LAZCLUZE
JANSSEN-CILAG
mesilato de lazertinibe
Antineoplásico.
Apresentações.
Comprimidos revestidos de 80 mg de lazertinibe em frasco com 90 comprimidos revestidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém: 80 mg de lazertinibe (equivalente a 96,48 mg de mesilato de lazertinibe monoidratado).
Lazcluze ® 80 mg
Excipientes: sílica coloidal hidrofóbica, celulose microcristalina, manitol, croscarmelose sódica, estearato de magnésio, copolímero enxertado de macrogol (PEG) e álcool polivinílico, talco, dióxido de titânio, monocaprilocaprato de glicerila tipo I, óxido de ferro amarelo, álcool polivinílico parcialmente hidrolisado.
Informações técnicas.
1. INDICAÇÕES
Lazcluze® em combinação com amivantamabe é indicado para o tratamento de primeira linha de pacientes adultos com câncer de pulmão de não pequenas células (CPNPC) localmente avançado ou metastático com mutações de deleção no éxon 19 ou de substituição L858R no éxon 21 do receptor do fator de crescimento epidérmico (EGFR).
2. RESULTADOS DE EFICÁCIA
O NSC3003 (MARIPOSA) é um estudo de fase 3 randomizado, controlado por ativo e multicêntrico que avalia a eficácia e a segurança de Lazcluze® em combinação com amivantamabe em comparação à monoterapia com osimertinibe como tratamento de primeira linha em pacientes com CPNPC localmente avançado ou metastático com mutação do EGFR não suscetível a tratamento curativo. Era necessário que as amostras dos pacientes apresentassem uma das duas mutações comuns do EGFR (deleção no éxon 19 ou mutação de substituição L858R no éxon 21), conforme identificado por testes locais. O teste de tecido cobas central (usando o Teste de Mutação do EGFR cobas® v2) ficaram em concordância com os exames locais em 96,3% das amostras com um resultado do exame de tecido cobas válido.
No total, 1074 pacientes foram randomizados (2:2:1) para receber Lazcluze® em combinação com amivantamabe, monoterapia com osimertinibe ou monoterapia com Lazcluze® (um regime não aprovado para CPNPC) até a progressão da doença ou toxicidade inaceitável. O Lazcluze® foi administrado a 240 mg por via oral uma vez ao dia. O amivantamabe foi administrado por via intravenosa a 1050 mg (para pacientes < 80 kg) ou 1400 mg (para pacientes ≥ 80 kg) uma vez por semana por 4 semanas e, em seguida, a cada 2 semanas, começando na Semana 5. O osimertinibe foi administrado com uma dose de 80 mg por via oral uma vez ao dia. A randomização foi estratificada por tipo de mutação do EGFR (deleção no éxon 19 ou mutação de substituição L858R no éxon 21), raça (asiáticos ou não asiáticos) e histórico de metástase cerebral (sim ou não).
Os dados demográficos e as características da doença no período basal foram equilibrados entre os braços de tratamento. A idade mediana foi de 63 (faixa: 25-88) anos, com 45% dos pacientes ≥ 65 anos; 62% eram do sexo feminino; e 59% eram asiáticos e 38% eram brancos. O status de desempenho do Grupo de Oncologia Cooperativo do Leste (ECOG) no período basal foi 0 (34%) ou 1 (66%); 69% nunca fumaram; 41% apresentaram metástases cerebrais anteriores; e 90% apresentaram câncer em Estágio IV no diagnóstico inicial. Em relação ao status de mutação do EGFR, 60% apresentavam deleções no éxon 19 e 40% apresentavam mutações de substituição L858R no éxon 21.
O Lazcluze® em combinação com amivantamabe demonstrou uma melhora estatisticamente significativa e clinicamente expressiva na sobrevida livre de progressão (SLP) por avaliação por BICR, com uma redução de 30% no risco de progressão ou óbito em comparação ao osimertinibe (RR (Risco Relativo) =0,70 [IC de 95%: 0,58, 0,85], p=0,0002). A SLP mediana correspondente foi de 23,72 meses (IC de 95%: 19,12, 27,66) para o braço de Lazcluze® em combinação com amivantamabe e de 16,59 meses (IC de 95%: 14,78, 18,46) para o braço de osimertinibe.
A análise final da sobrevida global (SG) demonstrou uma melhora estatisticamente significativa na SG para Lazcluze® em combinação com o amivantamabe em comparação com o osimertinibe (consulte a Tabela 1 e a Figura 1). Uma maior proporção de pacientes tratados com Lazcluze® em combinação com amivantamabe estava viva em 12 meses, 18 meses, 24 meses, 36 meses e 42 meses (90%, 82%, 75%, 60% e 56%, respectivamente) em comparação aos pacientes tratados com osimertinibe (88%, 79%, 70%, 51% e 44%, respectivamente). O Lazcluze® em combinação com amivantamabe também forneceu um benefício no tempo até a segunda progressão ou óbito (SLP2) (RR=0,75 [IC de 95%: 0,58, 0,98], p=0,0314). Embora a taxa de resposta objetiva (TRO) tenha sido comparável entre os braços, a duração da resposta (DOR) mediana entre os respondedores confirmados foi mais longa com o Lazcluze® em combinação com amivantamabe (25,76 vs. 16,76 meses). O Lazcluze® em combinação com amivantamabe também forneceu um benefício no tempo até a progressão sintomática (TTSP), uma medida da carga de sintomas de câncer de pulmão (RR=0,72 [IC de 95%: 0,57, 0,91], p=0,0049). A Tabela 1, a Figura 1 e a Figura 2 resumem os resultados de eficácia para o Lazcluze® em combinação com amivantamabe.
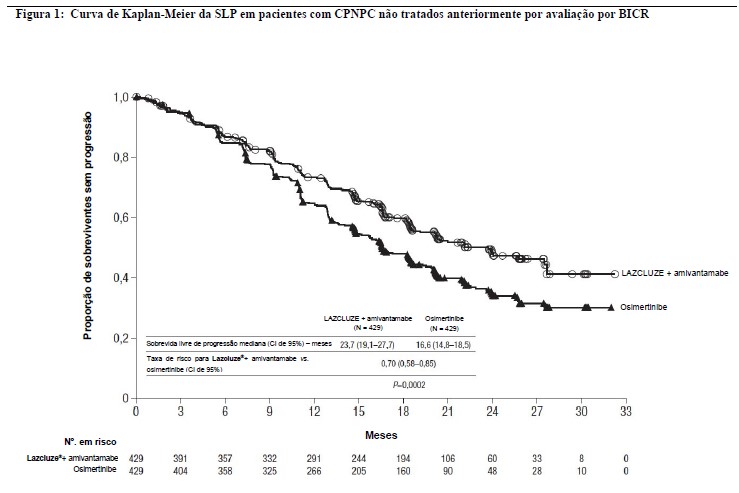
O benefício na SLP de Lazcluze® em combinação com amivantamabe em comparação ao osimertinibe foi geralmente consistente entre subgrupos clinicamente relevantes pré-especificados, incluindo faixa etária, sexo, raça, peso, tipo de mutação, status de desempenho do ECOG, histórico de tabagismo e histórico de metástase cerebral na entrada no estudo (Vide Figura 2).
O estudo MARIPOSA incluiu imagens de ressonância magnética (IRM) do cérebro exigidas pelo protocolo, que historicamente não foram usadas em estudos que avaliaram CPNPC com mutação do EGFR. Isso pode ter levado à detecção mais precoce de recorrências e a valores medianos mais curtos associados para a SLP. Para considerar isso, uma análise de sensibilidade foi realizada, por meio da qual foram censurados os pacientes com progressão somente cerebral como o local da primeira progressão. A SLP extracraniana baseada na avaliação por BICR foi consistente com o benefício do tratamento observado na análise primária. A SLP extracraniana mediana foi de 27,5 meses com o Lazcluze® em combinação com amivantamabe, em comparação a 18,37 meses com o osimertinibe (RR=0,68 [IC de 95%: 0,55, 0,83], p nominal=0,0001).
A análise estratificada da SLP avaliada pelo pesquisador mostra que o efeito melhorado do tratamento da combinação de Lazcluze® e amivantamabe em relação ao osimertinibe também foi observado quando avaliado pelo pesquisador. Os resultados para a análise da TRO com base na avaliação do pesquisador para comparação do braço de Lazcluze® em combinação com amivantamabe versus o braço de osimertinibe foram consistentes com os resultados para a TRO com base na avaliação por BICR.
Os resultados de análises exploratórias pré-especificadas da TRO e da DOR no sistema nervoso central (SNC) por BICR no subconjunto de pacientes com lesões intracranianas mensuráveis no período basal para a combinação de Lazcluze® e amivantamabe demonstraram uma TRO intracraniana semelhante àquela do controle. De acordo com o protocolo, todos os pacientes no MARIPOSA realizaram IRM do cérebro em série para avaliar a resposta intracraniana e a duração. Os resultados estão resumidos na Tabela 2.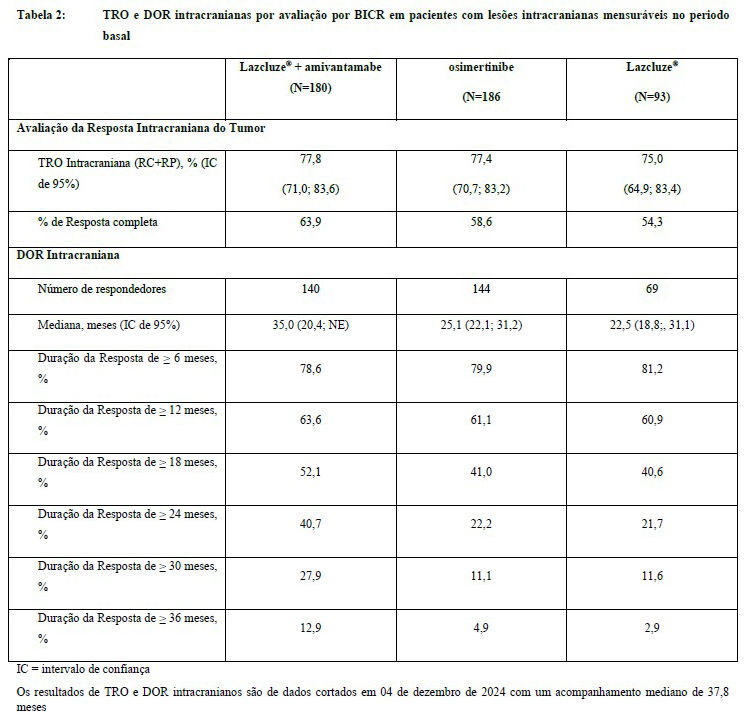
REFERÊNCIAS
Passaro A, Wang J, Wang Y, et al. Amivantamab plus chemotherapy with and without lazertinib in EGFR-mutant advanced NSCLC after disease progression on osimertinib: Primary results from the phase 3 MARIPOSA-2 study. Ann Oncol. 2023; DOI: https://doi.org/10.1016/j.annonc.2023.10.117.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodiâmicas
Mecanismo de ação
O lazertinibe é um inibidor da tirosina quinase (TKI) do EGFR de terceira geração altamente potente. Ele inibe seletivamente tanto mutações de ativação primária do EGFR (deleções no éxon 19 e mutações de substituição L858R no éxon 21) quanto a mutação de resistência T790M do EGFR, enquanto apresenta menos atividade contra o EGFR de tipo selvagem.
Efeitos Farmacodinâmicos
Com base nas análises de exposição-resposta quanto à eficácia, não foi observada nenhuma relação aparente entre a exposição ao lazertinibe e a sobrevida livre de progressão no regime de dose de 240 mg uma vez ao dia. Uma análise semelhante de exposição-resposta quanto à segurança concluiu que a parestesia e a estomatite pareceram mostrar uma tendência de aumento da ocorrência com o aumento da exposição ao lazertinibe.
Efeito sobre o intervalo QT/QTc e a eletrofisiologia cardíaca
Eletrofisiologia cardíaca
O potencial de prolongamento do intervalo QTc do lazertinibe foi avaliado por uma análise de exposição-resposta (E-R) conduzida com dados clínicos de 243 pacientes com CPNPC que receberam 20, 40, 80, 120, 160, 240 ou 320 mg de lazertinibe uma vez ao dia em um estudo de fases I/II. A análise de E-R não revelou nenhuma relação clinicamente relevante entre a concentração plasmática de lazertinibe e a alteração no intervalo QTc. O limite superior bilateral do IC de 90% com a Cmáx em estado de equilíbrio a partir da dose recomendada de 240 mg uma vez ao dia e da dose clínica mais alta testada de 320 mg uma vez ao dia foi de 5,83 e de 7,23 mseg, respectivamente.
Propriedades Farmacocinéticas
Após a administração oral única e múltipla uma vez ao dia, a concentração plasmática máxima (Cmáx) e a área sob a curva de concentração plasmática-tempo (AUC) de lazertinibe aumentaram aproximadamente de maneira proporcional à dose ao longo da faixa de doses de 20 a 320 mg. A exposição plasmática no estado de equilíbrio foi obtida no dia 15 de administração uma vez ao dia e um acúmulo de aproximadamente 2 vezes foi observado no estado de equilíbrio com a dose de 240 mg uma vez ao dia.
A exposição plasmática ao lazertinibe foi comparável quando o lazertinibe foi administrado em combinação com amivantamabe ou na forma de monoterapia.
Absorção
O tempo mediano para atingir a dose única e a Cmáx no estado de equilíbrio foi comparável e variou de 2 a 4 horas.
Após a administração de 240 mg de lazertinibe com uma refeição com alto teor de gordura (800~1000 kcal, teor de gordura de aproximadamente 50%), a Cmáx e a AUC de lazertinibe foram comparáveis àquelas sob condições de jejum, sugerindo que o lazertinibe pode ser tomado com ou sem alimentos.
Distribuição
O lazertinibe foi amplamente distribuído, com um volume de distribuição aparente médio (CV%) de 4264 (43,2%) L na dose de 240 mg. A ligação às proteínas plasmáticas média (CV%) do lazertinibe foi de aproximadamente 99,2% (0,13%) em humanos.
Eliminação
A depuração aparente média e a meia-vida de eliminação terminal (CV%) do lazertinibe na dose de 240 mg foram de 44,5 (29,5%) L/h e 64,7 (32,8%) horas, respectivamente.
Metabolismo
O lazertinibe é metabolizado primariamente por conjugação de glutationa mediada pela glutationa S-transferase mu 1 (GSTM1), com uma contribuição relativamente menor da via metabólica oxidativa mediada pela CYP3A4. Os metabólitos mais abundantes são catabólitos de glutationa e considerados clinicamente inativos. A exposição plasmática de lazertinibe foi afetada pelo metabolismo mediado pela GSTM1, levando a uma exposição mais baixa (diferença de menos de 2 vezes) em pacientes com GSTM1 não nula. Não é necessário ajuste de dose com base no status de GSTM1.
Excreção
Após uma dose oral única de lazertinibe radiomarcado, aproximadamente 86% da dose foi recuperada nas fezes ( < 5% como inalterado) e 4% na urina ( < 0,2% como inalterado).
Populações especiais
Pacientes pediátricos (17 anos de idade ou menos)
A farmacocinética do lazertinibe em pacientes pediátricos não foi investigada.
Idosos (65 anos de idade ou mais)
Baseada em uma análise farmacocinética (PK) da população, não foram observadas diferenças clinicamente significativas na farmacocinética do lazertinibe com base na idade.
Insuficiência renal
Com base em uma análise PK da população, não é necessário nenhum ajuste de dose para pacientes com insuficiência renal leve, moderada ou severa com uma taxa de filtração glomerular estimada (eGFR) de 15 a 89 mL/min. Os dados em pacientes com insuficiência renal severa (eGFR de 15 a 29 mL/min) são limitados (n=3), mas não há nenhuma evidência que sugira que um ajuste de dose seja necessário nesses pacientes. Não está disponível nenhum dado em pacientes com doença renal em estágio terminal (eGFR < 15 mL/min).
Insuficiência hepática
Com base nos achados de um estudo de farmacologia clínica, a insuficiência hepática moderada (Classe B de Child-Pugh) não teve nenhum efeito clinicamente significativo sobre a PK de dose única do lazertinibe. Com base em uma análise PK da população, não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve (bilirrubina total ≤ ULN e AST > ULN ou ULN < bilirrubina total ≤ 1,5×ULN e qualquer AST) ou moderado (1,5×ULN < bilirrubina total ≤ 3×ULN e qualquer AST). Não está disponível nenhum dado em pacientes com insuficiência hepática severa (bilirrubina total > 3×ULN e qualquer AST).
Outras populações
Não foi observada nenhuma diferença clinicamente significativa na PK do lazertinibe com base na idade, no sexo, no peso corporal, na raça, na etnia, na função hepática, na função renal, nas avaliações laboratoriais do período basal (depuração de creatinina, albumina, alaninaaminotransferase, fosfatase alcalina, aspartato-aminotransferase), no status de desempenho do ECOG, no tipo de mutação do EGFR, no estágio do câncer no diagnóstico inicial, nos tratamentos anteriores, na metástase cerebral e no histórico de tabagismo.
Informações não clínicas
Em estudos de toxicidade de doses repetidas com o lazertinibe em ratos e cães, órgãos e tecidos (olhos, pele, fígado, pulmões, rins, duodeno, medula óssea, ovário, vagina e testículo em ratos; e olhos, pele, pulmões, rins, duodeno, esôfago, jejuno e testículo em cães) contendo linhagens de células epiteliais foram afetados, com as alterações se estendendo de atrofia epitelial leve a erosões degenerativas, inflamação e necrose. Esses achados foram observados nos animais em faixas de exposições de 0,9-3,4x as exposições estimadas de pacientes com a administração da dose recomendada (240 mg) e foram totalmente ou parcialmente resolvidos durante as fases de recuperação.
Carcinogenicidade e Mutagenicidade
Não foi observada nenhuma evidência de genotoxicidade para o lazertinibe em testes de mutagenicidade bacteriana in vitro, aberração cromossômica in vitro e de micronúcleos in vivo em ratos. Não foram realizados estudos de longo prazo em animais para avaliar o potencial carcinogênico do lazertinibe.
Toxicologia Reprodutiva
Em um estudo de fertilidade e desenvolvimento embrionário inicial em ratos machos e fêmeas, o lazertinibe não afetou os parâmetros de ciclos estrais, de acasalamento, de fertilidade ou espermáticos, mas induziu um aumento na perda pós-implantação e um tamanho diminuído da ninhada viva com 30 mg/kg/dia, um nível de dose que se aproxima da exposição clínica humana na dose recomendada de 240 mg. Em estudos de desenvolvimento embriofetal, foram observadas diminuições nos pesos corporais fetais em associação a toxicidade materna em ratos com 60 mg/kg/dia, uma exposição materna aproximadamente 4 vezes mais alta do que a exposição clínica humana com 240 mg. Não houve nenhum efeito sobre o desenvolvimento embriofetal em coelhos com 45 mg/kg/dia, uma exposição materna que se aproxima da exposição clínica humana com 240 mg.
4. CONTRAINDICAÇÕES
Lazcluze® é contraindicado em pacientes com hipersensibilidade conhecida a lazertinibe ou a qualquer componente presente na formução.
5. ADVERTÊNCIAS E PRECAUÇÕES
Doença Pulmonar Intersticial (DPI)
Doença pulmonar intersticial (DPI)/pneumonite, incluindo eventos fatais, foram relatadas em pacientes que receberam Lazcluze® (vide "Reações Adversas"). Monitore os pacientes quanto a sintomas indicativos de DPI/pneumonite (p. ex., dispneia, tosse, febre). Se houver o desenvolvimento de sintomas, interrompa o tratamento com Lazcluze® aguardando a investigação desses sintomas. Avalie a suspeita de DPI e inicie o tratamento apropriado, conforme necessário. Descontinue o Lazcluze® em pacientes com confirmação de DPI (vide "Posologia e Modo de Usar").
Eventos Tromboembólicos Venosos (TEV)
Eventos tromboembólicos venosos (TEV), que abrangem trombose venosa profunda (TVP) e embolia pulmonar (EP), incluindo eventos fatais, foram relatados em pacientes que receberam Lazcluze® com amivantamabe. Os eventos TEV ocorreram predominantemente nos primeiros quatro meses de tratamento (vide "Reações Adversas"). Recomenda-se que anticoagulantes profiláticos sejam usados pelos primeiros quatro meses de tratamento. O uso de anticoagulantes deve estar de acordo com as diretrizes clínicas; o uso de antagonistas da vitamina K não é recomendado.
Monitore sinais e sintomas de eventos TEV. Trate pacientes com eventos TEV com anticoagulantes, conforme clinicamente indicado. Para eventos TEV associados a instabilidade clínica, o tratamento deve ser suspenso até que o paciente esteja clinicamente estável. Depois disso, tanto
o Lazcluze® quanto o amivantamabe podem ser retomados, a critério do médico responsável pelo tratamento.
No caso de recorrência apesar da anticoagulação apropriada, descontinue o Lazcluze® ou o amivantamabe. O tratamento pode continuar com o Lazcluze® ou o amivantamabe, mas não ambos, a critério do médico responsável pelo tratamento (vide "Posologia e Modo de Usar").
Reações Cutâneas e Ungueais
Reações cutâneas e ungueais podem ocorrer em pacientes tratados com Lazcluze® .
Erupção cutânea (incluindo dermatite acneiforme), prurido e pele seca ocorreram em pacientes que receberam Lazcluze® com amivantamabe (vide "Reações Adversas"). Instrua os pacientes a limitar a exposição ao sol durante e por 2 meses após o tratamento com Lazcluze® .
Ao iniciar o tratamento com Lazcluze® em combinação com o amivantamab, administrar um creme emoliente isento de álcool (por exemplo, isento de isopropanol, isento de etanol) para reduzir o risco de reações adversas dermatológicas. Instrua os pacientes a limitar a exposição solar durante e durante 2 meses após o tratamento com Lazcluze® em combinação com o amivantamab. Aconselhe os pacientes a usar roupas de proteção e usar protetor solar UVA/UVB de amplo espetro.
Considere medidas profiláticas (por exemplo, uso de antibióticos orais) para reduzir o risco de reações adversas dermatológicas. Se as reações de pele forem desenvolvidas, administrar corticosteroides tópicos e antibióticos tópicos e/ou orais. Para reações de grau 3, administrar esteroides orais e considerar a consulta dermatológica. Encaminhar prontamente pacientes que apresentam erupção cutânea grave, aparência ou distribuição atípicas ou falta de melhora dentro de 2 semanas para um dermatologista. Reter, reduzir a dose ou descontinuar permanentemente Lazcluze® e amivantamab com base na gravidade.
Distúrbios Oculares
Ceratite ocorreu em pacientes que receberam Lazcluze® com amivantamabe (vide "Reações Adversas"). Encaminhe imediatamente pacientes que apresentarem piora dos sintomas oculares a um oftalmologista e aconselhe a descontinuação de lentes de contato até que os sintomas sejam avaliados.
Efeitos sobre a Capacidade de Dirigir e Usar Máquinas
Não foi realizado nenhum estudo a respeito dos efeitos sobre a capacidade de dirigir e usar máquinas. Se os pacientes apresentarem sintomas relacionados ao tratamento que afetem a sua capacidade de se concentrar e reagir, recomenda-se que eles não dirijam ou usem máquinas até que o efeito desapareça.
Gravidez, Amamentação e Fertilidade
Gravidez (Categoria C)
Não há nenhum dado do uso de lazertinibe em mulheres grávidas. Estudos em animais mostraram toxicidade reprodutiva (diminuições no peso fetal) (vide "Informações Não Clínicas"). Com base no seu mecanismo de ação e em dados em animais, o lazertinibe pode causar dano fetal quando administrado a uma mulher grávida. O lazertinibe não deve ser usado durante a gravidez, a menos que a condição clínica da mulher necessite do tratamento com lazertinibe.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Amamentação
Não se sabe se o lazertinibe ou os seus metabólitos são excretados no leite humano ou afetam a produção de leite. Uma vez que o risco para a criança amamentada não pode ser excluído, aconselhe as mulheres a não amamentar durante o tratamento e por 3 semanas após a última dose de Lazcluze® .
Contracepção
Aconselhe as mulheres com potencial reprodutivo a usar contracepção eficaz durante o tratamento e por 3 semanas após a dose final de Lazcluze®. Aconselhe os pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo a usar contracepção eficaz (p. ex., preservativo) e a não doar ou armazenar sêmen durante o tratamento e por 3 semanas após a última dose de Lazcluze® .
Fertilidade
Não há nenhum dado a respeito do efeito do Lazcluze® sobre a fertilidade humana. Estudos em animais mostraram que o lazertinibe pode comprometer a fertilidade de fêmeas (vide "Informações Não Clínicas").
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de Outros Medicamentos sobre o lazertinibe
Fortes Indutores da CYP3A4
A administração concomitante de 240 mg de lazertinibe com rifampicina (forte indutor da CYP3A4) diminuiu a exposição plasmática ao lazertinibe. As razões médias geométricas (CI de 90%) do lazertinibe para a Cmáx e a AUC0-120h foram de 0,28 (0,23, 0,34) e 0,17 (0,14, 0,19), respectivamente, quando administrado concomitantemente com rifampicina em relação ao lazertinibe isolado. Com base na análise do modelo de PK baseada em fisiologia, não se espera nenhuma diminuição clinicamente relevante na exposição ao lazertinibe quando o Lazcluze® for administrado concomitantemente com indutores fracos ou moderados da CYP3A4. A administração concomitante de Lazcluze® com fortes indutores da CYP3A4 deve ser evitada.
Fortes Inibidores da CYP3A4
A administração concomitante de 160 mg de lazertinibe com itraconazol (forte inibidor da CYP3A4) aumentou a exposição plasmática ao lazertinibe em menos de 50%. As razões médias geométricas (CI de 90%) do lazertinibe para a Cmáx e a AUC0-120h foram de 1,19 (1,08, 1,30) e 1,46 (1,39, 1,53), respectivamente, quando administrado concomitantemente com itraconazol em relação ao lazertinibe isolado. Não é necessário nenhum ajuste de dose quando o Lazcluze® for usado com inibidores da CYP3A4.
Agentes redutores de ácido gástrico
Os resultados de uma análise PK retrospectiva de um estudo populacional de pacientes sugerem que não houve nenhuma alteração clinicamente relevante na exposição plasmática ao lazertinibe quando administrado concomitantemente com agentes redutores de ácido gástrico. Não é necessário nenhum ajuste de dose quando o Lazcluze® for usado com agentes redutores de ácido gástrico.
Efeito do Lazertinibe sobre Outros Medicamentos
A administração concomitante de midazolam (substrato da CYP3A4) com 160 mg de lazertinibe aumentou a exposição plasmática ao midazolam em menos de 50%. As razões médias geométricas (CI de 90%) do midazolam para a Cmáx e a AUC0-última foram de 1,39 (1,23, 1,58) e 1,47 (1,34, 1,60), respectivamente, quando administrado concomitantemente com lazertinibe em relação ao midazolam isolado. O lazertinibe é um inibidor da enzima CYP3A4. Para substratos sensíveis da CYP3A4 com um índice terapêutico estreito, monitore quanto a reações adversas, uma vez que a exposição plasmática aumentada de substratos da CYP3A4 administrados concomitantemente pode aumentar o risco de toxicidade relacionada à exposição.
A administração concomitante de rosuvastatina (substrato de BCRP) com 160 mg de lazertinibe aumentou a exposição plasmática à rosuvastatina em aproximadamente 2 vezes. As razões médias geométricas (CI de 90%) da rosuvastatina para a Cmáx e a AUC0-última foram de 2,24 (1,82, 2,76) e 2,02 (1,70, 2,40), respectivamente, quando administrada concomitantemente com lazertinibe em relação à rosuvastatina isolada. O lazertinibe é um inibidor do transportador BCRP. Para substratos sensíveis de BCRP com um índice terapêutico estreito, monitore quanto a reações adversas, uma vez que a exposição plasmática aumentada de substratos de BCRP administrados concomitantemente pode aumentar o risco de toxicidade relacionada à exposição.
A administração concomitante de metformina (substrato do OCT1) com 160 mg de lazertinibe não aumentou a exposição plasmática à metformina. As razões médias geométricas (CI de 90%) da metformina para a Cmáx e a AUC0-última foram de 0,81 (0,72, 0,91) e 0,94 (0,83, 1,06), respectivamente, quando administrada concomitantemente com lazertinibe em relação à metformina isolada. O lazertinibe não é um inibidor do transportador OCT1.
Achados in vitro sugerem que o lazertinibe pode inibir o UGT1A1; no entanto, em razão da falta de efeito sobre os níveis de bilirrubina indireta em estudos clínicos e na análise do modelo de PK baseada em fisiologia, não se espera nenhuma interação clinicamente relevante.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Lazcluze® deve ser conservado em temperatura ambiente (entre 15°C e 30°C).
Este medicamento tem validade de 24 meses a partir da data de sua fabricação. Após aberto, a apresentação de 80 mg é válida por 3 meses.
Aspecto físico
Cada comprimido revestido de Lazcluze® 80 mg é oval, amarelo, gravado em baixo relevo com "LZ" em um lado e "80" no outro lado.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Posologia -adultos (≥ 18 anos)
A dose recomendada de Lazcluze® é de 240 mg por via oral uma vez ao dia em combinação com amivantamabe até a progressão da doença ou até que o tratamento não seja mais tolerado pelo paciente.
Recomenda-se administrar o Lazcluze® a qualquer momento antes do amivantamabe quando administrados no mesmo dia. Consulte as informações de prescrição do amivantamabe para informações sobre a dosagem recomendada de amivantamabe.
Dose(s) esquecida(s)
Se uma dose de Lazcluze® for esquecida, ela pode ser administrada dentro de 12 horas. Se mais de 12 horas tiverem se passado desde o momento em que a dose deveria ser administrada, não administre a dose omitida e administre a próxima dose de acordo com o esquema posológico habitual.
Modificações de dose
As reduções de dose recomendadas para reações adversas são apresentadas na Tabela 3.
Modificações de dose para reações adversas específicas são apresentadas na Tabela 4.
Consulte as informações de prescrição do amivantamabe para informações sobre as modificações de dose para o amivantamabe.
Administração
Este medicamento é para uso oral. Engula os comprimidos inteiros, com ou sem alimentos. Não triture, divida ou mastigue o comprimido.
Se ocorrer vômito a qualquer momento após a administração de Lazcluze®, tome a próxima dose no dia seguinte.
Este medicamento não deve ser partido, aberto ou mastigado.
Populações especiais
Pacientes pediátricos (17 anos de idade ou menos)
A segurança e a eficácia do Lazcluze® não foram estabelecidas em pacientes pediátricos.
Idosos (65 anos de idade ou mais)
Entre 421 pacientes com câncer de pulmão de não pequenas células tratados com Lazcluze® em combinação com amivantamabe no NSC3003, 44,7% tinham 65 anos de idade ou mais e 11,6% tinham 75 anos ou mais. Não foi observada nenhuma diferença global na segurança ou eficácia em função da idade. Não é necessário nenhum ajuste de dose com base na idade (vide "Propriedades Farmacocinéticas").
Insuficiência renal
Não foi conduzido nenhum estudo formal de lazertinibe em pacientes com insuficiência renal.
Não é necessário nenhum ajuste de dose para pacientes com insuficiência renal leve, moderada ou severa. A PK do lazertinibe em pacientes com doença renal em estágio terminal é desconhecida (vide "Propriedades Farmacocinéticas").
Insuficiência hepática
Não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve ou moderada. A PK do lazertinibe em pacientes com insuficiência
hepática severa é desconhecida (vide "Propriedades Farmacocinéticas").
9. REAÇÕES ADVERSAS
Ao longo desta seção, reações adversas são apresentadas. Reações adversas são eventos adversos que foram considerados razoavelmente associados ao uso de lazertinibe com base na avaliação abrangente das informações sobre eventos adversos disponíveis. Uma relação causal com
o lazertinibe não pode ser confiavelmente estabelecida em casos individuais. Adicionalmente, uma vez que os estudos clínicos são conduzidas sob condições amplamente variáveis, as taxas de reações adversas observadas nos estudos clínicas de um medicamento não podem ser diretamente comparadas às taxas nos estudos clínicas de outro medicamento e podem não refletir as taxas observadas na prática clínica.
Os dados de segurança descritos abaixo refletem a exposição a Lazcluze® + amivantamabe em 421 pacientes não tratados anteriormente com CPNPC localmente avançado ou metastático cujos tumores apresentam mutações de deleção no éxon 19 ou de substituição L858R no éxon 21 do EGFR no Estudo NSC3003. A duração mediana do tratamento foi de 18,5 meses (faixa: 0,2 a 31,4 meses) para o braço de Lazcluze® + amivantamabe e a duração mediana do tratamento foi de 18 meses (faixa: 0,2 a 32,7) para o braço de osimertinibe.
As reações adversas graves em > 1% dos pacientes foram tromboembolismo venoso (11%), doença pulmonar intersticial (DPI) (2,8%), erupção cutânea (2,1%), alanina-aminotransferase aumentada (1,9%) e fadiga (1,2%). As reações adversas que levaram à descontinuação do Lazcluze® em ≥ 1%dos pacientes foram DPI (2,8%), tromboembolismo venoso (1,7%) e erupção cutânea (1,2%).
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Sintomas e sinais
A dose máxima tolerada de Lazcluze® não foi determinada. Em estudos clínicos, doses diárias de até 320 mg uma vez ao dia foram administradas.
Tratamento
Não há nenhum antídoto específico conhecido para superdosagem de Lazcluze®. No caso de uma superdosagem, interrompa o Lazcluze® e empreenda medidas gerais de suporte. Os pacientes devem ser monitorados rigorosamente quanto a sinais ou sintomas de reações adversas.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
Registro: 1.1236.3447
VENDA SOB PRESCRIÇÃO
Esta bula foi aprovada pela ANVISA em 02/07/2025