LIVTENCITY
TAKEDA
maribavir
Antiviral.
Apresentações.
Comprimido revestido 200 mg. Embalagem com 28 ou 56 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 200 mg de maribavir. Excipientes: Núcleo do comprimido: celulose microcristalina, amidoglicolato de sódio, estearato de magnésio. Revestimento: álcool polivinílico, macrogol, dióxido de titânio, talco, azul brilhante 133 laca de alumínio.
Informações técnicas.
1. INDICAÇÃO
LIVTENCITY é indicado para o tratamento de infecção e/ou doença por citomegalovírus (CMV) que são refratárias (com ou sem resistência) a uma ou mais terapias anteriores em pacientes adultos que foram submetidos a um transplante de células-tronco hematopoiéticas (TCTH) ou transplante de órgãos sólidos (TOS).
Deve-se considerar a orientação oficial sobre o uso adequado de agentes antivirais.
2. RESULTADOS DE EFICÁCIA
Um estudo de superioridade ativa controlada, multicêntrico, randomizado, de fase 3 (Estudo SHP620-303) avaliou a eficácia e segurança do tratamento com LIVTENCITY em comparação com o tratamento atribuído pelo investigador (IAT) em 352 receptores de TCTH e TOS com infecções por CMV que eram refratárias a tratamento com ganciclovir, valganciclovir, foscarnet ou cidofovir, incluindo infecções por CMV com ou sem resistência confirmada a 1 ou mais agentes anti CMV. A infecção refratária por CMV foi definida como falha documentada em atingir uma redução de > 1 log10 no nível de DNA de CMV no sangue total ou no plasma após um período de tratamento de 14 dias ou mais com ganciclovir/valganciclovir oral, foscarnet intravenoso ou cidofovir intravenoso. Essa definição foi aplicada à infecção atual por CMV e ao agente anti-CMV administrado mais recentemente. Os pacientes foram estratificados por tipo de transplante (TCTH ou TOS) e triagem dos níveis de DNA do CMV e, em seguida, randomizados em uma proporção de 2:1 para receber LIVTENCITY 400 mg duas vezes ao dia ou IAT (ganciclovir, valganciclovir, foscarnet ou cidofovir) para um período de tratamento de 8 semanas e uma fase de acompanhamento de 12 semanas.
A idade média dos indivíduos do estudo foi de 53 anos e a maioria dos indivíduos era do sexo masculino (61%), brancos (76%) e não hispânicos ou latinos (83%), com distribuições semelhantes nos dois braços de tratamento. As características da doença de linha de base estão resumidas na Tabela 1 abaixo.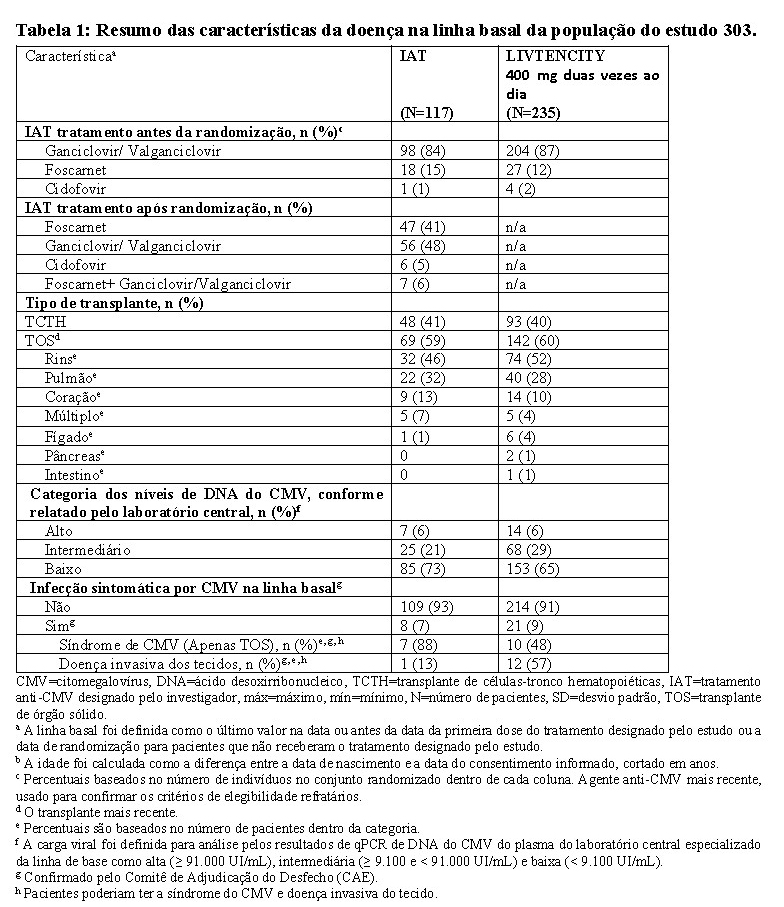
O desfecho primário de eficácia foi o clearance de viremia de CMV confirmado (concentração de DNA de CMV no plasma abaixo do limite inferior de quantificação ( < LLOQ; ou seja, < 137 UI/mL)) na Semana 8, independentemente de qualquer tratamento atribuído ao estudo ter sido descontinuado antes do final do período estipulado de 8 semanas de terapia. O desfecho secundário chave foi a eliminação da viremia por CMV e o controle dos sintomas da infecção por CMV na Semana 8 com manutenção deste efeito do tratamento até a Semana 16 do Estudo. O controle dos sintomas da infecção por CMV foi definido como resolução ou melhora da doença invasiva do tecido ou síndrome de CMV para pacientes sintomáticos no início do estudo, ou sem novos sintomas para pacientes assintomáticos na linha basal.
Para o desfecho primário, o LIVTENCITY foi superior ao IAT (56% vs. 24%, respectivamente, p < 0,001). Para o desfecho secundário chave, 19% vs. 10% alcançaram o clearance de viremia por CMV e o controle dos sintomas de infecção por CMV no grupo LIVTENCITY e IAT, respectivamente (p=0,013) (Ver Tabela 2).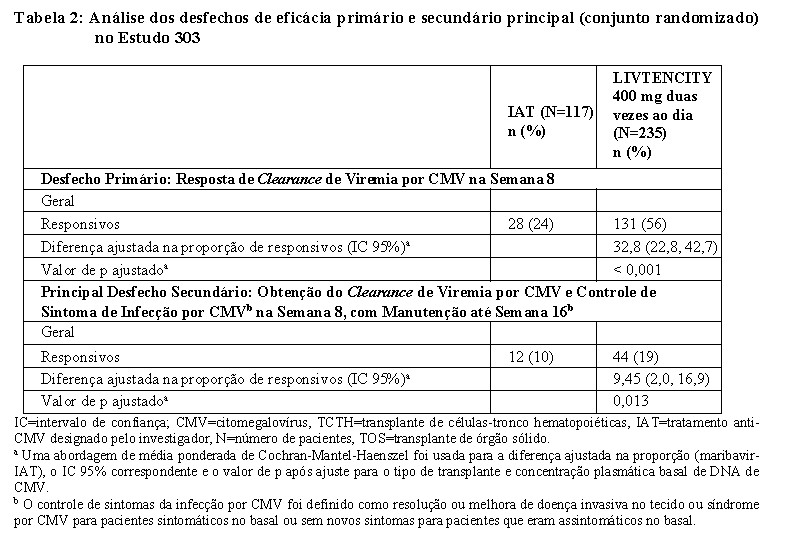
O efeito do tratamento foi consistente entre o tipo de transplante, a faixa etária e a presença de síndrome de CMV/doença na linha basal. Entretanto, LIVTENCITY foi menos efetivo em indivíduos com níveis aumentados no DNA de CMV (≥ 50.000 UI/mL) e pacientes com ausência de resistência genotípica (vide tabela 3).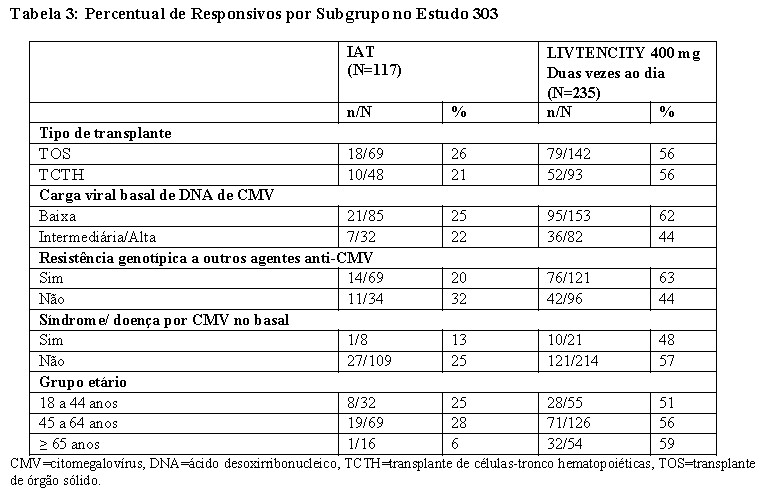
Recorrência
O desfecho secundário de recorrência de viremia por CMV foi relatado em 57% dos pacientes tratados com maribavir e em 34% dos pacientes tratados com IAT. Destes, 18% no grupo maribavir tiveram recorrência de viremia por CMV durante o tratamento em comparação com 12% no grupo IAT. A recorrência de viremia por CMV durante o seguimento foi observada em 39% dos pacientes no grupo maribavir e em 22% dos pacientes no grupo IAT.
Mortalidade geral: A mortalidade por todas as causas foi avaliada durante todo o período do estudo. Uma porcentagem semelhante de indivíduos em cada grupo de tratamento morreu durante o estudo (LIVTENCITY 11% [27/235]; IAT 11% [13/117]).
Braço resgate
Vinte e dois pacientes receberam LIVTENCITY como terapia resgate devido à piora de viremia por CMV ou infecções novas/ persistentes sintomáticas por CMV em 7 (31,8%) ou falta de melhora da infecção por CMV mais intolerância a IAT em 15 (68,2%) pacientes. Dos 22 pacientes, 11 (50,0%) pacientes obtiveram clearance de viremia por CMV confirmado na Semana 8 da fase de tratamento de resgate com LIVTENCITY e 11 (50,0%) pacientes foram não responsivos.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de Ação
Maribavir é um inibidor competitivo da proteína quinase UL97. A inibição do UL97 ocorre na fase de replicação do DNA viral, inibindo a serina/treonina quinase UL97 pela inibição competitiva da ligação do ATP ao local de ligação do ATP da quinase, sem afetar o processo de maturação do concatâmero, abolindo a fosfotransferase inibindo a replicação e maturação do DNA do CMV, encapsidação do DNA do CMV e egresso nuclear de DNA de CMV.
Atividade antiviral
Maribavir inibiu a replicação do CMV humano em ensaios de redução de rendimento de vírus, hibridização de DNA e redução em placa em células de fibroblastos de pulmão humano (MRC-5), rim embrionário humano (HEK) e fibroblastos de prepúcio humano (MRHF). Os valores de EC50 variaram de 0,03 a 2,2 mM, dependendo da linhagem celular e do ponto final do ensaio. A atividade antiviral de cultura de células do maribavir também foi avaliada contra isolados clínicos de CMV. Os valores medianos de EC50 foram 0,1 mM (n = 10, intervalo 0,03-0,13 mM) e 0,28 mM (n = 10, intervalo 0,12-0,56 mM) usando ensaios de hibridização de DNA e redução em placa, respectivamente. Nenhuma diferença significativa nos valores de EC50 entre os quatro genótipos de glicoproteína B de CMV humano (N = 2, 1, 4 e 1 para gB1, gB2, gB3 e gB4, respectivamente) foi observada.
Atividade antiviral combinada
Quando o maribavir foi testado em combinação in vitro com outros compostos antivirais, observou-se forte antagonismo com o ganciclovir.
Nenhum antagonismo foi observado em combinação com cidofovir, foscarnet e letermovir.
Resistência viral
Em Cultura de Células
Maribavir não afeta a polimerase de DNA codificada por UL54 que, quando apresenta determinadas mutações, confere resistência ao ganciclovir/valganciclovir, foscarnet e/ou cidofovir. As mutações conferindo resistência ao maribavir foram identificadas no gene UL97: L337M, F342Y, V353A, V356G, L397R, T409M, H411L/N/Y, D456N, V466G C480F, P521L e Y617del. Essas mutações conferem resistência que varia de 3,5 vezes a > 200 vezes de aumento nos valores de EC50. As variantes do gene UL27 (R233S, W362R, W153R, L193F, A269T, V353E, L426F, E22stop, W362stop, 218delC e 301-311del) conferiram apenas resistência leve ao maribavir (aumento < 5 vezes no EC50), enquanto L335P conferiu alta resistência ao maribavir.
Em Estudos Clínicos
Nos estudos 202 e 203 de fase 2, avaliando maribavir em 279 receptores de TCTH ou TOS, dados de genotipagem pUL97 após o tratamento de 23 de 29 pacientes que inicialmente obtiveram clearance de viremia e posteriormente apresentaram recidiva da infecção por CMV, enquanto em uso de maribavir, demonstraram 17 pacientes com mutações T409M ou H411Y e 6 pacientes com mutação C480F. Entre os 25 pacientes que não responderam a > 14 dias de terapia
com maribavir, 9 tiveram mutações T409M ou H411Y e 5 pacientes tiveram mutação C480F. A genotipagem pUL27 adicional foi realizada em 39 pacientes no Estudo 202 e 43 pacientes no Estudo 203. A única substituição de aminoácido associada com resistência no pUL27 que não foi detectada no basal foi G344D. A análise fenotípica de recombinantes pUL27 e pUL97 demonstrou que mutações pUL97 T409M, H411Y e C480F conferiram aumentos 78 vezes, 15 vezes e 224 vezes, respectivamente, no EC50 de maribavir em comparação com a cepa tipo selvagem, enquanto a mutação pUL27 G344D não demonstrou conferir resistência ao maribavir em comparação com a cepa tipo selvagem.
Na Fase 3, o Estudo 303 avaliando maribavir em pacientes com resistência fenotípica ao valganciclovir/ganciclovir, a análise da sequência de DNA de todas as regiões codificadoras de pUL97 e pUL27 foi realizada em 134 sequências pareadas de pacientes tratados com maribavir. As substituições de pUL97 emergentes do tratamento F342Y (4,5 vezes), T409M (78 vezes), H411L/N/Y (69, 9 e 12 vezes, respectivamente) e/ou C480F (224 vezes) foram detectados em 60 indivíduos e foram associados à não resposta (47 indivíduos foram insucessos no tratamento e 13 indivíduos foram reincidentes). Um indivíduo com a substituição pUL27 L193F (suscetibilidade reduzida em 2,6 vezes ao maribavir) no início do estudo não atingiu o desfecho primário. Além disso, as seguintes mutações múltiplas foram associadas à não resposta; F342Y+T409M+H411N (78 vezes), C480F+H411L+H411Y (224 vezes), F342Y+H411Y (56 vezes), T409M+C480F (224 vezes) e H411Y+C480F (224 vezes).
Resistência Cruzada
Foi observada resistência cruzada entre maribavir e ganciclovir/valganciclovir (vGCV/GCV) em cultura de células e em estudos clínicos. Na Fase 3 do Estudo 303, um total de 44 pacientes no braço de maribavir teve um tratamento emergente de substituição associada a resistência (RAS) ao tratamento atribuído pelo investigador (IAT). Destes, 24 tinham C480F ou F342Y RAS emergente ao tratamento, ambos são resistentes ao ganciclovir/valganciclovir e maribavir. Desses 24 pacientes, 1 (4%) atingiu o desfecho primário. No geral, apenas oito desses 44 pacientes atingiram o desfecho primário. pUL97 vGCV/GCV substituições associadas à resistência F342S/Y, K355del, V356G, D456N, V466G, C480R, P521L e Y617del reduzem a suscetibilidade ao maribavir > 4,5 vezes. Outras vias de resistência a vGCV/GCV não foram avaliadas quanto à resistência cruzada ao maribavir. As substituições de polimerase de DNA pUL54 que conferem resistência a vGCV/GCV, cidofovir ou foscarnet permaneceram suscetíveis ao maribavir.
As substituições pUL97 F342Y e C480F são substituições associadas à resistência emergente do tratamento com maribavir que conferem suscetibilidade > 1,5 vezes reduzida ao vGCV/GCV, uma redução de vezes que está associada à resistência fenotípica ao vGCV/GCV. O significado clínico desta resistência cruzada ao vGCV/GCV para estas substituições não foi determinado. O vírus resistente ao maribavir permaneceu suscetível ao cidofovir e foscarnet. Além disso, não há relatos de quaisquer substituições associadas à resistência ao pUL27 maribavir sendo avaliadas para resistência cruzada a vGCV/GCV, cidofovir ou foscarnet. Dada a falta de substituições associadas à resistência para esses medicamentos mapeados para pUL27, não é esperada resistência cruzada para substituições de pUL27 maribavir.
Propriedades Farmacocinéticas
A atividade farmacológica de maribavir é devida ao medicamento de origem. A farmacocinética de maribavir foi caracterizada após a administração oral em indivíduos sadios e pacientes transplantados. A exposição ao maribavir aumentou aproximadamente proporcionalmente à dose. Em indivíduos sadios, a média geométrica no estado de equilíbrio nos valores de AUC0-t, Cmáx e Cconcentração mínima antes da próxima dose foram 101 mg*h/mL, 16,4 mg/mL e 2,89 mg/mL, respectivamente, após doses orais de 400 mg duas vezes ao dia de maribavir.
Em receptores de transplante, a exposição ao maribavir no estado de equilíbrio após administração oral de doses de 400 mg duas vezes ao dia é fornecida abaixo, com base em uma análise farmacocinética populacional. O estado de equilíbrio foi atingido em 2 dias, com uma razão de acúmulo de 1,47 para AUC e 1,37 para Cmax. A variabilidade intraindividual ( < 22%) e a variabilidade interindividual ( < 37%) nos parâmetros farmacocinéticos de maribavir são baixas a moderadas.
Absorção
Maribavir foi rapidamente absorvido com concentrações plasmáticas de pico ocorrendo 1,0 a 3,0 horas após a dose. A exposição ao maribavir não é afetada ao se esmagar o comprimido, administrar o comprimido esmagado através de tubo nasogástrico (NG)/ orogástrico ou na administração concomitante com inibidores da bomba de próton (IBP), antagonistas do receptor H2 histamina (bloqueadores H2) ou antiácidos.
A variabilidade intraindivíduo ( < 22%) e variabilidade interindivíduo ( < 37%) nos parâmetros PK (farmacocinéticos) de maribavir são baixas a moderadas.
Efeito do Alimento
Em indivíduos sadios, a administração oral de uma dose única de 400 mg de maribavir com refeição rica em calorias não teve qualquer efeito estatisticamente significante na exposição geral (AUC) e resultou em 28% de diminuição na Cmax de maribavir, o que não foi considerado clinicamente relevante.
Distribuição
Com base na análise farmacocinética populacional, a média aparente no volume de distribuição no estado de equilíbrio é estimada como sendo 24,9 L.
A ligação in vitro de maribavir às proteínas plasmáticas humanas foi 98,0% ao longo do intervalo de concentração de 0,05-200 mg/mL. A ligação ex vivo de maribavir às proteínas (98,5%-99,0%) foi consistente com os dados in vitro, sem diferença aparente observada entre indivíduos sadios, indivíduos com comprometimento hepático (moderado) ou renal (leve, moderado ou grave), pacientes com o vírus da imunodeficiência humana ou pacientes transplantados.
Maribavir pode atravessar a barreira hematoencefálica em humanos, mas espera-se que a penetração no SNC seja baixa em comparação com os níveis plasmáticos (veja 5. ADVERTÊNCIAS E PRECAUÇÕES).
Os dados in vitro indicam que o maribavir é um substrato da glicoproteína-P (P-gp), proteína de resistência ao câncer da mama (BCRP) e transportador de cátions orgânicos 1 (OCT1). As alterações nas concentrações plasmáticas de maribavir devido à inibição de P-gp/BCRP/OCT1 não foram clinicamente relevantes.
Biotransformação
Maribavir é eliminado principalmente pelo metabolismo hepático via CYP3A4 (via metabólica primária - fração metabolizada estimada como sendo pelo menos 35%), com contribuição secundária de CYP1A2 (fração metabolizada estimada como não mais que 25%). O principal metabólito de maribavir é formado por N-dealquilação da porção isopropil e é considerado como farmacologicamente inativo. A razão metabólica para esse metabólito principal no plasma foi 0,15-0,20. Múltiplas enzimas UGT, nomeadamente UGT1A1, UGT1A3, UGT2B7 e possivelmente UGT1A9, estão envolvidas na glucuronidação de maribavir em humanos, no entanto, a contribuição da glucuronidação para a depuração global de maribavir é baixa com base em dados in vitro.
Em estudos in vitro, o metabolismo de maribavir não foi mediado por CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A5, 1A4, UGT1A6, UGT1A10 ou UGT2B15.
Eliminação
A meia-vida de eliminação terminal e clearance oral de maribavir são 4,3 horas e 2,67 L/h, respectivamente, em pacientes transplantados. Após a administração de dose única oral de [14C]-maribavir, aproximadamente 61% e 14% da radioatividade foram recuperadas na urina e fezes, respectivamente, principalmente como o metabólito principal e inativo. A excreção urinária de maribavir inalterado é mínima.
Populações Especiais
Insuficiência Renal
Nenhum efeito clinicamente significativo de insuficiência renal leve, moderada ou grave (depuração de creatinina medida variando de 12 a 70 mL/min) foi observado nos parâmetros farmacocinéticos totais de maribavir após uma dose única de 400 mg de maribavir. A diferença nos parâmetros farmacocinéticos do maribavir entre indivíduos com insuficiência renal leve/moderada ou grave e indivíduos com função renal normal foi < 9%. Como o maribavir está fortemente ligado às proteínas plasmáticas, é improvável que o maribavir seja removido de forma significativa por hemodiálise ou diálise peritoneal.
Insuficiência Hepática
Nenhum efeito clinicamente significativo de insuficiência hepática moderada (Child-Pugh Classe B, escore de 7-9) foi observado nos parâmetros farmacocinéticos de maribavir totais ou não ligados após uma dose única de 200 mg de maribavir. Em comparação com indivíduos saudáveis de controle, AUC e Cmáx foram 26% e 35% maiores, respectivamente, em indivíduos com insuficiência hepática moderada. Não se sabe se a exposição ao maribavir aumentará em pacientes com insuficiência hepática grave.
Idade, Gênero, Raça, Etnia e Peso
Idade (18-79 anos), gênero, raça (caucasiana, negra, asiática ou outras), etnia (hispânica/latina ou não hispânica/não latina) e peso corporal (36 a 141 kg) não tiveram efeito clinicamente significativo na farmacocinética de maribavir com base na análise de farmacocinética na população.
Tipos de Transplante
Os tipos de transplante (TCTH vs. TOS) ou entre tipos de TOS (fígado, pulmão, rim ou coração) ou presença de doença do enxerto contra o hospedeiro (GvHD) gastrointestinal (GI) não tem impacto clinicamente significativo na farmacocinética de maribavir.
Dados pré-clínicos de segurança
Geral
Anemia regenerativa e hiperplasia de célula de mucosa no trato intestinal, observadas com desidratação foram notadas em ratos e macacos, em conjunto com observações clínicas de fezes moles à líquidas e a alterações de eletrólitos (apenas em macacos). Um nível sem efeito adverso observado (NOAEL) não foi estabelecido em macacos e foi < 100 mg/kg/dia, que é aproximadamente 0,25 da exposição humana na dose humana recomendada (DHR). Em ratos, o NOAEL foi 25 mg/kg/dia, em exposições que foram 0,05 e 0,1 vezes a exposição na DHR em machos e fêmeas, respectivamente.
Maribavir não demonstrou fototoxicidade in vitro, portanto, o potencial para fototoxicidade em humanos é considerado improvável (veja Propriedades Farmacocinéticas e 5. ADVERTÊNCIAS E PRECAUÇÕES).
Maribavir foi detectado em níveis baixos no plexo coroide de camundongos e no cérebro e CSF de macacos.
Carcinogênese
Não foi identificado potencial carcinogênico em ratos até 100 mg/kg/dia, nos quais as exposições em machos e fêmeas foram 0,2 e 0,36 vezes, respectivamente, a exposição em humanos na dose humana recomendada. Em camundongos machos, uma elevação suspeita na incidência de hemangioma, hemangiossarcoma e hemangioma/ hemangiossarcoma combinados em diversos tecidos na dose de 150 mg/kg/dia é de relevância incerta em termos da sua tradução para risco em humanos, considerando a ausência de um efeito em camundongos fêmeas ou em ratos após 104 semanas de administração, a ausência de efeitos neoplásicos proliferativos em camundongos machos e fêmeas após 13 semanas de administração, o pacote de genotoxicidade negativo e a diferença na duração de administração em humanos. Não houve achados carcinogênicos na próxima dose mais baixa de 75 mg/kg/dia, que é aproximadamente 0,35 e 0,25 em machos e fêmeas, respectivamente, a exposição em humanos na dose humana recomendada.
Mutagêneses
Maribavir não foi mutagênico em um ensaio de mutação bacteriana, nem clastogênico no ensaio de micronúcleo na medula óssea. Em ensaios de linfoma em camundongo, maribavir demonstrou potencial mutagênico na ausência da ativação metabólica e os resultados foram incertos na presença da ativação metabólica. No geral o peso da evidência indica que maribavir não exibe potencial genotóxico.
Reprodução
Fertilidade
No estudo combinado de fertilidade e desenvolvimento embriofetal em ratos, não houve efeitos de maribavir na fertilidade. No entanto, em ratos machos, foram observadas diminuições na velocidade do esperma em linha reta, em doses ≥ 100 mg/kg/dia (que é estimada como sendo menor do que a exposição em humanos na dose humana recomendada), mas sem qualquer impacto na fertilidade masculina.
Desenvolvimento pré-natal e pós-natal
Em um estudo combinado de fertilidade e desenvolvimento embriofetal em ratos, maribavir não foi teratogênico e não teve efeito no crescimento e desenvolvimento embriofetal em doses até 400 mg/kg/dia. Uma redução no número de
fetos viáveis devido ao aumento nas reabsorções precoces e perdas após implantação foi observada em fêmeas em todas as doses testadas de maribavir que também foram tóxicas para a mãe. A dose mais baixa correspondeu a aproximadamente metade da exposição humana no DHR. No estudo de toxicidade de desenvolvimento pré e pós-natal realizado em ratos, observou-se diminuição da sobrevida dos filhotes devido a cuidados maternos inadequados e ganho de peso corporal reduzido associado a um atraso nos marcos do desenvolvimento (descolamento do pavilhão auricular, abertura ocular e separação prepucial) com doses de maribavir ≥ 150 mg/kg/dia. O desenvolvimento pós-natal não foi afetado com 50 mg/kg/dia. A fertilidade e o desempenho de acasalamento da geração F1 e sua capacidade de manter a gravidez e gerar descendentes vivos não foram afetados até 400 mg/kg/dia.
Em coelhos, o maribavir não foi teratogênico em doses de até 100 mg/kg/dia (aproximadamente 0,45 vezes a exposição humana no DHR).
4. CONTRAINDICAÇÕES
Hipersensibilidade ao maribavir ou a qualquer um dos excipientes listados no item Composição.
Administração concomitante com ganciclovir ou valganciclovir (veja 6. INTERAÇÕES MEDICAMENTOSAS).
5. ADVERTÊNCIAS E PRECAUÇÕES
Falha virológica durante o tratamento e recidiva após o tratamento
A falha virológica pode ocorrer durante e após o tratamento com LIVTENCITY. A recidiva virológica durante o período de pós-tratamento ocorreu, normalmente, dentro de 4-8 semanas após a descontinuação do tratamento. Algumas substituições associadas à resistência ao pUL97 de maribavir conferem resistência cruzada ao ganciclovir e valganciclovir. Os níveis de DNA do CMV devem ser monitorados e as mutações de resistência devem ser investigadas em pacientes que não respondem ao tratamento. O tratamento deve ser descontinuado caso sejam detectadas mutações de resistência ao maribavir.
Doença por CMV com envolvimento no Sistema Nervoso Central (SNC)
LIVTENCITY não foi estudado em pacientes com infecção por CMV no SNC. Com base nos dados não-clínicos, espera-se que a penetração no SNC de maribavir seja baixa em comparação com os níveis de plasma (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS). Portanto, não se espera que LIVTENCITY seja eficaz no tratamento de infecções por CMV no SNC (por exemplo, meningoencefalite).
Uso com imunossupressores
LIVTENCITY tem o potencial para aumentar as concentrações de imunossupressores que são substratos do citocromo P450 (CYP)3A/P-gp com intervalos terapêuticos estreitos (incluindo tacrolimo, ciclosporina, sirolimo e everolimo). Os níveis de plasma desses imunossupressores devem ser frequentemente monitorados durante todo o tratamento com LIVTENCITY, especialmente após a iniciação e após a descontinuação de LIVTENCITY, e as doses devem ser ajustadas, conforme necessário (veja 6. INTERAÇÕES MEDICAMENTOSAS, 9. REAÇÕES ADVERSAS e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Risco de reações adversas ou efeito terapêutico reduzido devido a interações medicamentosas
O uso concomitante de LIVTENCITY e determinados medicamentos pode resultar em interações medicamentosas conhecidas ou potencialmente significativas, algumas das quais podem levar a:
• Possíveis reações adversas clinicamente significativas decorrentes da maior exposição aos medicamentos concomitantes;
• Efeito terapêutico reduzido de LIVTENCITY.
Veja a Tabela 5 para ações para prevenir ou controlar essas interações medicamentosas conhecidas ou potencialmente significantes, incluindo recomendações de dosagem (veja 4. CONTRAINDICAÇÕES e 6. INTERAÇÕES MEDICAMENTOSAS).
Conteúdo de Sódio
Esse medicamento contém menos de 1 mmol de sódio (23 mg) por comprimido, ou seja, essencialmente "livre de sódio".
Uso durante a gravidez e a lactação
Gravidez
Não há dados sobre o uso de maribavir em mulheres grávidas. Estudos em animais demonstraram toxicidade reprodutiva (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS). LIVTENCITY não é recomendado durante a gravidez e em mulheres com potencial para engravidarem que não fazem uso de contracepção.
Não é esperado que maribavir afete as concentrações plasmáticas de contraceptivos esteroides orais de ação sistêmica (veja 6. INTERAÇÕES MEDICAMENTOSAS).
Categoria C de Risco na Gravidez -Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Não se sabe se maribavir ou seus metabólitos são excretados no leite humano. Um risco para o lactente não pode ser excluído. A amamentação deve ser descontinuada durante o tratamento com LIVTENCITY.
Fertilidade
Estudos de fertilidade não foram realizados em humanos com LIVTENCITY. Não foram observados efeitos na fertilidade ou desempenho reprodutivo em ratos em um estudo combinado de fertilidade e desenvolvimento embriofetal, no entanto, uma redução na velocidade de linha reta dos espermatozoides foi observada em doses ≥ 100 mg/kg/dia (que é estimada como sendo < 1 vez a exposição em humanos na dose humana recomendada). Não houve efeitos sobre órgãos reprodutivos em machos ou fêmeas nos estudos não clínicos em ratos e macacos (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Efeitos na capacidade de dirigir veículos e operar máquinas
LIVTENCITY não tem influência na capacidade de dirigir e operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Efeitos de outros medicamentos sobre maribavir
Maribavir é metabolizado, principalmente, pela CYP3A, e espera-se que medicamentos que induzem ou inibem a CYP3A afetem o clearance de maribavir (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS).
A coadministração de LIVTENCITY e medicamentos que são inibidores de CYP3A pode resultar em aumento nas concentrações de maribavir no plasma (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS). Entretanto, nenhum ajuste da dose é necessário quando maribavir é coadministrado com inibidores de CYP3A.
Espera-se que a administração concomitante de indutores CYP3A potentes ou moderados (como rifampicina, rifabutina, carbamazepina, fenobarbital, fenitoína, efavirenz e Erva-de-São-João) reduza significativamente as concentrações no plasma de maribavir, o que pode resultar em redução da eficácia. Portanto, medicamentos alternativos sem potencial de indução de CYP3A devem ser considerados. A coadministração de maribavir com fortes indutores do citocromo P450 3A (CYP3A), como a rifampicina, rifabutina ou a Erva-de-São-João, não é recomendada.
Caso a coadministração de maribavir com outros indutores potentes ou moderados de CYP3A (por exemplo, carbamazepina, efavirenz, fenobarbital e fenitoína) não possa ser evitada, a dose de maribavir deve ser aumentada para 1200 mg duas vezes ao dia (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS e 8. POSOLOGIA E MODO DE USAR).
Efeito de maribavir sobre outros medicamentos
A coadministração de maribavir com valganciclovir/ganciclovir é contraindicado (veja 4. CONTRAINDICAÇÕES). LIVTENCITY pode antagonizar o efeito antiviral de ganciclovir e valganciclovir ao inibir a serina/treonina quinase humana do CMV UL97, que é necessária para a ativação/fosforilação do ganciclovir e valganciclovir (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS e 4. CONTRAINDICAÇÕES).
Em concentrações terapêuticas, não se espera interações clinicamente significativas quando maribavir é administrado concomitantemente com substratos da CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2E1, 2D6 e 3A4; UGT1A1, 1A4, 1A6, 1A9, 2B7; bomba de exportação de sais biliares (BSEP); proteína de extrusão de múltiplos medicamentos e toxina (MATE)1/2K; transportadores de ânions orgânicos (OAT)1; transportadores catiônicos orgânicos (OCT)1 e OCT2; polipeptídio transportando ânion orgânico (OATP)1B1 e OATP1B3 com base em resultados de interação clínica e in vitro (veja Tabela 5 e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Maribavir atuou como um indutor da enzima CYP1A2 in vitro. Não há dados clínicos disponíveis para excluir um risco de interação via indução de CYP1A2 in vivo. Portanto, a administração concomitante de maribavir e medicamentos que são substratos sensíveis do CYP1A2 com uma janela terapêutica estreita (por exemplo, tizanidina e teofilina) deve ser evitada devido ao risco de falta de eficácia dos substratos do CYP1A2.
A coadministração de maribavir aumentou as concentrações de plasma de tacrolimo (veja Tabela 5). Quando os imunossupressores tacrolimo, ciclosporina, everolimo ou sirolimo são coadministrados com maribavir, os níveis de imunossupressores devem ser monitorados frequentemente durante o tratamento com maribavir, especialmente após o início e após a descontinuação de maribavir e ajuste de dose, quando necessário (veja 5. ADVERTÊNCIAS E PRECAUÇÕES e Tabela 5).
Maribavir inibiu o transportador P-gp in vitro em concentrações clinicamente relevantes. Em um estudo clínico, a coadministração de maribavir aumentou as concentrações de plasma de digoxina (veja Tabela 5). Assim, deve-se ter cuidado quando maribavir e substratos sensíveis de P-gp (por exemplo, digoxina e dabigatrana) são coadministrados. As concentrações séricas de digoxina devem ser monitoradas e a dose de digoxina pode precisar ser reduzida, conforme necessário (veja Tabela 5).
Maribavir inibiu o transportador BCRP in vitro em concentrações clinicamente relevantes. Assim, espera-se que a coadministração de maribavir com substratos de BCRP, como a rosuvastatina, aumente sua exposição e leve a efeitos indesejáveis.
In vitro, o maribavir inibe o OAT3, portanto, as concentrações plasmáticas dos medicamentos transportados pelo OAT3 podem ser aumentadas (por exemplo: ciprofloxacina, imipenem e cilastatina).
In vitro, maribavir inibe o MATE1. Não há dados clínicos disponíveis sobre se a coadministração de maribavir com substratos sensíveis de MATE1 (por exemplo, metformina) poderia potencialmente levar a interações clinicamente relevantes.
Informações gerais
Se os ajustes de dose dos medicamentos concomitantes forem feitos devido ao tratamento com maribavir, as doses devem ser reajustadas após o tratamento com maribavir ser concluído. A Tabela 5 fornece uma lista de interações medicamentosas estabelecidas ou potencialmente clinicamente significativas. As interações medicamentosas descritas são baseadas nos estudos realizados com maribavir ou são interações medicamentosas previstas que podem ocorrer com maribavir (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS e 5. ADVERTÊNCIAS E PRECAUÇÕES).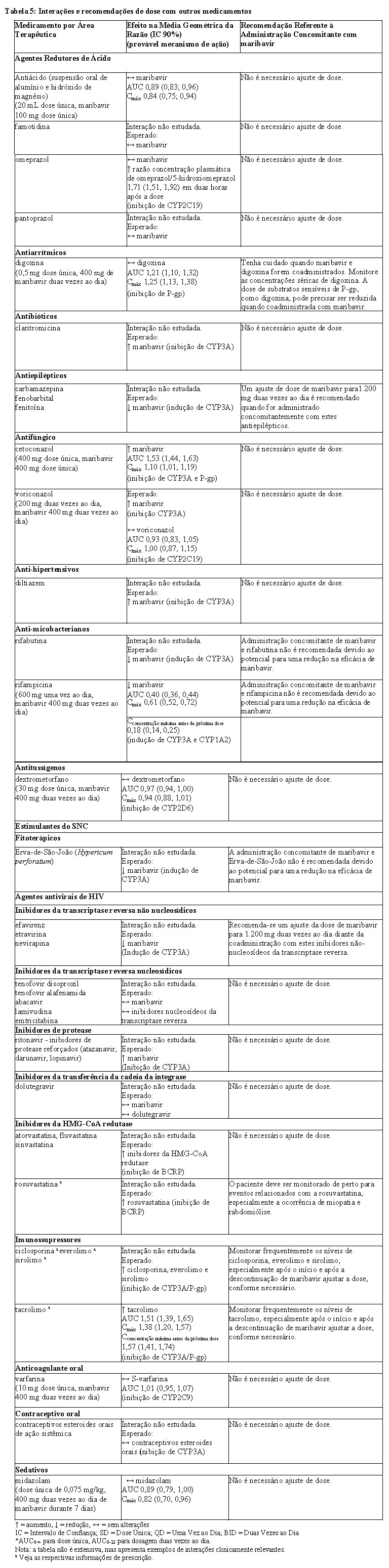
População pediátrica Estudos de interação só foram realizados em adultos.
7. CONDIÇÕES DE ARMAZENAMENTO
Conservar em temperatura ambiente entre 15°C e 30°C.
LIVTENCITY, comprimidos revestidos tem validade de 30 meses a partir da data de sua fabricação.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
LIVTENCITY é fornecido em comprimidos revestidos convexos, ovais, azuis com "SHP" impresso de um lado e "620" no outro.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
LIVTENCITY deve ser iniciado por um médico experiente no controle de pacientes que foram submetidos a um transplante de órgãos sólidos ou a um transplante de células-tronco hematopoiéticas.
Posologia
A dose recomendada de LIVTENCITY é de 400 mg (dois comprimidos de 200 mg) duas vezes ao dia, resultando em uma dose diária de 800 mg durante oito semanas. A duração do tratamento pode precisar ser individualizada com base nas características clínicas de cada paciente.
Coadministração com indutores de CYP3A
A coadministração de LIVTENCITY com fortes indutores do citocromo P450 3A (CYP3A), como a rifampicina, rifabutina ou a Erva-de-São-João, não é recomendada devido ao potencial para uma redução na eficácia de maribavir.
Caso a coadministração de LIVTENCITY com outros indutores potentes ou moderados de CYP3A (por exemplo, carbamazepina, efavirenz, fenobarbital e fenitoína) não possa ser evitada, a dose de LIVTENCITY deve ser aumentada para 1.200 mg duas vezes ao dia (veja 5. ADVERTÊNCIAS E PRECAUÇÕES, 6. INTERAÇÕES MEDICAMENTOSAS e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Doses perdidas
Os pacientes devem ser orientados para, caso percam uma dose de LIVTENCITY e a próxima dose estiver prevista para as próximas três horas, eles devem pular a dose perdida e continuar com a programação regular. Os pacientes não devem dobrar sua próxima dose ou tomar além da dose prescrita.
Populações Especiais
Pacientes Idosos
Não é necessário ajuste de dose para pacientes maiores que 65 anos (veja 2. RESULTADOS DE EFICÁCIA e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência Renal
Não é necessário ajuste de dose de LIVTENCITY para pacientes com insuficiência renal leve, moderada ou grave. A administração de LIVTENCITY em pacientes com doença renal em estágio terminal (DRT), incluindo pacientes em diálise, não foi estudada. Não é esperado que ajustes da dose sejam necessários para pacientes em diálise devido à alta ligação de proteína de plasma de maribavir (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência Hepática
Não é necessário ajuste de dose de LIVTENCITY para pacientes com insuficiência hepática leve (Child-Pugh Classe A) ou moderada (Child-Pugh Classe B). A administração de LIVTENCITY em pacientes com insuficiência hepática grave (Child-Pugh Classe C) não foi estudada. Não se sabe se a exposição a maribavir aumentará de forma significativa em pacientes com insuficiência hepática grave. Assim, deve-se tomar cuidado quando LIVTENCITY é administrado em pacientes com insuficiência hepática grave (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS).
População Pediátrica
A segurança e a eficácia de LIVTENCITY em pacientes menores de 18 anos de idade não foram estabelecidas. Não há dados disponíveis.
Método de Administração
LIVTENCITY é destinado apenas para uso oral e pode ser administrado com ou sem alimento. Os comprimidos revestidos podem ser administrados inteiros, esmagados ou esmagados através de um tubo nasogástrico ou orogástrico.
Este medicamento não deve ser mastigado.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
Os eventos adversos foram coletados durante a fase de tratamento e a fase de acompanhamento até a semana de estudo 20 no estudo de Fase 3 (veja 3. CARACTERÍSTICAS FARMACOLÓGICAS). A exposição média (SD) para LIVTENCITY foi de 48,6 (13,82) dias com um máximo de 60 dias. As reações adversas mais frequentemente notificadas que ocorreram em pelo menos 10% dos indivíduos do grupo LIVTENCITY foram: alteração do paladar (46%), náuseas (21%), diarreia (19%), vômitos (14%) e fadiga (12%). As reações adversas graves mais comumente relatadas foram diarreia (2%) e náusea, diminuição de peso, fadiga, aumento do nível do medicamento imunossupressor e vômito (todos ocorrendo em < 1%).
Lista tabulada de reações adversas
As reações adversas estão listadas abaixo por classe de sistema de órgãos e frequência. As frequências são definidas
da seguinte forma: muito comum (≥ 1/10), comum (≥ 1/100 a < 1/10), incomum (≥ 1/1.000 a < 1/100), raro (≥ 1/10.000
a < 1/1.000) ou muito raro ( < 1/10.000).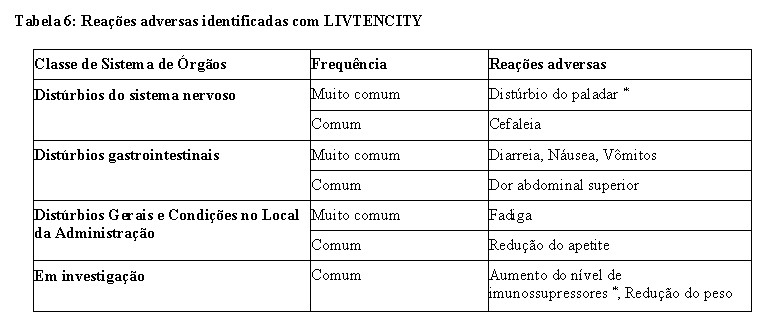
Descrição de reações adversas selecionadas*
Distúrbio do paladar
Distúrbio do paladar (consistindo dos termos preferidos reportados de ageusia, disgeusia, hipogeusia e distúrbio do paladar) ocorreu em 46% dos pacientes tratados com LIVTENCITY. Esses eventos raramente levaram à descontinuação de LIVTENCITY (0,9%) e, para a maioria dos pacientes, resolveram-se enquanto os pacientes permaneceram em terapia (37%) ou dentro de uma mediana de 7 dias (estimativa de Kaplan-Meier, IC 95%: 4-8 dias) após a descontinuação do tratamento.
Aumento no Nível de Imunossupressores no plasma
O aumento no nível de medicamento imunossupressor (composto pelos termos preferidos, nível do medicamento imunossupressor aumentado e nível do medicamento aumentado) ocorreu em 9% dos pacientes tratados com LIVTENCITY. LIVTENCITY tem o potencial para aumentar a concentração de imunossupressores que são substratos de CYP3A e/ou P-gp com faixas terapêuticas estreitas (incluindo tacrolimo, ciclosporina, sirolimo e everolimo). (veja
3. CARACTERÍSTICAS FARMACOLÓGICAS, 5. ADVERTÊNCIAS E PRECAUÇÕES e 6. INTERAÇÕES MEDICAMENTOSAS).
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
No Estudo 303, uma superdose acidental de uma dose extra única ocorreu em 1 indivíduo tratado com LIVTENCITY no Dia 13 (dose diária total de 1200 mg). Não houve relato de reações adversas. No Estudo 202, 40 indivíduos foram expostos a doses de 800 mg de duas vezes ao dia e 40 indivíduos foram expostos a 1200 mg duas vezes ao dia por uma média de aproximadamente 90 dias. No Estudo 203, 40 indivíduos foram expostos a doses de 800 mg duas vezes ao dia e 39 indivíduos foram expostos a 1200 mg duas vezes ao dia por um máximo de 177 dias. Não houve diferenças observadas no perfil de segurança em qualquer estudo em comparação com o grupo de 400 mg duas vezes ao dia no Estudo 303, no qual indivíduos receberam maribavir por um máximo de 60 dias.
Não há antídoto específico conhecido para o maribavir. No caso de superdose, recomenda-se monitorar o paciente para reações adversas e instituir tratamento sintomático adequado. Devido à alta ligação às proteínas plasmáticas ao maribavir, é imp