo de usar).
• Sexo, raça, peso corporal e fenótipo
Análises farmacocinéticas populacionais em pacientes com CPNPC avançado e voluntários saudáveis indicam que não há efeitos clinicamente relevantes de idade, sexo, raça, peso corporal ou fenótipos para CYP3A5 e CYP2C19.
Dados de Segurança Pré-Clínicos
• Toxicidade por dose repetida
As principais toxicidades observadas foram inflamação em vários tecidos (com aumento de glóbulos brancos) e alterações no pâncreas (com aumento de amilase e lipase), sistema hepatobiliar (com aumento de enzimas hepáticas), sistema reprodutor masculino, sistema cardiovascular, rins e trato gastrointestinal e, nervos periféricos e sistema nervoso central (potencial para comprometimento funcional cognitivo) (aproximadamente 4,6 a 21 vezes a exposição clínica humana a 100 mg com base na AUC para todas as toxicidades). Foram também observadas alterações na pressão arterial e frequência cardíaca, e prolongamento do intervalo QRS e PR em animais após administração aguda (aproximadamente 2,6 vezes a exposição clínica humana a 100 mg após uma dose única com base na Cmax). Todas as descobertas dos órgãos-alvo, com exceção da hiperplasia do duto biliar hepático (aproximadamente 7,1 a 21 vezes a exposição clínica humana a 100 mg, com base na AUC), foram parcialmente para totalmente reversíveis.
• Genotoxicidade
O lorlatinibe não foi mutagênico em um ensaio de mutação reversa bacteriana (Ames). O lorlatinibe induziu micronúcleos via mecanismo aneugênico em células TK6 linfoblastoides humanas in vitro e na medula óssea de ratos. A exposição de animais ao nível de efeito não observado para aneugenicidade foi de aproximadamente 16,5 vezes a exposição clínica humana a 100 mg com base na AUC.
• Carcinogenicidade
Não foram conduzidos estudos de carcinogenicidade com lorlatinibe.
• Toxicidade reprodutiva
Foram observados efeitos nos órgãos reprodutores masculinos (testículos, epidídimos e próstata) em animais (aproximadamente 3,9 a 1,6 vezes a exposição clínica humana a 100 mg com base na AUC). Os efeitos nos órgãos reprodutores masculinos foram total ou parcialmente reversíveis.
Em estudos de toxicidade embrionária e fetal, aumento da mortalidade embrionária e pesos corporais fetais baixos foram observados. Anormalidades morfológicas fetais incluíram membros rotacionados, dígitos supranumerários, gastrosquise, rins mal formados, cabeça em cúpula, palato aumentado arqueado e dilatação dos ventrículos do cérebro. As doses mais baixas com efeitos embrionários e fetais em animais correlacionaram-se com 0,6 a 1,1 vezes a exposição clínica humana a 100 mg, com base na AUC.
4. CONTRAINDICAÇÕES
O uso concomitante de indutores potentes do CYP3A com o lorlatinibe é contraindicado devido à potencialidade de hepatotoxicidade grave (elevações da aspartato aminotransferase [AST] e da alanina aminotransferase [ALT]) (vide item 6. Interações Medicamentosas).
Lorbrena® é contraindicado em pacientes com hipersensibilidades ao lorlatinibe ou a qualquer componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hiperlipidemia
O uso de lorlatinibe tem sido associado ao aumento de colesterol sérico e triglicerídeos (vide item 9. Reações Adversas). O colesterol sérico e os triglicerídeos devem ser monitorados antes do início de lorlatinibe; 2, 4 e 8 semanas após o início de lorlatinibe; e periodicamente depois disso. É necessário iniciar ou aumentar a dose de agentes hipolipemiantes (vide item 8. Posologia e Modo de usar).
Efeitos do sistema nervoso central
Foram observados efeitos no sistema nervoso central (SNC) em pacientes que tomavam lorlatinibe, incluindo efeitos psicóticos, alterações na função cognitiva, no humor, na fala, e no estado mental (vide item 9. Reações Adversas). A modificação ou suspensão da dose pode ser necessária para os pacientes que desenvolvem efeitos no SNC (vide item 8. Posologia e Modo de usar).
Bloqueio atrioventricular
Prolongamento do intervalo PR e eventos de bloqueio atrioventricular (AV) foram relatados em pacientes que receberam Lorbrena®. Monitorar o eletrocardiograma (ECG) antes de iniciar o lorlatinibe e após, mensalmente, particularmente em pacientes com condições predisponentes para a ocorrência de eventos cardíacos clinicamente significativos. A modificação da dose pode ser necessária para pacientes que desenvolvem bloqueio AV (vide item 8. Posologia e Modo de usar).
Pneumonite
Reações adversas graves ou fatais consistentes com pneumonite ocorreram com o lorlatinibe (vide item 9. Reações Adversas). Qualquer paciente que apresentar um agravamento dos sintomas respiratórios indicativos de pneumonite (por exemplo, dispneia, tosse e febre) deve ser prontamente avaliado para pneumonite. O lorlatinibe deve ser suspenso e/ou descontinuado permanentemente com base na gravidade (vide item 8. Posologia e Modo de usar).
Interações medicamentosas:
Em um estudo realizado em voluntários saudáveis, a utilização concomitante de lorlatinibe e rifampicina, um indutor potente de CYP3A, estava associada a aumentos de ALT e AST sem aumento da bilirrubina total e da fosfatase alcalina. O uso concomitante de qualquer indutor potente de CYP3A é contraindicado (vide item 4. Contraindicações). Quaisquer indutores potentes de CYP3A têm de ser descontinuados durante pelo menos 3 meias-vidas plasmáticas do indutor potente de CYP3A antes de iniciar o tratamento com lorlatinibe.
Fertilidade, gravidez e lactação
Fertilidade e gravidez
Com base em dados de animais e mecanismo de ação, existe risco de dano fetal se exposta ao Lorbrena® (vide item 3. Características Farmacológicas - Propriedades Farmacodinâmicas e Dados de Segurança Pré-clínicos). Mulheres em idade reprodutiva devem ser orientadas a evitar a gravidez durante o uso de Lorbrena®. É necessário um método anticoncepcional não hormonal altamente eficaz para pacientes do sexo feminino durante o tratamento com Lorbrena®, porque o lorlatinibe pode tornar os contraceptivos hormonais ineficazes (vide item 6. Interações Medicamentosas). Se um método hormonal de contracepção for inevitável, deve ser usado preservativo em combinação com o método hormonal. A contracepção eficaz deve ser continuada por pelo menos 21 dias após a conclusão da terapia.
Durante o tratamento com Lorbrena® e durante pelo menos 97 dias após a dose final, os pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo devem usar métodos contraceptivos eficazes, incluindo preservativo, e os pacientes do sexo masculino com parceiras grávidas devem usar preservativos. A fertilidade masculina pode ser comprometida durante o tratamento com lorlatinibe (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos). Os homens devem procurar aconselhamento sobre a preservação efetiva da fertilidade antes do tratamento.
Mulheres em idade fértil/Contracepção em homens e mulheres
Mulheres em idade reprodutiva devem ser orientadas a evitar a gravidez durante o uso de Lorbrena®. É necessário um método anticoncepcional não hormonal altamente eficaz para pacientes do sexo feminino durante o tratamento com Lorbrena®, porque o lorlatinibe pode tornar os contraceptivos hormonais ineficazes (vide item 6. Interações Medicamentosas). Se um método hormonal de contracepção for inevitável, um preservativo deve ser usado em combinação com o método hormonal. A contracepção eficaz deve ser continuada por pelo menos 21 dias após a conclusão da terapia.
Durante o tratamento com Lorbrena® e por pelo menos 97 dias após a dose final, aconselhar pacientes do sexo masculino com parceiras do sexo feminino de potencial reprodutivo a usar contracepção eficaz, incluindo preservativo, e aconselhar pacientes do sexo masculino com parceiras grávidas a usarem preservativos.
Gravidez
Estudos em animais mostraram toxicidade embrionária (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos). Não existem dados de mulheres grávidas que utilizam Lorbrena®. Lorbrena® pode causar dano fetal quando administrado a uma mulher grávida.
Lorbrena® não é recomendado durante a gravidez ou para mulheres em idade fértil que não estejam usando métodos contraceptivos.
Lorbrena® é um medicamento classificado na categoria C de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Amamentação
Não se sabe se o lorlatinibe e os seus metabólitos são excretados no leite humano. Não se pode excluir o risco a recém-nascidos.
Lorbrena® não deve ser usado durante a amamentação. A amamentação deve ser descontinuada durante o tratamento com Lorbrena® e durante 7 dias após a última dose.
Fertilidade
Com base nos resultados de segurança não clínicos, a fertilidade masculina pode ficar comprometida durante o tratamento com Lorbrena® (vide item 3. Características Farmacológicas - Dados de Segurança Pré-clínicos). Não se sabe se Lorbrena® afeta a fertilidade feminina. Os homens devem procurar aconselhamento sobre a preservação efetiva da fertilidade antes do tratamento.
Efeitos na capacidade de dirigir e operar máquinas
O lorlatinibe tem influência moderada na capacidade de dirigir e operar máquinas. Recomenda-se precaução ao conduzir ou operar máquinas, uma vez que os pacientes podem apresentar efeitos no SNC (vide item 9. Reações Adversas).
6. INTERAÇÕES MEDICAMENTOSAS
Os dados in vitro indicam que o lorlatinibe é principalmente metabolizado pelo CYP3A4 e pela uridina difosfato glucuronosiltransferase (UGT) 1A4, com contribuições menores do CYP2C8, CYP2C19, CYP3A5 e UGT1A3 CYP2C8, CYP2C19, CYP3A5 e UGT1A3.
Inibidores de CYP3A
O itraconazol, um inibidor potente de CYP3A, administrado em dose de 200 mg uma vez ao dia durante 5 dias, aumentou a área média sob a curva (AUC) em 42% e Cmax em 24% de dose oral única de 100 mg de lorlatinibe em voluntários saudáveis. Administração concomitante de lorlatinibe com inibidores potentes de CYP3A (ex. boceprevir, cobicistate, conivaptan, itraconazol, cetoconazol, posaconazol, telaprevir, troleandomicina, voriconazol, ritonavir, paritaprevir em combinação com ritonavir e ombitasvir e/ou dasabuvir e ritonavir em combinação com danoprevir, elvitegravir, indinavir, lopinavir, saquinavir ou tipranavir) pode aumentar as concentrações plasmáticas de lorlatinibe. Os produtos à base de toranja também podem aumentar as concentrações plasmáticas de lorlatinibe. Deve-se considerar um medicamento concomitante alternativo com menor potencial para inibir o CYP3A. Se um inibidor potente de CYP3A tiver de ser administrado concomitantemente, recomenda-se uma redução da dose de lorlatinibe (vide item 8. Posologia e Modo de usar).
Indutores de CYP3A
A rifampicina, um indutor potente de CYP3A, administrado em uma dose de 600 mg uma vez por dia durante 9 dias reduziu a AUC média de lorlatinibe em 85% e Cmax em 76% de uma dose única de 100 mg de lorlatinibe em voluntários saudáveis; elevações nos testes de função hepática (AST e ALT) também foram observados. A administração concomitante de lorlatinibe com indutores potentes de CYP3A (por exemplo, rifampicina, carbamazepina, enzalutamida, mitotano, fenitoína e hipericão) pode diminuir as concentrações plasmáticas de lorlatinibe. O uso de um indutor potente de CYP3A com lorlatinibe é contraindicado (vide item 4. Contraindicações). Quaisquer indutores potentes de CYP3A têm de ser descontinuados durante pelo menos 3 meias-vidas plasmáticas do indutor potente de CYP3A antes de iniciar o tratamento com lorlatinibe. A utilização concomitante com indutores moderados de CYP3A deve ser evitada, se possível, uma vez que também pode reduzir as concentrações plasmáticas do lorlatinibe.
Inibidores da bomba de prótons, antagonistas dos receptores H2 ou antiácidos de ação local
O rabeprazol, inibidor da bomba de prótons, teve um efeito mínimo sobre a exposição plasmática ao lorlatinibe (intervalo de confiança [IC] de 90% para a razão AUCinf, expressa em porcentagem: 97,6%, 104,3%). Nenhum ajuste de dose é necessário quando lorlatinibe é administrado com inibidores da bomba de prótons, antagonistas dos receptores H2 ou antiácidos de ação local.
Medicamentos nos quais as concentrações plasmáticas podem ser alteradas por lorlatinibe
• Substratos de CYP3A
O lorlatinibe tem um efeito líquido de indução no CYP3A tanto in vitro quanto in vivo. O lorlatinibe 150 mg por via oral uma vez ao dia por 15 dias diminuiu a AUCinf em 64% e Cmax em 50% com uma dose oral única de 2 mg de midazolam (um substrato sensível de CYP3A). Assim, a administração concomitante de lorlatinibe com substratos de CYP3A com índices terapêuticos estreitos, incluindo, mas não limitados a contraceptivos hormonais, alfentanil, ciclosporina, diidroergotamina, ergotamina, fentanil, pimozida, quinidina, sirolimus e tacrolimus, deve ser evitada já que a concentração desses medicamentos pode ser reduzida pelo lorlatinibe.
• Estudos in vitro de outras inibições e induções de CYP
Estudos in vitro indicaram que não é provável que ocorram interações medicamentosas clínicas resultantes da inibição do metabolismo de substratos para CYP1A2, CYP2B6, CYP2C8, CYP2C19 e CYP2D6 mediadas pelo lorlatinibe.
Estudos in vitro indicaram que o lorlatinibe é um inibidor do CYP2C9 e que ativa o receptor pregnano X (PXR) humano, com o efeito líquido in vivo sendo indução fraca do CYP2C9. Estudos in vitro também indicaram que o lorlatinibe é um inibidor dependente do tempo, bem como um indutor de CYP3A, com o efeito líquido in vivo sendo induzido. Estudos in vitro também indicaram que o lorlatinibe é um indutor de CYP2B6 e ativa o receptor androstano constitutivo (CAR) humano e in vivo o lorlatinibe é um indutor fraco do CYP2B6. In vitro, o lorlatinibe tem baixo potencial para causar interações medicamentosas por indução de CYP1A2.
In vitro, o principal metabólito circulante do lorlatinibe mostrou um baixo potencial para causar interações medicamentosas inibindo CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A, ou induzindo CYP1A2, CYP2B6 e CYP3A.
• Estudos in vitro da inibição de UDP-glucuronisiltransferase (UGT)
Estudos in vitro indicaram que não é provável que ocorram interações medicamentosas clínicas resultantes da inibição do metabolismo de substratos para UGT1A4, UGT1A6, UGT1A9, UGT2B7 e UGT2B15 mediadas pelo lorlatinibe. Estudos in vitro indicaram que o lorlatinibe é um inibidor da UGT1A1 e que ativa a PXR, com o efeito líquido in vivo sendo fraca indução da UGT.
Estudos in vitro indicaram que não é provável que ocorram interações medicamentosas clínicas resultantes de inibição de substratos para UGT1A1, UGT1A1, UGT1A4, UGT1A6, UGT1A9 e UGT2B15 pelo principal metabólito circulante de lorlatinibe.
• Estudos in vitro com transportadores de medicamentos
Estudos in vitro indicaram que interações medicamentosas clínicas como resultado da inibição, mediada por lorlatinibe, da proteína de resistência ao câncer de mama (BCRP, sistemicamente), da proteína de extrusão de vários medicamentos e toxinas (MATE)2K, do transportador de ânions orgânicos (OAT)1 e do transportador de cátions orgânicos (OCT)2 são improváveis. Estudos in vitro indicaram que o lorlatinibe é um inibidor da glicoproteína P (P-gp) e que ativa a PXR, com o efeito líquido in vivo sendo indução moderada. O lorlatinibe pode ter o potencial de inibir BCRP (trato GI), OATP1B1, OATP1B3, OCT1, MATE1 e OAT3 em concentrações clinicamente relevantes.
Estudos in vitro indicaram que não é provável que ocorram interações medicamentosas clínicas resultantes de inibição de substratos para Pgp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1 e MATE2K pelo principal metabólito circulante de lorlatinibe.
• Estudos in vivo com transportadores de medicamentos
Um estudo de interação medicamentosa realizado em pacientes com CPNPC indicou que o lorlatinibe é um indutor moderado de P-gp. Os substratos da P-gp com índice terapêutico estreito (por exemplo, digoxina) deve ser usado com cautela em combinação com o lorlatinibe devido à probabilidade de concentrações plasmáticas reduzidas desses substratos.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Lorbrena® deve ser conservado em temperatura ambiente (entre 15 °C e 30 °C) e pode ser utilizado por 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas:
Lorbrena® 25 mg
Comprimido rosa claro, redondo, revestido por película de liberação imediata, gravado com a palavra "Pfizer" em uma das faces e "25 LLN" na outra face.
Lorbrena® 100 mg
Comprimido rosa escuro, oval, revestido por película de liberação imediata, gravado com a palavra "Pfizer" em uma das faces e "100 LLN" na outra face.
8. POSOLOGIA E MODO DE USAR
Dosagem recomendada
O esquema da dose recomendada de Lorbrena® é de 100 mg por via oral, uma vez ao dia, de forma contínua. O tratamento deve ser mantido enquanto o paciente estiver obtendo benefício clínico com a terapia.
Lorbrena® pode ser administrado com ou sem alimento (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
Os pacientes devem ser encorajados a tomar a sua dose de lorlatinibe aproximadamente à mesma hora todos os dias. Os comprimidos devem ser engolidos inteiros (não devem ser mastigados, esmagados ou partidos antes de serem engolidos). Nenhum comprimido deve ser ingerido se estiver quebrado, rachado, ou de outro modo não intacto.
Este medicamento não deve ser partido, aberto ou mastigado.
Se uma dose do lorlatinibe for esquecida, o paciente deve toma-la tão logo se lembrar, a menos que falte menos de 4 horas até a próxima dose, e nesse caso, o paciente não deve tomar a dose esquecida. Os pacientes não devem tomar 2 doses ao mesmo tempo para compensar a dose esquecida.
Modificações de dose
A interrupção do tratamento e/ou redução da dose pode ser necessária com base na segurança e tolerância individual. Os níveis de redução da dose estão resumidos abaixo.
• Primeira redução da dose: 75 mg de Lorbrena® por via oral uma vez ao dia.
• Segunda redução da dose: 50 mg de Lorbrena® por via oral uma vez ao dia.
O Lorbrena® deve ser descontinuado permanentemente se o paciente não conseguir tolerar 50 mg por via oral uma vez ao dia.
As recomendações de modificação da dose para toxicidades e para pacientes que desenvolvem bloqueio atrioventricular (AV) de primeiro grau, segundo grau ou completo são fornecidas na Tabela 3.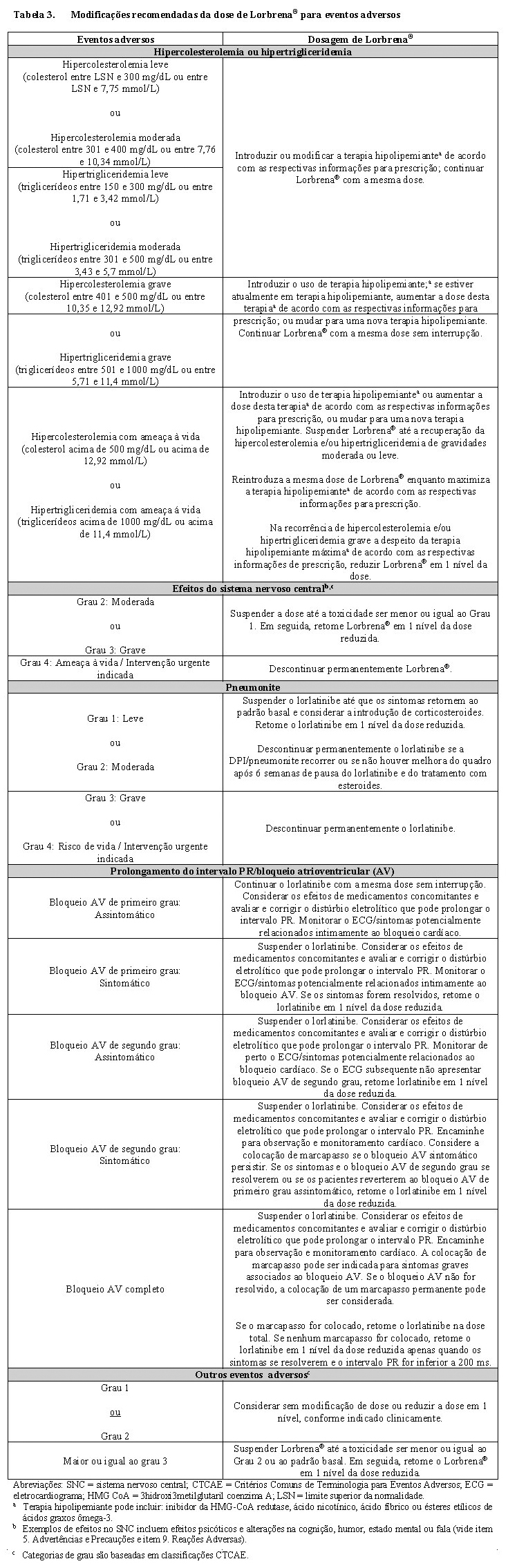
• Inibidores potentes do citocromo P-450 (CYP) 3A
O uso concomitante de Lorbrena® com inibidores potentes do CYP3A pode aumentar as concentrações plasmáticas de lorlatinibe. Deve-se considerar um medicamento concomitante alternativo com menor potencial para inibir o CYP3A (vide item 6. Interações Medicamentosas e item 3. Características Farmacológicas - Propriedades Farmacocinéticas). Se um inibidor potente do CYP3A tiver que ser administrado concomitantemente, a dose inicial diária de 100 mg de Lorbrena® deve ser reduzida para uma dose diária de 75 mg. Se o uso concomitante de um inibidor potente do CYP3A for descontinuado, o Lorbrena® deve ser retomado com a dose utilizada antes do início do inibidor potente do CYP3A e após um período de washout de 3 a 5 meias-vidas do inibidor potente do CYP3A.
• Insuficiência hepática
Nenhum ajuste de dose é recomendado em pacientes com insuficiência hepática leve. Informações limitadas sobre o perfil de segurança em pacientes com insuficiência hepática moderada ou grave estão disponíveis para lorlatinibe. Portanto, o Lorbrena® não é recomendado em pacientes com insuficiência hepática moderada a grave (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
• Disfunção renal
Nenhum ajuste de dose é recomendado em pacientes com insuficiência renal leve ou moderada (clearance de creatinina [CLcr]: ≥30 mL/min) com base em uma análise populacional farmacocinética. Informações para uso de lorlatinibe em pacientes com insuficiência renal grave (CLcr: < 30 mL/min) são limitadas (n=1). Portanto, o Lorbrena® não é recomendado em pacientes com insuficiência renal grave (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
• Idosos (≥65 anos)
Os dados limitados sobre a segurança e a eficácia do lorlatinibe em pacientes com 65 anos ou mais não sugerem que um ajuste da dose seja necessário em pacientes idosos (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
• Pacientes pediátricos
A segurança e a eficácia do lorlatinibe em pacientes pediátricos não foram estabelecidas.
9. REAÇÕES ADVERSAS
Os dados descritos abaixo refletem a exposição ao Lorbrena® em 295 pacientes adultos com CPNPC metastático positivo para ALK ou positivo para c-ros oncogene 1 (ROS1) que receberam Lorbrena® 100 mg por via oral uma vez ao dia no Estudo B7461001. A tabela 4 apresenta as RAMs de lorlatinib listadas em ordem descendente de gravidade médica ou importância clínica dentro de cada categoria de frequência do CIOMS e da SOC.
A duração mediana do tratamento foi de 16,3 meses (faixa de variação: 1 dia a 39,7 meses), a idade média era de 53 anos (faixa de variação: 19 a 85 anos), e 18% dos pacientes tinham mais de 65 anos. Um total de 58% dos pacientes eram do sexo feminino, 49% dos pacientes eram brancos e 37% dos pacientes eram asiáticos.
Os eventos adversos mais frequentemente notificados foram hipercolesterolemia (84,4%), hipertrigliceridemia (67,1%), edema (54,6%), neuropatia periférica (47,8%), efeitos cognitivos (28,8%), fadiga (28,1%), aumento de peso (26,4%), artralgia (24,7%), efeitos de humor (22,7%) e diarreia (22,7%).
Reduções de dose devido a eventos adversos ocorreram em 23,4% dos pacientes que receberam lorlatinibe. Os eventos adversos mais comuns que levaram a reduções de dose foram edema e neuropatia periférica. Descontinuação permanente do tratamento associada a reações adversas ocorreu em 3,1% dos pacientes que receberam lorlatinibe. Os eventos adversos mais frequentes que levaram à descontinuação permanente foram efeitos cognitivos e efeitos psicóticos.
Reações adversas graves foram relatadas em 7,8% dos pacientes que receberam lorlatinibe. Os eventos adversos graves mais frequentes relatados foram efeitos cognitivos e pneumonite.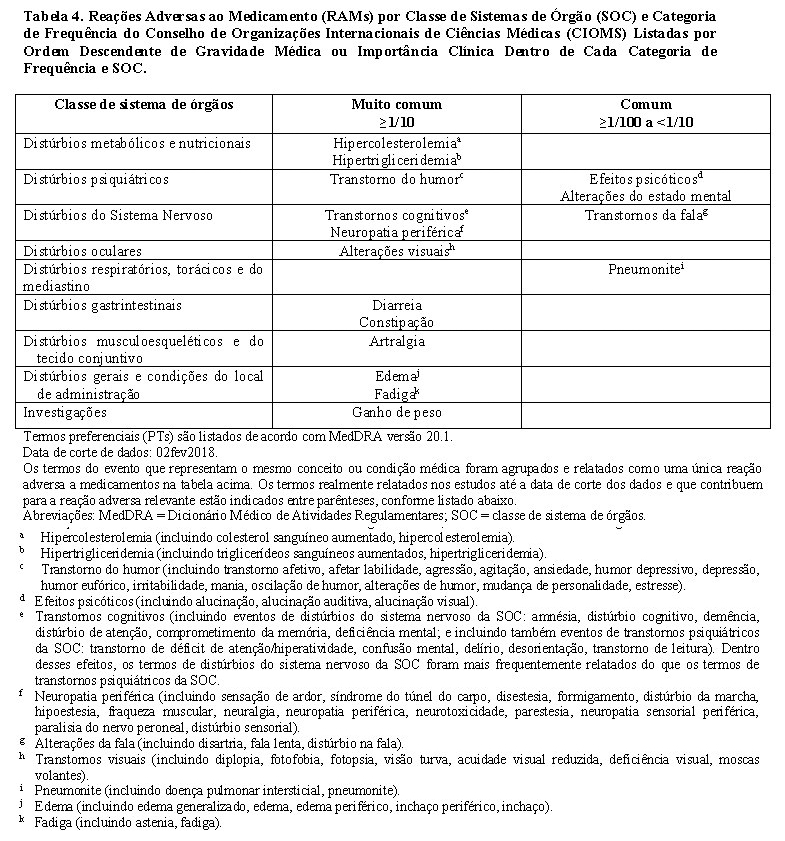
Descrição das reações adversas selecionadas
• Hipercolesterolemia/hipertrigliceridemia
No Estudo B7461001, reações adversas de aumento do colesterol sérico ou triglicerídeos foram relatadas em 84,4% e 67,1% dos pacientes, respectivamente. Reações adversas leves ou moderadas de hipercolesterolemia ou hipertrigliceridemia ocorreram em 67,8% e 50,5% dos pacientes, respectivamente. Nenhum paciente teve o tratamento com lorlatinibe descontinuado devido a hipercolesterolemia ou hipertrigliceridemia (vide item 8. Posologia e Modo de usar e item 5. Advertências e Precauções). O tempo médio para início de hipercolesterolemia e hipertrigliceridemia foi de 15 dias. A duração mediana de hipercolesterolemia e hipertrigliceridemia foi de 381 e 405 dias, respectivamente.
• Efeitos do Sistema Nervoso Central
No Estudo B7461001, as reações adversas no SNC foram principalmente distúrbios cognitivos (28,8%), alterações no humor (22,7%), na fala (9,8%), e efeitos psicóticos (7,8%) foram geralmente leves, transitórias e reversíveis após o atraso e/ou redução da dose (vide item 8. Posologia e Modo de usar e item 5. Advertências e Precauções). O distúrbio cognitivo mais comum de qualquer grau foi o comprometimento da memória (11,5%) e as reações de grau 3 ou 4 mais comuns foram transtorno cognitivo e confusão mental (0,7% cada). A alteração do humor mais comum de qualquer grau foi a irritabilidade (6,1%), que também foi a reação de grau 3 ou 4 mais comum (1,0%). A alteração na fala mais comum de qualquer grau foi disartria (4,1%), e a reação de grau 3 ou 4 mais comum foi fala lenta (0,3%). O efeito psicótico mais comum de qualquer grau foi alucinação (3,7%) e as reações mais comuns de Grau 3 ou 4 foram alucinações, alucinação auditiva e alucinação visual (0,3% cada). O tempo mediano para o início das alterações cognitivas, no humor, na fala, e psicóticas foi de 92, 44, 42 e 23 dias, respectivamente. A duração média das alterações cognitivas no humor, na fala e psicóticas foi de 224, 83, 106 e 74 dias, respectivamente.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificação de Eventos Adversos a Medicamentos - VigiMed, disponível em http://portal.anvisa.gov.br/vigimed, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
O tratamento de superdosagem com o medicamento consiste em medidas de suporte em geral. Dado o efeito dependente da dose no intervalo PR, recomenda-se a monitorização eletrocardiográfica. Não há antídoto para o lorlatinibe.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.2110.0476
VENDA SOB PRESCRIÇÃO MÉDICA