LOKELMA
ASTRAZENECA
ciclossilicato de zircônio sódico
Tratamento da hipercalemia em adultos.
Apresentações.
Sachês com 5 g de pó para suspensão oral em embalagens contendo 30 ou 3 sachês.
VIA ORAL
USO ADULTO
Composição.
LOKELMA® 5 g
Cada sachê contém 5 g de ciclossilicato de zircônio sódico hidratado (contém aproximadamente 400 mg de sódio). Não há excipientes.
Informações técnicas.
1. INDICAÇÕES
LOKELMA® é indicado para o tratamento da hipercalemia em pacientes adultos.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Os efeitos redutores de potássio de LOKELMA® foram demonstrados em três ensaios randomizados, duplo-cegos, controlados com placebo em pacientes com hipercalemia. Os três estudos avaliaram o efeito inicial de LOKELMA® para corrigir a hipercalemia durante um período de 48 horas e dois dos estudos também avaliaram a manutenção do efeito da normocalemia obtida. Os estudos de manutenção incluíram pacientes com doença renal crônica (58%), insuficiência cardíaca (10%), diabetes mellitus (62%) e em terapia com inibidores do sistema renina-angiotensina-aldosterona - SRAA (68%). O total de 1760 pacientes receberam doses de LOKELMA®, sendo 507 pacientes expostos por pelo menos 360 dias. Além disso, a eficácia e segurança de LOKELMA® foi estudada em um estudo duplo-cego controlado por placebo de 196 pacientes em hemodiálise crônica com hipercalemia, que receberam doses de LOKELMA® por 8 semanas. Nos estudos, LOKELMA® reduziu o potássio sérico e manteve a normocalemia independentemente da causa subjacente da hipercalemia, idade, sexo, raça, comorbidades ou utilização concomitante de inibidores do SRAA. Não foram impostas restrições na dieta; os pacientes foram instruídos a continuar a sua dieta normal sem quaisquer alterações específicas.
Um estudo com duas etapas randomizado, duplo-cego, controlado por placebo
Neste estudo, 753 pacientes (idade média de 66 anos, intervalo de 22 a 93 anos) com hipercalemia (5,0 - ≤6,5 mmol/L, valor basal médio de potássio de 5,3 mmol/L) foram randomizados para receber LOKELMA® 1,25 g, 2,5 g, 5 g ou 10 g ou placebo três vezes ao dia nas 48 horas iniciais.
LOKELMA® mostrou reduções dose-dependentes nos níveis de potássio sérico nas concentrações de 2,5 g, 5 g e 10 g dentro de horas da administração da primeira dose (Tabela 1). Reduções estatisticamente significantes nos níveis de potássio foram observadas 1 hora após a primeira dose de 10 g de LOKELMA®. A redução média no potássio sérico foi de -0.7 mmol/L e 86% dos pacientes apresentaram valores de potássio normais dentro de 48 horas com uma dose de 10 g. Os pacientes com níveis basais de potássio mais elevados apresentaram uma maior resposta a LOKELMA®. Os pacientes com níveis de potássio pré-tratamento superiores a 5,5 mmol/L (média do valor basal de 5,8 mmol/L) obtiveram um decréscimo médio de 1,1 mmol/l em 48 horas, enquanto os pacientes com níveis de potássio basais inferiores ou iguais a 5,3 mmol/L obtiveram um decréscimo médio de 0,6 mmol/l com a dose mais elevada. A redução dos níveis de potássio foi semelhante entre os pacientes com doença renal crônica, insuficiência cardíaca, diabetes mellitus e entre aqueles em terapia com inibidores do SRAA (bloqueadores dos receptores da angiotensina, inibidores da enzima conversora da angiotensina, antagonistas da aldosterona).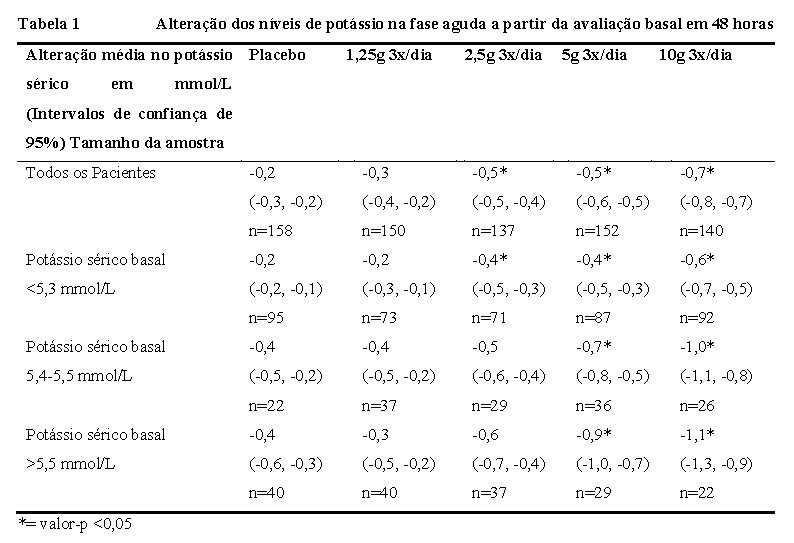
Os pacientes que atingiram a normocalemia (níveis de potássio entre 3,5 e 5,0 mmol/L) foram novamente randomizados para a mesma concentração da droga ativa ou para placebo administrados uma vez por dia durante 12 dias. Esta etapa do estudo atendeu aos desfechos de eficácia pré-definidos com as doses de 2,5 g, 5 g e 10 g comparado aos seus respectivos grupos de placebo. A eficácia foi consistente entre os subgrupos pré-especificados com insuficiência cardíaca, doença renal crônica e diabetes mellitus ou em pacientes sob terapia com inibidores do SRAA. Ao final do período de tratamento, quando LOKELMA® deixou de ser administrado, os níveis de potássio aumentaram para valores próximos aos basais.
Um estudo de manutenção com extensão, com múltiplas etapas, controlado com placebo
Na etapa de correção do estudo, 258 pacientes com hipercalemia (média do valor basal de 5,6; intervalo de 4,1-7,2 mmol/L) receberam 10 g de LOKELMA® administrado três vezes ao dia durante 48 horas. Foram observadas reduções no potássio 1 hora após a administração da primeira dose de 10 g de LOKELMA®. O tempo médio para atingir a normocalemia foi de 2,2 horas, com 84% dos pacientes atingindo a normocalemia dentro de 24 horas e 98% dentro de 48 horas. As respostas foram maiores em pacientes com hipercalemia mais grave; o potássio sérico diminuiu 0,8, 1,2 e 1,5 mmol/L em pacientes com valores basais de potássio sérico < 5,5, 5,5-5,9 e ≥ 6,0 mmol/L, respectivamente.
Os pacientes que atingiram a normocalemia (níveis de potássio entre 3,5 e 5,0 mmol/L) foram randomizados, de forma duplo-cega, para uma das três doses de LOKELMA® (5 g (n=45), 10 g (n=51) ou 15 g (n=56)) ou placebo (n=85) administrado uma vez ao dia durante 28 dias (etapa de descontinuação da randomização duplo-cega).
A proporção de indivíduos com média de potássio sérico < 5,1 mmol/L do 8° ao 29° Dia do Estudo foi maior com doses de 5 g, 10 g e 15 g de LOKELMA® uma vez ao dia (80%, 90% e 94%, respectivamente), comparado com o placebo (46%). Houve uma redução média no potássio sérico de 0,77 mmol/L, 1,10 mmol/L, 1,19 mmol/L e 0,44 mmol/L nos grupos tratados com as doses de 5 g, 10 g, 15 g de LOKELMA® uma vez por dia e placebo, respectivamente. Resultados da etapa de manutenção estendida (aberta): 123 pacientes participaram da etapa aberta de 11 meses. Os níveis médios de potássio sérico foram de 4,66 mmol/L ao longo do período de extensão. O tratamento foi descontinuado no fim do estudo (Dia 365). A Figura 1 apresenta os níveis médios de potássio sérico durante as etapas de correção e manutenção do estudo.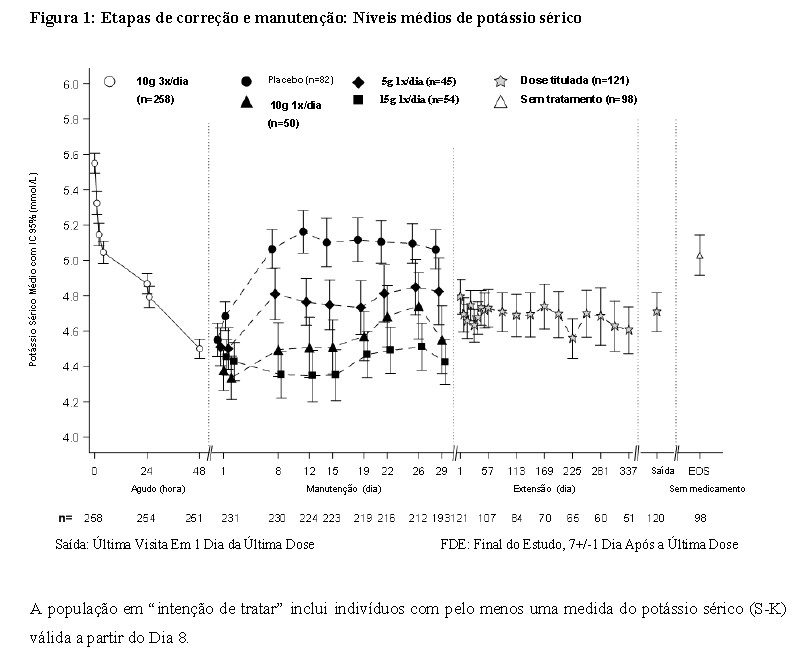
Um estudo de eficácia e segurança, em duas etapas, multicêntrico, aberto, com doses múltiplas
Os efeitos a longo prazo (até 12 meses) de LOKELMA® foram avaliados neste estudo em 751 indivíduos com hipercalemia (média basal de 5,59 mmol/L; intervalo de 4,3 - 7,6 mmol/L). As condições de comorbidade incluíram doença renal crônica (65%), diabetes mellitus (64%), insuficiência cardíaca (15%) e hipertensão (83%). O uso de diuréticos e inibidores do SRAA foi reportado por 51% e 70% dos indivíduos, respectivamente. Durante a etapa de correção, LOKELMA® foi administrado em uma dose de 10 g 3 vezes ao dia por um período de no mínimo 24 horas e até 72 horas. Os indivíduos que atingiram normocalemia (3,5-5,0 mmol/L, inclusive) dentro de 72 horas (n=746; 99%) foram incluídos na etapa de manutenção do estudo. Todos os indivíduos na etapa de manutenção receberam LOKELMA® em uma dose inicial diária de 5 g, que pode ser aumentada em incrementos de 5 g ao dia (até o máximo de 15 g uma vez ao dia) ou diminuída (até o mínimo de 5 g em dias alternados) com base no regime de titulação.
A redução média do potássio sérico foi de 0,81 mmol/L, 1,02 mmol/L e 1,10 mmol/L em 24 (n=748), 48 (n=104) e 72 (n=28) horas, respectivamente. O total de 126 pacientes apresentavam níveis basais de potássio sérico ≥ 6.0 mmol/L (potássio basal médio de 6,28 mmol/L) e estes pacientes apresentaram uma redução média de 1.37 mmol/L ao final da etapa aguda.
A proporção de indivíduos com média de potássio sérico ≤ 5,1 mmol/L entre os dias 85-365 da etapa de manutenção foi de 88% (IC 95% 0,857, 0,908) e ≤ 5,5 mmol/L entre os dias 85-365 da etapa de manutenção foi de 99% (IC 95% 0,976, 0,995). A normocalemia foi mantida enquanto os pacientes permaneceram em tratamento com o medicamento e a média do potássio sérico aumentou após a descontinuação. Entre aqueles pacientes em terapia com inibidores do SRAA na avaliação basal, 89% não descontinuaram a terapia com inibidores do SRAA, 74% puderam manter a mesma dose durante a etapa de manutenção, e entre aqueles que não estavam em terapia com inibidores do SRAA na avaliação basal, 14% puderam iniciar esta terapia.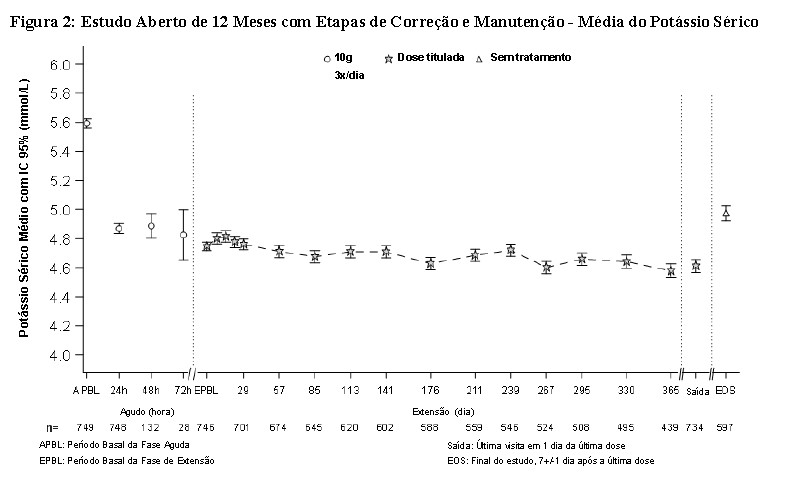
A população em "intenção de tratar" inclui indivíduos com pelo menos uma medida do potássio sérico (S-K) válida a partir do Dia 8.
Um estudo em pacientes com doença renal crônica com hipercalemia
Este estudo foi um estudo de escalonamento de dose, duplo-cego, controlado com placebo em 90 pacientes (60 pacientes com LOKELMA®; 30 controles) com TFGe entre 30-60 ml/min/1,73 m2 e hipercalemia (valor basal de potássio sérico de 5,2 mmol/l, intervalo de 4,6-6,0 mmol/L). Os pacientes foram randomizados para receber doses crescentes de LOKELMA® (0,3 g, 3 g e 10 g) ou placebo, administrado três vezes ao dia com alimentos durante dois a quatro dias. O desfecho primário foi a taxa de alteração no potássio sérico a partir da avaliação basal até os 2 primeiros dias de tratamento. O estudo atingiu o desfecho primário de eficácia com as doses de 3 g e 10 g de LOKELMA® comparado com o placebo. LOKELMA® na dose de 10 g e na dose de 3 g resultou em reduções máximas médias de 0,92 mmol/l e 0,43 mmol/l, respectivamente. As coletas de urina de 24 horas mostraram que LOKELMA® diminuiu a excreção urinária de potássio a partir da avaliação basal em 15,8 mmol/24 h em comparação com o aumento do placebo de 8,9 mmol/24 horas (p < 0,001). A excreção de sódio manteve-se inalterada relativamente ao placebo (10 g 3x/dia, aumento de 25,4 mmol/24 horas comparado com o placebo que aumentou em 36,9 mmol/24 horas (não significativo)).
Um estudo randomizado, duplo-cego, controlado por placebo em pacientes em hemodiálise crônica
Neste estudo, 196 pacientes (idade média de 58 anos, faixa de 20 a 86 anos) com doença renal estágio terminal em diálise estável por pelo menos 3 meses e hipercalemia pré-diálise persistente foram randomizados para receber LOKELMA® 5 g ou placebo uma vez ao dia em dias em que não realizam diálise. Na randomização, os níveis séricos médios de potássio foram 5,8 mmol/L (variação de 4,2 - 7,3 mmol/L) no grupo de LOKELMA® e 5,9 mmol/L (variação de 4,2 - 7,3 mmol/L) no grupo do placebo. Para alcançar o nível sérico de potássio pré-diálise entre 4,0 - 5,0 mmol/L durante o período de ajuste da dose (4 semanas iniciais), a dose poderia ser ajustada semanalmente em incrementos de 5 g até 15 g uma vez ao dia com base na dosagem de potássio pré-diálise aferida após o longo intervalo interdialítico. A dose atingida ao final do período de ajuste de dose foi mantida ao longo do período de avaliação subsequente de 4 semanas. A proporção de respondedores, definidos como aqueles que mantiveram um potássio sérico pré-diálise entre 4,0 e 5,0 mmol/L em pelo menos 3 de 4 tratamentos de diálise após o longo intervalo interdialítico e que não receberam terapia de resgate durante o período de avaliação, foi de 41% no grupo de LOKELMA® e 1% no grupo do placebo (p < 0,001) (veja a Figura 3).
Em análises post-hoc, o número de vezes que os pacientes apresentaram potássio sérico entre 4,0 e 5,0 mmol/L após o longo intervalo interdialítico e durante o período de avaliação, foi maior no grupo de LOKELMA®. 24% dos pacientes estavam dentro deste intervalo em todas as 4 visitas no grupo de LOKELMA® e nenhum no grupo do placebo. O número de pacientes que mantiveram o nível sérico de potássio entre 3,5 e 5,5 mmol/L após o longo intervalo interdialítico e durante o período de avaliação foi maior no grupo de LOKELMA®. Em todas as 4 visitas, o valor sérico de potássio estava dentro dessa faixa para 52% dos pacientes no grupo de LOKELMA® e para 5% dos pacientes no grupo do placebo, e por pelo menos 3 visitas o valor sérico de potássio estava dentro desse intervalo para 70% dos pacientes do grupo de LOKELMA® e 21% dos pacientes no grupo do placebo.
No final do tratamento, o nível sérico médio de potássio após a diálise foi de 3,6 mmol/L (intervalo 2,6 - 5,7 mmol/L) no grupo de LOKELMA® e de 3,9 mmol/L (intervalo 2,2 - 7,3 mmol/L) no grupo do placebo. Não houve diferença entre os grupos de LOKELMA® e de placebo no ganho de peso interdialítico, um marcador da retenção de sódio e líquido. O ganho de peso interdialítico foi definido como o peso pré-diálise menos o peso pós-diálise da sessão de diálise anterior e foi medido após o longo intervalo interdialítico.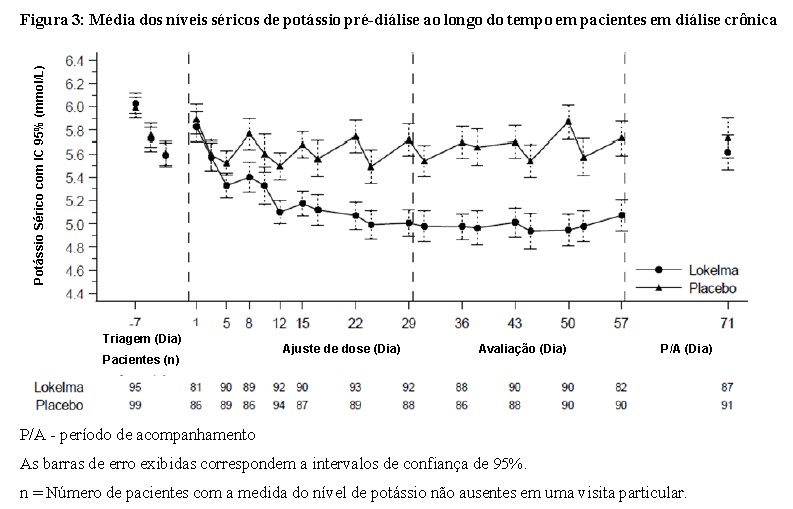
Referências Bibliográficas
Ash SR, Singh B, Lavin PT,. Strovos F, Rasmussen HS. A phase 2 study on the treatment of hyperkalemia in patients with chronic kidney disease suggests that the selective potassium trap, ZS-9, is safe and efficient. Kidney Int. 2015;88:404-411.
Packham DK, Rasmussen HS, Lavin PT, El-Shahawy MA, Roger SA, Block G, Quinibi WY, Pergola P, Singh B. Sodium Zirconium Cyclosilicate in Hyperkalemia. N Engl J Med. 2015;372:222-231.
Kosiborod M, Rasmussen HS, Lavin PT, Quinibi WY, Spinowitz B, Pakcham D, Roger S, yang A, Lerma E, Singh B. Effect of Sodium Zirconium Cyclosilicate on Potassium Lowering for 28 Days Among Outpatients With HyperkalemiaThe HARMONIZE Randomized Clinical Trial. JAMA. 2014;312:2223-2233.
Fishbane S, Pergola P, Packham DK, Roger S, Lerma EV, Butler J, Von Haehling S, Spinowitz BS, Block G, Adler SH, Singh B, Lavin P, McCullough P, Kosiborod M. Maintained efficacy and safety of sodium zirconium cyclosilicate for hyperkalemia: 12-month, open-label, phase 3 study. J Amer Soc Nephrol 2017 Oct;28(Supl):390. (Poster session)
Fishbane, Steven et al. A Phase 3b, Randomized, Double-Blind, Placebo-Controlled Study of Sodium Zirconium Cyclosilicate for Reducing the Incidence of Predialysis Hyperkalemia. Journal of the American Society of Nephrology, p. ASN. 2019050450, 2019.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
LOKELMA® é um pó inorgânico não polimérico, não absorvido, com uma estrutura microporosa uniforme que captura preferencialmente o potássio em troca de cátions de hidrogênio e sódio. LOKELMA® é altamente seletivo para os íons de potássio, mesmo na presença de outros cátions, tais como cálcio e magnésio, in vitro. LOKELMA® captura o potássio ao longo de todo o trato gastrointestinal (GI) e reduz a concentração de potássio livre no lúmen GI, diminuindo, desta forma, os níveis de potássio sérico e aumentando a excreção fecal de potássio para resolver a hipercalemia.
Propriedades Farmacodinâmicas
LOKELMA® reduz as concentrações de potássio sérico em apenas 1 hora após ingestão e as concentrações de potássio sérico continuam diminuindo durante o período de tratamento de 48 horas. O ciclossilicato de zircônio sódico hidratado não afeta as concentrações séricas de cálcio, magnésio e sódio. Em pacientes que não continuam o tratamento, os níveis de potássio aumentam. Há uma forte correlação entre os níveis iniciais de potássio sérico e a dimensão do efeito de LOKELMA®; os pacientes com níveis iniciais mais elevados de potássio sérico têm maior redução do potássio sérico.
Em um estudo realizado em indivíduos saudáveis aos quais foi dado LOKELMA® 5 g ou 10 g, uma vez ao dia durante quatro dias, uma redução dose-dependente na concentração de potássio sérico e na excreção urinária de potássio total foi acompanhada por aumentos médios na excreção fecal de potássio. Não foram observadas alterações estatisticamente significantes na excreção urinária de sódio.
Foi também demonstrado que LOKELMA® se liga ao amônio, in vitro e in vivo, removendo, deste modo, o amônio e aumentando os níveis séricos de bicarbonato. Os pacientes tratados com LOKELMA® apresentaram um aumento de bicarbonato de 1,1 mmol/L com 5 g uma vez por dia, de 2,3 mmol/L com 10 g uma vez por dia e de 2,6 mmol/L com 15 g uma vez por dia em comparação com um aumento médio de 0,6 mmol/L no grupo tratado com placebo. LOKELMA® demonstrou uma alteração nos níveis médios de aldosterona sérica (variação: -30% a -31%) em comparação com o grupo tratado com placebo (+14%). Não foram observados efeitos sobre a pressão arterial sistólica e diastólica.
Além disso, foram observadas reduções médias do nitrogênio ureico sanguíneo (NUS) nos grupos de 5 g (-1,1 mg/dl) e de 10 g (-2,0 mg/dl) três vezes por dia, em comparação com pequenos aumentos médios nos grupos tratados com placebo (0,8 mg/dl) e com dose baixa de LOKELMA® (0,3 mg/dl).
Propriedades Farmacocinéticas
LOKELMA® reduz as concentrações de potássio sérico em apenas 1 hora após ingestão e as concentrações de potássio sérico continuam diminuindo durante o período de tratamento de 48 horas.
Absorção
LOKELMA® é um composto inorgânico, insolúvel, que não é sujeito a metabolismo enzimático. Além disso, estudos clínicos demonstraram que não é absorvido sistemicamente. Um estudo de balanço de massa in vivo, em ratos, mostrou que o ciclossilicato de zircônio sódico hidratado foi recuperado nas fezes sem evidência de absorção sistêmica. Devido a estes fatores e à sua insolubilidade, não foram realizados estudos in vivo ou in vitro para avaliar o seu efeito nas enzimas do citocromo P450 (CYP450) ou na atividade de transporte.
Eliminação
LOKELMA® é eliminado pela via fecal.
Interações Medicamentosas
Uma vez que LOKELMA® não é absorvido ou metabolizado pelo organismo, não são esperados efeitos de outros medicamentos sobre a ação farmacológica de LOKELMA® (vide item "Interações Medicamentosas").
Dados de segurança pré-clínica
Os dados pré-clínicos não revelam riscos especiais para humanos, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico, toxicidade reprodutiva e desenvolvimento. Não foram conduzidos estudos de carcinogenicidade.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado para pacientes com hipersensibilidade a substância ativa.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipocalemia
Pode ser observada hipocalemia. A titulação de dose, como descrito na posologia de manutenção, pode ser necessária nestes casos para prevenir hipocalemia moderada ou grave. Em pacientes com níveis de potássio sérico < 3,0 mmol/L, LOKELMA® deve ser descontinuado e o paciente deve ser reavaliado. O potássio sérico deve ser monitorado quando for clinicamente indicado, por exemplo após alterações nos medicamentos que afetam a concentração de potássio sérico (como o uso de inibidores do sistema renina-angiotensina-aldosterona (SRAA) ou diuréticos), e a dose de LOKELMA® titulada, se necessário.
Interferência em imagem de Raio-X
LOKELMA® pode ser opaco aos raios X e, portanto, pode afetar a interpretação dos resultados radiográficos.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não foram realizados estudos dos efeitos sobre a capacidade de dirigir veículos e operar máquinas.
Uso durante a gravidez e lactação
Não foram conduzidos estudos clínicos em mulheres grávidas ou lactantes.
Estudos de reprodução conduzidos em doses equivalentes à humana de 115 g/dia em coelhos e 58 g/dia em ratos (presumindo-se uma massa corporal de 60 kg) não indicaram efeitos nocivos diretos relacionados à gravidez, desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal. Uma vez que estudos de reprodução animal nem sempre são preditivos de uma resposta humana, LOKELMA® deve ser utilizado durante a gravidez apenas se o benefício potencial para a mãe justificar quaisquer riscos para o feto.
Devido às suas propriedades físico-químicas, o ciclossilicato de zircônio sódico hidratado não é absorvido sistemicamente e não se espera a sua excreção no leite materno.
Categoria B
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
6. INTERAÇÕES MEDICAMENTOSAS
Interações Medicamentosas
Uma vez que LOKELMA® não é absorvido ou metabolizado pelo organismo, não são esperados efeitos de outros medicamentos sobre a ação farmacológica de LOKELMA®.
Efeito do LOKELMA® em outros medicamentos
Uma vez que LOKELMA® não é absorvido ou metabolizado pelo organismo e não se liga significativamente a outros medicamentos, os efeitos sobre outros medicamentos são limitados.
LOKELMA® pode aumentar transitoriamente o pH gástrico através da absorção dos íons de hidrogênio, o que pode levar a alterações na solubilidade e na cinética de absorção de medicamentos administrados concomitantemente com biodisponibilidade dependente do pH. Portanto, LOKELMA® deve ser administrado pelo menos 2 horas antes ou 2 horas depois de medicamentos orais com biodisponibilidade dependente do pH gástrico clinicamente significativa.
Exemplos de medicamentos que devem ser tomados 2 horas antes ou depois de LOKELMA® para evitar a interação medicamentosa do possível aumento do pH gástrico são listados abaixo: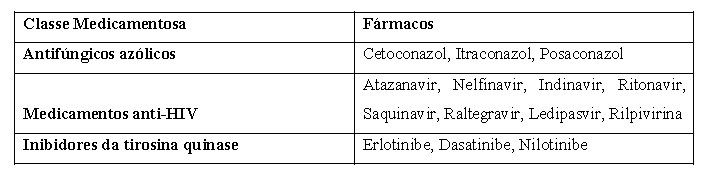
LOKELMA® pode ser administrado concomitantemente, sem espaçamento dos horários das doses, com medicamentos orais que não exibem biodisponibilidade dependente de pH.
Em um estudo clínico de interação medicamentosa conduzido em indivíduos saudáveis, a administração concomitante de LOKELMA® com anlodipino, dabigatrana, clopidogrel, atorvastatina, furosemida, glipizida, varfarina, losartana ou levotiroxina não resultou em interações medicamentosas clinicamente significativas. Não foram necessários ajustes de dose ou separação do tempo de administração para estes medicamentos.
Em um outro estudo clínico de interação medicamentosa em indivíduos saudáveis, a administração concomitante de LOKELMA® 15 g com tacrolimo 5 mg resultou em um decréscimo da AUC e Cmax em 37% e 29%, respectivamente. Portanto, tacrolimo deve ser tomado pelo menos 2 horas antes ou depois de LOKELMA®. No mesmo estudo, a administração concomitante de LOKELMA® e ciclosporina não mostrou interação clinicamente significativa.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (entre 15 e 30°C).
LOKELMA® tem validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, administrar imediatamente (vide item "Modo de usar").
Aspecto físico
LOKELMA® pó para suspensão oral é apresentado da seguinte maneira: pó cristalino, insolúvel, de cor branca a cinza.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Modo de usar
Os pacientes devem ser instruídos a esvaziar o conteúdo total do sachê, ou sachês, com base na dose prescrita, em um copo contendo aproximadamente 45 ml de água. Mexer bem e beber enquanto o pó ainda estiver suspenso, já que o produto não se dissolve. A suspensão é insípida e tem aspecto de um líquido turvo. Se o pó assentar, a água deve ser mexida novamente. Se necessário, o paciente pode enxaguar o copo com um pouco de água e beber até tomar todo o medicamento.
Deve-se assegurar que todo o produto seja ingerido.
LOKELMA® pode ser ingerido com ou sem alimentos.
Posologia
Fase de correção do tratamento de hipercalemia
Em pacientes cujos níveis de potássio sérico são > 5,0 (mmol/L), a dose inicial recomendada de LOKELMA® para atingir a normocalemia (níveis de potássio normais entre 3,5 e 5,0 mmol/L) é 10 g, administrada três vezes ao dia, sob a forma de suspensão oral em água. Em geral, a normocalemia é obtida dentro de 24 a 48 horas. Se o potássio sérico medido permanecer superior a 5,0 mmol/L ao final de 48 horas, um dia adicional (24 horas) de administração de 10 g três vezes por dia pode ser administrado, antes de iniciar a dose de manutenção. Se a normocalemia não for obtida ao final do dia 3, devem ser consideradas outras abordagens terapêuticas.
Fase de manutenção do tratamento de hipercalemia
Para o tratamento de manutenção contínuo, a dose mínima efetiva para prevenir a recorrência de hipercalemia deve ser estabelecida. Recomenda-se uma dose de 5 g uma vez ao dia, com possibilidade de titulação até 10 g uma vez por dia ou redução de até 5 g em dias alternados (dia sim/dia não), conforme necessário, para manter um nível de potássio normal. Não deve ser utilizado mais de 10 g por dia durante a terapia de manutenção.
Os níveis séricos de potássio devem ser monitorados regularmente durante o tratamento. A frequência de monitoramento depende de uma variedade de fatores, incluindo outros medicamentos concomitantes, progressão da doença renal crônica e ingestão dietética de potássio.
Esquecimento de dose
Se um paciente esquecer uma dose, deve ser instruído a tomar a próxima dose no horário habitual.
População especial
Pacientes com insuficiência renal
Não são necessárias alterações nas doses normais para pacientes com insuficiência renal que não estejam em hemodiálise crônica.
Para pacientes em diálise, LOKELMA® deve ser administrado somente nos dias em que o paciente não for submetido à diálise. A dose inicial recomendada é de 5 g uma vez ao dia. Para estabelecer a normocalemia (4,0-5,0 mmol/L), a dose pode ser aumentada ou diminuída semanalmente com base no valor de potássio sérico pré-diálise após um longo intervalo interdialítico. A dose pode ser ajustada em intervalos de uma semana com incrementos de 5 a 15 g, uma vez ao dia, em dias em que o paciente não é submetido à diálise. Para manter a normocalemia, recomenda-se a monitorização regular do potássio sérico (por exemplo, mensalmente).
Pacientes com insuficiência hepática
Não é necessário ajuste de dose para pacientes com insuficiência hepática.
Pacientes idosos
Não é necessário ajuste de dose para pacientes idosos.
Pacientes pediátricos
A eficácia e segurança de LOKELMA® não foram estabelecidas em pacientes pediátricos.
9.REAÇÕES ADVERSAS
Estudos clínicos
A segurança de LOKELMA® foi avaliada em estudos clínicos para a redução de hipercalemia que envolveram mais de 1.500 pacientes.
As reações adversas mais frequentemente relatadas foram eventos relacionados a edema, os quais foram reportados por 5,7% dos pacientes tratados com LOKELMA® (1,7%, 2,7%, 5,2% e 14,3% dos pacientes randomizados para placebo, LOKELMA® 5 g, 10 g ou 15 g uma vez ao dia por até um mês, respectivamente). Cinquenta e três por cento destes pacientes foram tratados com a introdução de diuréticos ou com o ajuste da dose de um diurético; os outros não precisaram de tratamento.
Nos estudos clínicos, 4,1% dos pacientes tratados com LOKELMA® desenvolveram hipocalemia com um valor de potássio sérico inferior a 3,5 mmol/L, o que foi resolvido com ajuste de dose ou com a descontinuação de LOKELMA®.
Lista tabulada de reações adversas
Foi utilizada a seguinte convenção para a frequência das reações adversas: Muito comum (≥ 1/10); Comum (≥ 1/100 a < 1/10); Incomum (≥ 1/1.000 a < 1/100); Rara (≥ 1/10.000 a < 1/1.000); Muito rara ( < 1/10.000); Desconhecido (não pode ser estimada a partir dos dados disponíveis).
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10.SUPERDOSE
A superdose de LOKELMA® pode originar hipocalemia. O potássio sérico deve ser verificado e pode ser dado um suplemento de potássio, conforme necessário.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.1618.0282
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela ANVISA em 26/01/2024.