LADIZAC
DR. REDDY'S
dasatinibe
Antineoplásico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
O LADIZAC® é apresentado na forma farmacêutica de comprimidos revestidos, nas concentrações de 20 mg em embalagens contendo 60 comprimidos e 100 mg em embalagens contendo 30 comprimidos, vendidos em frasco plástico.
USO ORAL
USO ADULTO
Composição.
Cada comprimido contém 20 mg ou 100 mg de dasatinibe e os seguintes ingredientes inativos: óleo de ricino hidrogenado, celulose microcritalina, dióxido de silício, lactose, croscarmelose sódica, hiprolose e estearato de magnésio. O comprimido é revestido por hipromelose, dióxido de titânio e triacetina.
Informações técnicas.
1. INDICAÇÕES
O LADIZAC® é indicado para o tratamento de adultos com leucemia mieloide crônica¹ cromossomo Philadelphia-positivo (LMC Ph+) na fase crônica recém-diagnosticada.
O LADIZAC® é indicado para o tratamento de adultos com leucemia mieloide crônica¹ cromossomo Philadelphia-positivo (LMC Ph+) nas fases crônica, acelerada ou blástica mieloide /linfoide com resistência ou intolerância à terapia anterior incluindo imatinibe.
O LADIZAC® também é indicado para o tratamento de adultos com leucemia linfoblástica aguda² cromossomo Philadelphia- positivo (LLA Ph+) com resistência ou intolerância à terapia anterior.
¹CID C92.1 - Leucemia mieloide crônica
²CID C91.0 - Leucemia linfoblástica aguda
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança do dasatinibe foram investigadas em pacientes adultos com LMC Ph+ ou LLA Ph+, resistentes ou intolerantes ao imatinibe: 1158 pacientes tinham LMC Ph+ na fase crônica, 858 pacientes tinham LMC Ph+ na fase acelerada, fase mieloide blástica, ou fase linfoide blástica, e 130 pacientes tinham LLA Ph+. Em um estudo clínico de LMC Ph+ na fase crônica, a resistência ao imatinibe incluiu falha ao atingir a resposta hematológica completa (RHC; depois de 3 meses), resposta citogenética maior (RCM; depois de 6 meses), ou resposta citogenética completa (RCC; depois de 12 meses); ou perda de uma resposta molecular prévia (com aumento concomitante ≥ 10% em metáfases de Ph+), resposta citogenética, ou resposta hematológica. A intolerância ao imatinibe incluiu a incapacidade para tolerar 400 mg ou mais de imatinibe por dia ou a descontinuação do imatinibe devido à toxicidade.
Os resultados descritos abaixo são baseados em um mínimo de 2 anos de acompanhamento após o início do tratamento com dasatinibe em pacientes com uma média de tempo desde o início do diagnóstico de aproximadamente 5 anos. Em todos os estudos, 48% dos pacientes eram mulheres, 81% eram brancos, 15% negros ou asiáticos, 25% tinham 65 anos de idade ou mais, e 5% tinham 75 anos de idade ou mais. A maioria dos pacientes possuía um longo histórico da doença com tratamentos anteriores extensos, incluindo imatinibe, quimioterapia citotóxica, interferon, e transplante de medula óssea. Em geral, 80% dos pacientes eram resistentes ao imatinibe e 20% eram intolerantes ao imatinibe. A dose máxima de imatinibe havia sido de 400- 600 mg/dia em aproximadamente 60% dos pacientes e > 600 mg/dia em 40% dos pacientes.
O objetivo principal de eficácia na fase crônica da LMC Ph+ foi a resposta citogenética maior (RCM), definida como eliminação (resposta citogenética completa, RCC) ou diminuição substancial (de pelo menos 65%, resposta citogenética parcial) das células hematopoiéticas Ph+. O objetivo principal de eficácia na fase acelerada, fase mieloide blástica, fase linfoide blástica da LMC Ph+, e na LLA Ph+ foi a resposta hematológica maior (RHM), definida como uma resposta hematológica completa (RHC), ou como nenhuma evidência de leucemia (NEL).
LMC Ph+ em fase crônica recém-diagnosticada
Um estudo Fase 3, aberto, multicêntrico, internacional, randomizado foi realizado em pacientes adultos com LMC Ph+ fase crônica recém-diagnosticada. Os pacientes foram randomizados para receber dasatinibe 100 mg uma vez ao dia ou imatinibe 400 mg uma vez ao dia. O desfecho primário foi a taxa de resposta citogenética completa confirmada (RCCc) em 12 meses. Os desfechos secundários incluem o tempo em RCCc (medida da duração da resposta), tempo para RCCc, taxa de resposta molecular maior (RMM), tempo para RMM, sobrevida livre de progressão (SLP) e sobrevida global (SG). Outros resultados de eficácia relevantes incluem RCC e taxa de resposta molecular completa (RMC).
Um total de 519 pacientes foram randomizados em um grupo de tratamento: 259 para dasatinibe e 260 para imatinibe. As características basais foram bem balanceadas entre os dois grupos de tratamento com respeito à idade (a mediana foi de 46 anos para o grupo de dasatinibe e 49 anos para o grupo de imatinibe com 10% e 11% dos pacientes com 65 anos de idade ou mais, respectivamente), gênero (44% e 37% de mulheres, respectivamente) e raça (51% e 55% de caucasianos, 42% e 36% de asiáticos, respectivamente). O tempo mediano do diagnóstico inicial de LMC Ph+ até a randomização foi 1 mês (0,03 - 9,72) para dasatinibe e 1 mês (0,10 - 8,02) para imatinibe. Pacientes com história de doença cardíaca grave foram incluídos neste estudo, exceto aqueles que tiveram infarto do miocárdio nos últimos 6 meses, insuficiência cardíaca congestiva ou angina descontrolada nos últimos 3 meses, arritmias significantes ou prolongamento do intervalo QT. Na base, a distribuição dos escores de Hasford foi similar nos grupos de tratamento com dasatinibe e imatinibe (baixo risco: 33% e 34%, risco intermediário: 48% e 47% e alto risco: 19% e 19%, respectivamente). O status de Performance (ECOG) foi também similar nos grupos de tratamento com dasatinibe e imatinibe (ECOG 0 = 82% e 79%, ECOG 1 = 18% e 20% e ECOG 2 = 0 e 1%, respectivamente).
Com 12 meses de acompanhamento mínimo, 85% dos pacientes randomizados para o grupo de dasatinibe e 81% dos pacientes randomizados no grupo do imatinibe ainda estavam recebendo tratamento em primeira linha. A descontinuação devido a progressão da doença ocorreu em 3% dos pacientes tratados com dasatinibe e 5% dos pacientes tratados com imatinibe. Com acompanhamento mínimo de 60 meses, 61% dos pacientes randomizados para o tratamento com dasatinibe e 63% dos pacientes randomizados para o tratamento com o imatinibe ainda estavam recebendo tratamento em primeira linha. A descontinuação devido a progressão da doença ocorreu em 7% dos pacientes tratados com dasatinibe e 9% em pacientes tratados com imatinibe. Descontinuação devido à falha ao tratamento ocorreu em 4% dos pacientes tratados com dasatinibe e 5% dos pacientes tratados com imatinibe. Descontinuação devido à intolerância ao tratamento ocorreu em 16% dos pacientes tratados com dasatinibe e 7% dos pacientes tratados com imatinibe.
Os resultados de eficácia estão apresentados na Tabela 1. Uma proporção maior de paciente estatisticamente significativa no grupo de dasatinibe alcançou a RCCc comparado com pacientes no grupo de imatinibe dentro dos primeiros 12 meses de tratamento. A eficácia de dasatinibe foi consistentemente demonstrada entre os diferentes subgrupos, incluindo idade, gênero e escores basais de Hasford.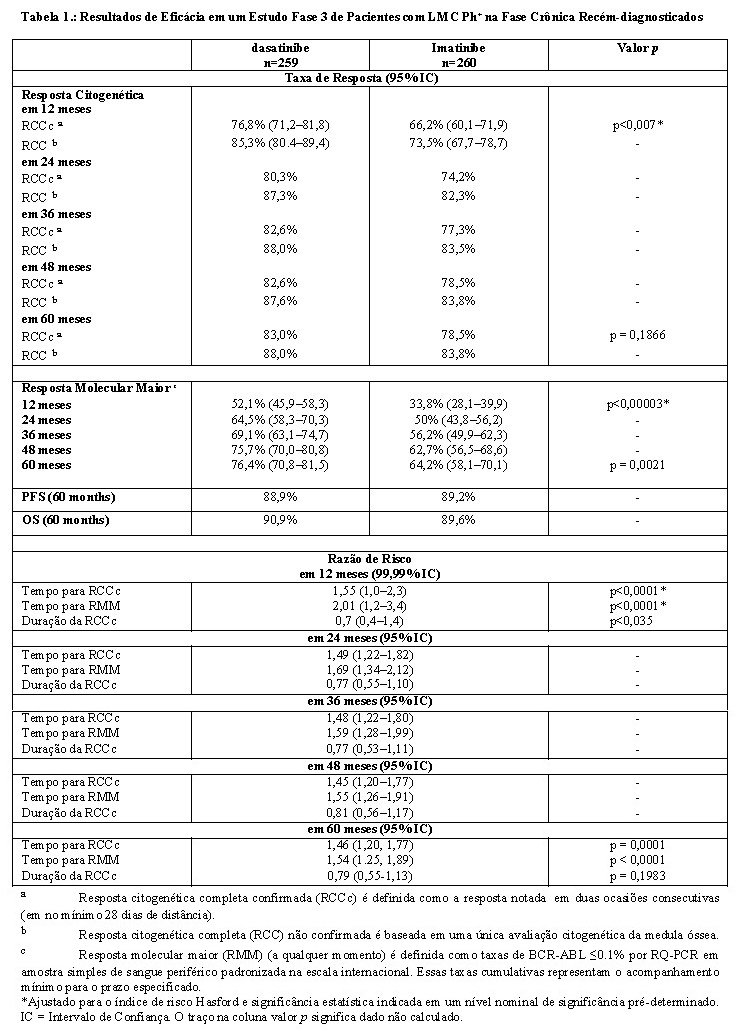
A progressão da doença foi definida como o aumento das células brancas do sangue apesar de manejo terapêutico apropriado, perda de RHC, perda de RCM, progressão para fase acelerada ou fase blástica, ou morte. Com um mínimo de 60 meses de acompanhamento, transformação para fase blástica ou acelerada ocorreu menos frequentemente com dasatinibe (n=8, 3,1%) do que com imatinibe (n=15, 5,8%).
Após 60 meses de acompanhamento, o tempo médio para RCCc foi 3,1 meses em 214 respondentes ao dasatinibe e 5,8 meses em 204 respondentes ao imatinibe. O tempo médio para RMM após 60 meses de acompanhamento foi 9,3 meses em 196 respondentes ao dasatinibe e 15,0 meses em 163 respondentes ao imatinibe. O tempo para RMM foi consistentemente menor com pacientes tratados com dasatinibe comparado com pacientes tratados com imatinibe. Com um acompanhamento mínimo de 60 meses, a taxa de RMC (isto é, ao menos uma redução de 4,5-log a partir do valor base padronizado de relação BCR-ABL ≤0.0032%) a qualquer momento foi 44% e 34% no grupo tratado com dasatinibe e com imatinibe, respectivamente.
A taxa de RMM a qualquer momento, em cada grupo de risco determinado pelo escore de Hasford, foi maior no grupo de dasatinibe comparada ao grupo de imatinibe (baixo risco: 90% e 69%; risco intermediário: 71% e 65%; alto risco: 67% e 54%, respectivamente).
A taxa de SLP foi consistentemente maior em pacientes tratados com dasatinibe que atingiram o nível de BCR-ABL ≤10% em 3
meses dos que não atingiram.
A taxa de SG foi consistentemente maior em pacientes tratados com dasatinibe que atingiram nível de BCR-ABL≤10% do que
aqueles que não atingiram.
Taxas de RMM foram consistentemente maiores nos pacientes tratados com dasatinibe comparado com os pacientes tratados com imatinibe.
A taxa estimada de SLP de 60 meses foi de 88,9% (IC: 84,0%-92,4%) e 89,2% (IC: 84,3%-92,7%) para o grupo de tratamento com o dasatinibe e com o imatinibe, respectivamente. As taxas estimadas de sobrevida de 60 meses para os pacientes tratados com dasatinibe e imatinibe foram 90,9% (IC: 86,6%-93,8%) e 89,6% (IC: 85,2%-92,8%), respectivamente. No acompanhamento mínimo de 60 meses, não houve diferença entre dasatinibe e imatinibe na SG (HR 1,01, 95% IC: 0,58-1,73, p=0,9800) ou SLP (HR 1,00, 95% IC 0,58-1,72, p=0,9998).
O número de pacientes no grupo dasatinibe e imatinibe que apresentaram progressão/falha ao tratamento foi 34 (13,1%) e 39 (15%)
pacientes, respectivamente.
Após 5 anos de acompanhamento, menos pacientes tratados com dasatinibe transformaram para fase acelerada ou blástica comparado aos pacientes tratados com imatinibe. Após 12, 24, 36, 48 e 60 meses de tratamento, transformação foi reportada em um total de 5 (1,9%), 6 (2,3%), 8 (3,1%), 8 (3,1%) e 8 (3,1%) pacientes tratados com dasatinibe comparado com 9 (3,5%), 13 (5,0%), 13 (5,0%), 14(5,4%) e 15 (5,8%) pacientes tratados com imatinibe, respectivamente. Notavelmente, não houve transformação adicional após 36 meses no grupo com dasatinibe, 2 pacientes adicionais apresentaram transformação no grupo com imatinibe após 36 meses (ambos entre o Ano 4 e 5).
O Tempo até o Benefício Clínico Máximo (TBCM) avalia as razões cumulativas para a descontinuação do tratamento. No estudo CA180056, TBCM foi definido como o tempo desde a randomização até o último dia de medicação no estudo para os pacientes que descontinuaram devido à toxicidade, ou a primeira data na qual o critério para falha ao tratamento foi atingido. Com um período de acompanhamento mínimo de 5 anos, um maior número de indivíduos foi descontinuado do estudo por causa de progressão, falha do tratamento ou intolerância no grupo de dasatinibe em comparação com o grupo de imatinibe (74 vs. 55, respectivamente). Tal diferença do tratamento não se manifestou até depois de 2 anos, momento no qual as curvas de Kaplan-Meier começaram a se separar resultando em um risco maior de eventos de progressão, falha do tratamento ou intolerância ao medicamento para o grupo dasatinibe em comparação ao grupo imatinibe [HR (IC de 95%): 1,35 (0,95-1,91)].
No estudo de Fase 3 de LMC Ph+ na fase crônica recém-diagnosticada, o sequenciamento de BCR-ABL foi realizado em amostras de sangue de pacientes que descontinuaram a terapia com dasatinibe e imatinibe. Entre os pacientes tratados com dasatinibe, as mutações detectadas foram T315I (4%), F317I/L (2%) e V299L (3%).
O dasatinibe, imatinibe e nilotinibe não parecem ser ativos contra a mutação T315I, baseado em dados in vitro.
Adicionalmente, mais pacientes tratados com dasatinibe (84%) atingiram a resposta molecular precoce (definida como níveis de BCR-ABL ≤10% em 3 meses) comparado com os sujeitos tratados com imatinibe (64%). Os pacientes que atingiram resposta molecular precoce em ambos os grupos tiveram menor risco de transformação, maior taxa de sobrevida livre de progressão (SLP) e maior taxa de sobrevida global (SG), como demonstrado nas Tabelas 2 e 3.

Outros estudos de longo prazo com pacientes com leucemia mieloide crônica recém diagnosticados estão em andamento.
LMC Ph+ fase crônica
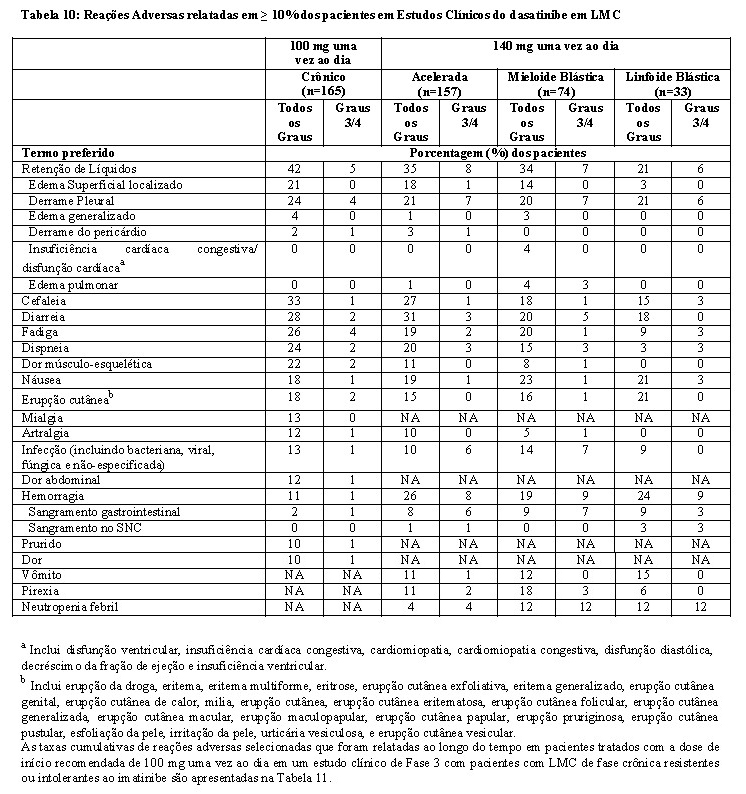
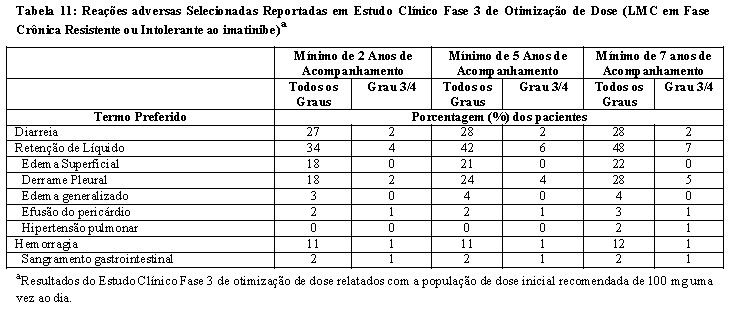
- Estudo de otimização da dose: Um estudo Fase 3 randomizado, aberto, foi conduzido em pacientes com LMC Ph+ em fase crônica para avaliar a segurança e eficácia de dasatinibe administrado uma vez ao dia comparado com dasatinibe administrado duas vezes ao dia. Pacientes com doenças cardíacas significantes incluindo o infarto do miocárdio em 6 meses, insuficiência cardíaca congestiva em 3 meses, arritmias significantes ou prolongamento intervalo QTc foram excluídos do estudo. O objetivo principal de eficácia foi resposta citogenética maior (RCM) em pacientes com LMC Ph+ resistentes ao imatinibe. Um total de 670 pacientes, dos quais 497 eram resistentes ao imatinibe, foram randomizados nos seguintes grupos: 100 mg uma vez ao dia, 140 mg uma vez ao dia, 50 mg duas vezes ao dia ou 70 mg de dasatinibe duas vezes ao dia. A média da duração do tratamento foi de 22 meses (faixa de < 1 a 31 meses).
A eficácia foi atingida em todos os grupos tratados com dasatinibe uma vez ao dia demonstrando eficácia comparável (não inferioridade) ao tratamento com dasatinibe duas vezes ao dia para o objetivo principal de eficácia (diferença da RCM 1,9%; 95% de intervalo de confiança [- 6,8% a 10,6%].), entretanto, o regime de 100 mg uma vez ao dia demonstrou segurança e tolerabilidade melhorada. Resultados de eficácia para pacientes com LMC Ph+ na fase crônica que receberam a dose inicial recomendada de 100 mg uma vez ao dia estão demonstrados na Tabela 4 e 5.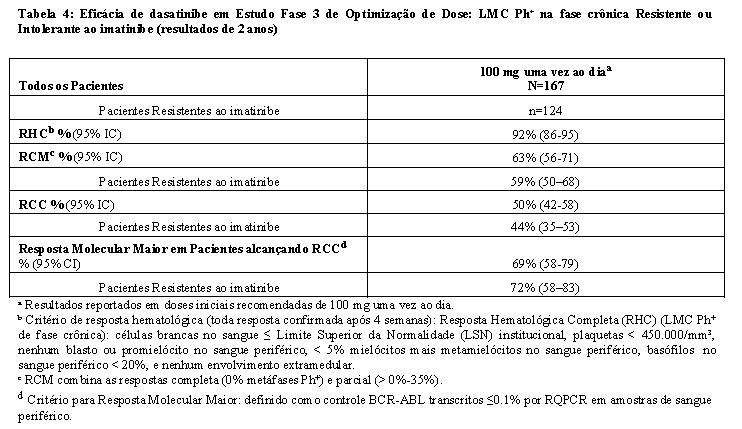
A eficácia foi avaliada também em pacientes que eram intolerantes ao imatinibe. Nesta população de paciente que recebeu 100 mg
uma vez ao dia, RCM foi alcançada em 77% e RCC em 67% de pacientes com acompanhamento mínimo de 2 anos.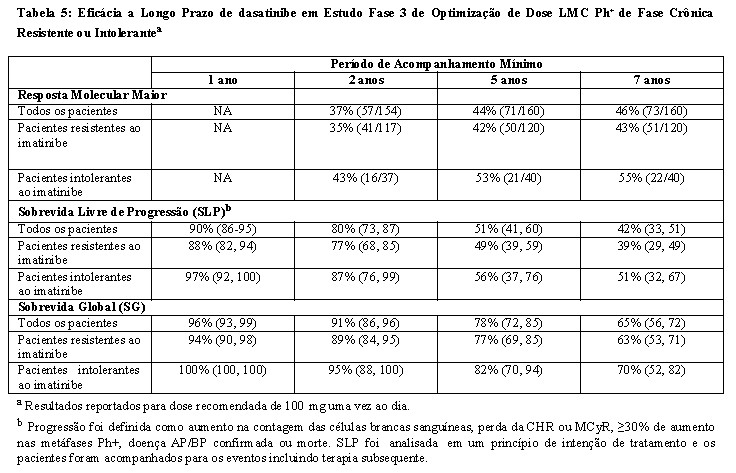
Por sete anos, a transformação quer para a fase acelerada ou para fase blástica ocorreram em nove pacientes em tratamento.
LMC Ph+ fase avançada e LLA Ph+
- Estudo de otimização da dose: Um estudo Fase 3 randomizado, aberto, foi conduzido em pacientes com LMC Ph+ em fase avançada (LMC Ph+ na fase acelerada, LMC Ph+ na fase mieloide blástica, ou LMC Ph+ na fase linfoide blástica) ou LLA Ph+ para avaliar a eficácia e segurança de dasatinibe administrado uma vez ao dia comparado com dasatinibe administrado duas vezes ao dia. O objetivo principal de eficácia foi resposta hematológica maior (RHM). Um total de 611 pacientes foram randomizados nos grupos de 140 mg de dasatinibe uma vez ao dia ou de 70 mg de dasatinibe duas vezes ao dia. A média da duração do tratamento foi de aproximadamente 6 meses para os dois grupos de tratamento (faixa de < 1 a 31 meses). O grupo tratado com dasatinibe uma vez ao dia demonstrou eficácia comparável (não inferioridade) ao tratamento com dasatinibe duas vezes ao dia para o objetivo principal de eficácia, entretanto, o regime de 140 mg uma vez ao dia demonstrou segurança e tolerabilidade melhorada.
As taxas de resposta para pacientes no grupo de 140 mg uma vez ao dia estão apresentadas na Tabela 6.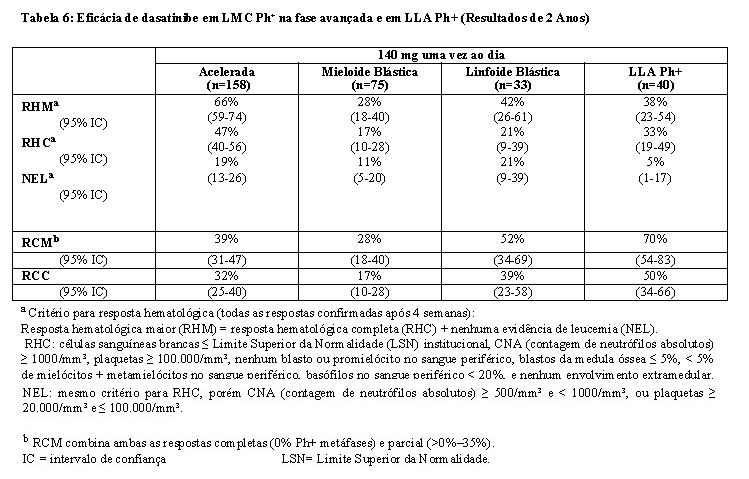
Em paciente com LMC Ph+ na fase acelerada tratados com o regime de 140 mg uma vez ao dia, a duração média da RHM e a sobrevida global (SG) média não foi atingida; a sobrevida livre de progressão (SLP) média foi de 25 meses. Em pacientes com LMC Ph+ na fase blástica mielóide tratados com o regime de 140 mg uma vez ao dia, a duração média da RHM foi 8 meses, a média da SLP foi 4 meses e a média de sobrevida global foi 8 meses. Em pacientes com LMC Ph+ na fase blástica linfóide, a duração média da RHM foi 5 meses, a média da SLP foi 5 meses e a sobrevida global média foi 11 meses. Em pacientes com LLA Ph+ tratados com o regime de 140 mg uma vez ao dia, a duração da RHM foi 5 meses, a média de SLP foi 4 meses e a média de sobrevida global foi 7 meses.
3. CARACTERÍSTICAS FARMACOLÓGICAS
O dasatinibe é inibidor de quinase. O nome químico do dasatinibe é N-(2-cloro-6-metilfenil)-2-[[6-[4-(2-hidroxietil)-1-piperazinil]- 2-metil-4-pirimidinil]amino]-5-tiazolecarboxamida, monoidratado. A fórmula molecular é C22H26CIN7O2S • H2O, o que corresponde a um peso de 506,02 (monoidratado). A base anidra livre possui um peso molecular de 488,01. O dasatinibe possui a seguinte estrutura química: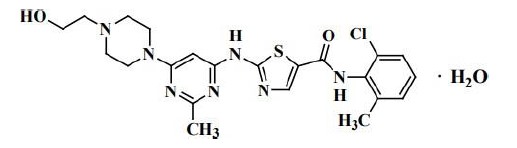
O dasatinibe é um pó branco a quase branco. O fármaco é insolúvel em água e levemente solúvel em etanol e metanol.
Mecanismo de Ação
O dasatinibe, em concentrações nanomolares, inibe as seguintes quinases: BCR-ABL, família SRC (SRC, LCK, YES, FYN), c-KIT, EPHA2, e PDGFRb. Com base em estudos modelo, o dasatinibe previsivelmente liga-se a conformações múltiplas da quinase ABL. In vitro, o dasatinibe é ativo em linhagens celulares leucêmicas representando variações da doença sensível e resistente ao mesilato de imatinibe. O dasatinibe inibiu o crescimento de linhagens celulares de Leucemia Mieloide Crônica (LMC) e Leucemia Linfoblástica Aguda Cromossomo Philadelphia-positivo (LLA Ph+) com superexpressão de BCR-ABL. Sob as condições dos ensaios, o dasatinibe foi capaz de superar a resistência ao imatinibe resultante das mutações no domínio da quinase do BCR-ABL, ativação das etapas de sinalização alternativas envolvendo as quinases da família SRC (LYN, HCK) e a superexpressão do gene de resistência a múltiplos medicamentos.
Farmacocinética
-Absorção
As concentrações plasmáticas máximas (Cmax) de dasatinibe são observadas entre 0,5 e 6 horas (TRmax), após a administração oral. O dasatinibe exibe aumentos na AUC proporcionais à dose e características de eliminação lineares na faixa de dose de 15 mg a 240 mg/dia. A média da meia-vida terminal geral do dasatinibe é de 3 a 5 horas.
Dados de um estudo realizado com 54 indivíduos sadios que receberam uma dose única de 100 mg de dasatinibe 30 minutos após o consumo de uma refeição rica em gorduras indicaram um aumento de 14% na AUC média do dasatinibe. O consumo de uma refeição com baixo teor de gordura 30 minutos antes do dasatinibe resultou num aumento de 21% na AUC média do dasatinibe. Os efeitos dos alimentos não foram clinicamente relevantes. A variabilidade da exposição ao dasatinibe é mais elevada em condições de jejum (47% CV) em comparação à refeição com baixo teor de gordura (39% CV) e à refeição rica em gorduras (32% CV).
Com base na análise farmacocinética da população de pacientes, estimou-se que a variabilidade da exposição ao dasatinibe se deve principalmente à variabilidade inter-ocasião da biodisponibilidade (44% CV) e, em menor medida, à variabilidade interindividual da biodisponibilidade e do clearance (32% e 30% CV, respectivamente). Não é esperado que a variabilidade aleatória inter-ocasião na exposição afete a exposição e a eficácia cumulativas.
- Distribuição
Nos pacientes, o dasatinibe apresenta um volume aparente de distribuição de 2505 L (CV% 93%), sugerindo que a droga é extensamente distribuída para o espaço extravascular. A ligação do dasatinibe e o seu metabólito ativo às proteínas plasmáticas humanas in vitro foi aproximadamente 96% e 93%, respectivamente, sem dependência da concentração no intervalo de 100 - 500 ng/ mL.
- Metabolismo
O dasatinibe é extensamente metabolizado em humanos, principalmente pela enzima 3A4 do citocromo P450. CYP3A4 é a principal enzima responsável pela formação do metabólito ativo. As enzimas mono-oxigenase 3 flavina (FMO-3) e a difosfato uridina glucuronosiltransferase (UGT) também estão envolvidas na formação dos metabólitos do dasatinibe.
A exposição do metabólito ativo, a qual é equipotente ao dasatinibe, representa aproximadamente 5% da AUC do dasatinibe. Isso indica que o metabólito ativo do dasatinibe dificilmente terá alguma função importante na farmacologia observada da droga. O dasatinibe apresenta diversos outros metabólitos oxidativos inativos.
O dasatinibe é um fraco inibidor da CYP3A4 dependente do tempo. Em concentrações clinicamente relevantes, dasatinibe não inibe CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, ou 2E1. O dasatinibe não é um indutor das enzimas CYP humanas.
- Eliminação
A meia-vida terminal média de dasatinibe é de 3 a 5 horas. O clearance oral médio aparente é de 363,8 L/h (CV% 81,3%). A eliminação dá-se principalmente pelas fezes. Após uma dose oral única de dasatinibe marcado com [14C], aproximadamente 4% e 85% da radioatividade administrada foi recuperada na urina e nas fezes, respectivamente, dentro de 10 dias. O dasatinibe inalterado contabilizou 0,1% e 19% da dose administrada na urina e fezes, respectivamente, sendo que o restante da dose corresponde aos metabólitos.
- Efeitos da idade e gênero
Análises farmacocinéticas dos dados demográficos indicam que não há efeitos clinicamente relevantes sobre a idade e sexo da farmacocinética do dasatinibe.
A farmacocinética do dasatinibe não foi avaliada em pacientes pediátricos.
- Insuficiência Hepática
Doses de 50 mg e 20 mg de dasatinibe foram avaliadas em oito pacientes com insuficiência hepática moderada (Child-Pugh Classe B) e sete pacientes com insuficiência hepática grave (Child-Pugh Classe C), respectivamente. Controles combinados com função hepática normal (n=15) também foram avaliados e receberam uma dose de 70 mg de dasatinibe. Comparados a sujeitos com função hepática normal, pacientes com insuficiência hepática moderada tiveram decréscimos na Cmax de normalização da dose e AUC de 47% e 8%, respectivamente. Pacientes com insuficiência hepática grave tiveram um decréscimo de 43% na Cmax de normalização da dose e de 28% na AUC comparado aos controles normais.
Estas diferenças na Cmax e AUC não são clinicamente relevantes. O ajuste de dose não é necessário em pacientes com insuficiência hepática. Devido a limitações deste estudo clínico, deve-se ter cautela ao administrar dasatinibe a pacientes com insuficiência hepática.
4. CONTRAINDICAÇÕES
O LADIZAC® é contraindicado em pacientes com hipersensibilidade ao dasatinibe ou a qualquer outro componente da formulação.
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais
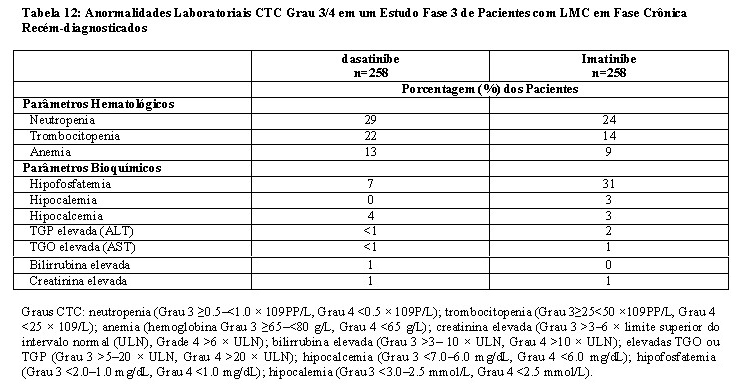
Mielossupressão
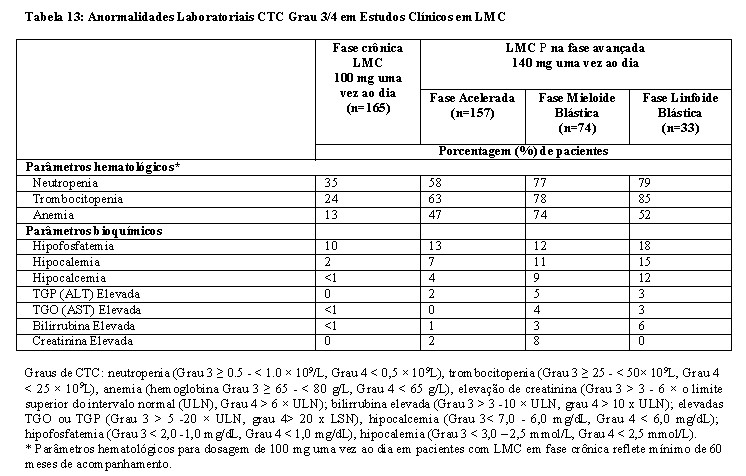
O tratamento com dasatinibe está associado com trombocitopenia, neutropenia e anemia graves (graus 3 e 4), que ocorrem mais cedo e mais frequentemente em pacientes com LMC na fase avançada ou LLA Ph+ do que em pacientes com LMC na fase crônica. Em pacientes adultos com LMC na fase avançada ou LLA Ph+ tratados com LADIZAC®, hemogramas completos devem ser realizados semanalmente pelos 2 primeiros meses, e mensalmente depois disso ou conforme indicado clinicamente. Em pacientes adultos com LMC na fase crônica, hemogramas completos devem ser realizados a cada 2 semanas por 12 semanas, e então a cada 3 meses depois disso ou conforme indicado clinicamente. A mielossupressão é geralmente reversível e tratada suspendendo-se temporariamente o LADIZAC® ou reduzindo-se a dose (vide 8. POSOLOGIA E MODO DE USAR e 9. REAÇÕES ADVERSAS).
Eventos Relacionados com Sangramento
Em pacientes com LMC fase crônica, hemorragia severa ocorreu em 5 pacientes (1%) tomando dasatinibe na dose recomendada (n=548).
Além de causar trombocitopenia em humanos, o dasatinibe causou disfunção plaquetária in vitro. Em todos os estudos clínicos de LMC ou LLA Ph+, hemorragias graves (graus 3 e 4) do SNC (sistema nervoso central), incluindo casos fatais, ocorreram em 1% dos pacientes tomando dasatinibe. Hemorragia gastrintestinal grave ocorreu em 6% dos pacientes e geralmente exigiu interrupções do tratamento e transfusões. Outros casos de hemorragia grave ocorreram em 2% dos pacientes. A maioria dos eventos relacionados com sangramento nos estudos clínicos foi tipicamente associada com trombocitopenia grave. Em pacientes com LMC em fase crônica recém-diagnosticados tratados com dasatinibe, dos 6 pacientes que apresentaram eventos de sangramento Grau 3 ou 4, 2 apresentaram evento de sangramento dentro de 3 dias do relato de trombocitopenia severa (Grau 3 ou 4). Estes eventos com contagem de plaquetas associada foram hemorragia GI baixa no dia antes a contagem de plaqueta Grau 4 e hemorragia GI no mesmo dia da contagem de plaqueta Grau 3.
Os pacientes tomando medicamentos que inibem a função plaquetária ou anticoagulantes foram excluídos da participação em estudos clínicos iniciais com dasatinibe. Em estudos posteriores, o uso de anticoagulantes, aspirina, e antiinflamatórios não esteroidais (AINEs) foi permitido concomitantemente com dasatinibe se a contagem de plaquetas fosse > 50000-75000 mm³. Deve- se ter cautela se os pacientes necessitarem tomar medicamentos que inibem a função plaquetária ou anticoagulantes.
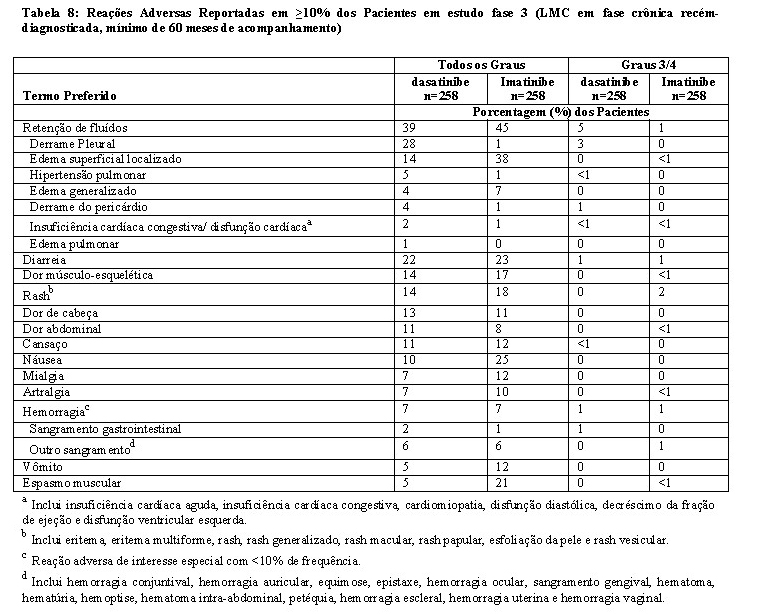
Retenção de Líquidos
O dasatinibe está associado com a retenção de líquidos. Após 5 anos de acompanhamento do estudo Fase 3 de LMC em fase crônica recém-diagnosticada (n=258), retenção de líquidos grave foi relatada em 13 pacientes (5%) tomando dasatinibe comparado com 2 pacientes (1%) tomando imatinibe (n=258). O evento adverso relacionado com retenção de líquidos mais comumente relatado foi derrame pleural (28,3%) no grupo com dasatinibe. O tempo mediano para a primeira ocorrência de derrame pleural Grau 1 ou 2 relacionado ao medicamento foi 114 semanas (faixa entre 4 - 299 semanas). Em pacientes com derrame pleural ≥ Grau 3 relacionado ao medicamento, o tempo até a primeira ocorrência foi 175 semanas (faixa entre 114 a 274 semanas). Pacientes com derrame pleural relacionados ao medicamento foram manejados com uma ou mais das opções seguintes: interrupção da dose (61,6%); redução da dose (41,1%); diuréticos (46,6%); corticosteroides (31,5%); combinação de diuréticos e corticosteroides (27,4%); e toracocentese terapêutica (12,3%).
Em todos os pacientes com LMC na fase crônica recém-diagnosticados ou resistentes ou intolerantes ao imatinibe (n=548), a retenção de líquidos grave ocorreu em 32 (6%) dos pacientes tratados com dasatinibe na dose recomendada (n=548). Em pacientes com LMC ou LLA Ph+ tratados com dasatinibe na dose recomendada (n=304), retenção de líquido grave foi relatada em 8% dos pacientes, incluindo derrame pleural e do pericárdio grave relatados em 7% e 1% dos pacientes, respectivamente. Eventos de edema pulmonar grave e hipertensão pulmonar grave foram relatados, cada um, em 1% dos pacientes.
Os pacientes que desenvolverem sintomas sugestivos de derrame pleural ou outra retenção de líquido, como dispneia nova ou piorada durante esforço ou em repouso, dor pleurítica ou tosse seca devem ser avaliados imediatamente por radiografia de tórax ou exame de imagem adicional conforme apropriado. Derrame pleural grave pode necessitar toracentese e terapia com oxigênio. Modificação da dose deve ser considerada. (vide 8.POSOLOGIA E MODO DE USAR). Eventos de retenção de líquidos foram tipicamente tratados com medidas de suporte, que pode incluir diuréticos ou administrações de esteróides por períodos curtos.
Prolongamento do intervalo QT
Dados in vitro sugerem que o dasatinibe tem potencial para prolongar a repolarização ventricular cardíaca (intervalo QT).
Após 5 anos de acompanhamento do estudo clínico Fase 3 de LMC em fase crônica recém-diagnosticada, 1 paciente ( < 1%) em cada grupo de tratamento com dasatinibe (n=258) e com imatinibe (n=258) teve prolongamento do intervalo QTc reportado com reação adversa. As alterações médias no QTcF foram 3,0 mseg em pacientes tratados com dasatinibe comparado com 8,2 mseg em pacientes tratados com imatinibe. Um paciente ( < 1%) em cada grupo apresentou um QTcF > 500 mseg.
Em 865 pacientes com leucemia tratados com dasatinibe em estudos de Fase II, as alterações médias em QTcF da linha de base, usando o método de Fridericia (QTcF) foram de 4-6 mseg, os intervalos de confiança (ICs) superiores a 95% para todas as alterações médias da linha de base foram 500 mseg.
O LADIZAC® deve ser administrado com cuidado em pacientes que apresentam ou que podem apresentar prolongamento do intervalo QTc. Estes incluem pacientes com hipocalemia ou hipomagnesemia, pacientes com síndrome congênita de QT longo, pacientes tomando medicamentos antiarrítmicos ou outros medicamentos que possam levar ao prolongamento do intervalo QT e terapia cumulativa com altas doses de antraciclina. Hipocalemia ou hipomagnesemia devem ser corrigidas anteriormente à administração de LADIZAC®.
Reativação da hepatite B
TKIs BCR-ABL estão sendo associados com a reativação do vírus da hepatite B (HBV) incluindo casos individuais reportados para dasatinibe. Em algumas instâncias, a reativação do HBV ocorrendo em conjunto com a terapia com outros TKIs BCR-ABL resultou em falência hepática aguda ou hepatite fulminante levando a transplante de fígado ou a um desfecho fatal.
A pesquisa para presença de HBV deve ser considerada de acordo com as diretrizes publicadas antes de iniciar a terapia com LADIZAC®. A consulta com um médico experiente no tratamento de hepatite B é recomendada para pacientes que tiveram sorologia positiva para HBV.
Pacientes que carregam o HBV e requerem tratamento com TKIs BCR-ABL devem ser cuidadosamente monitorados para sinais clínicos e laboratoriais de infecção ativa de HBV durante o tratamento e por vários meses após o término da terapia. Em pacientes que desenvolveram reativação do HBV enquanto estavam tomando dasatinibe, recomenda-se uma consulta imediata com um médico experiente no tratamento de HBV.
Reações adversas cardíacas
O dasatinibe foi estudado em um estudo clínico randomizado de 519 pacientes com LCM na fase crônica recém-diagnosticados que incluíam pacientes com doença cardíaca prévia. As reações adversas cardíacas de falência cardíaca congestiva/ disfunção cardíaca, derrame do pericárdio, arritmias, palpitações, prolongamento do intervalo QT e infarto do miocárdio (incluindo fatal) foram reportadas em pacientes tomando dasatinibe. Eventos adversos cardíacos foram mais frequentes em pacientes com fatores de risco ou com histórica médica prévia de doença cardíaca. Pacientes com fatores de risco (como hipertensão, hiperlipidemia, ou diabetes) ou história de doença cardíaca (como intervenção coronária percutânea prévia ou doença arterial coronariana) devem ser monitorados cuidadosamente para sinais e sintomas consistentes com disfunção cardíaca como dor no peito, falta de ar e diaforese. Se estes sinais e sintomas clínicos ocorrerem, aconselha-se a interrupção da administração de LADIZAC®. Após a resolução, uma avaliação funcional deve ser realizada antes da retomada do tratamento com LADIZAC®. O tratamento com LADIZAC® pode ser retomado nas doses originais se os eventos forem leves ou moderados (≤ Grau 2) ou retomado com redução de dose se o evento for severo (≥ Grau 3). Pacientes com doença cardiovascular significante ou descontrolada não foram incluídos nos estudos clínicos.
Reações Dermatológicas Graves
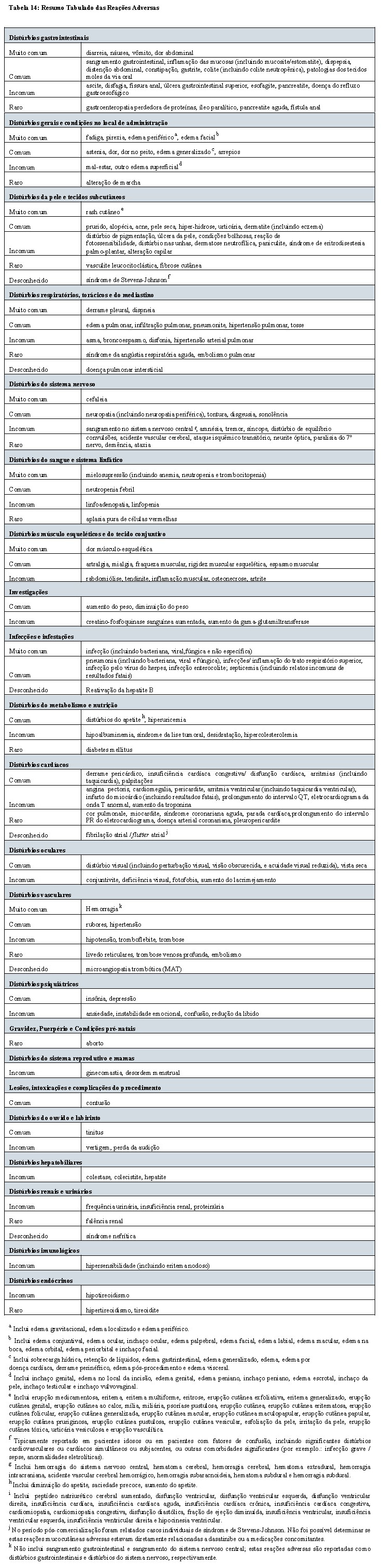
Casos individuais de reações dermatológicas mucocutâneas graves, incluindo síndrome de Stevens-Johnson e eritema multiforme, foram reportadas com o uso de dasatinibe.
O LADIZAC® deve ser descontinuado permanentemente em pacientes com experiência de reação mucocutânea grave durante o tratamento se nenhuma outra etiologia for identificada.
Hipertensão Arterial Pulmonar
Hipertensão arterial pulmonar (HAP), confirmada por cateterização cardíaca direita, foi relatada em associação ao tratamento com dasatinibe. Nestes casos, a HAP foi relatada após o início da terapia de dasatinibe, incluindo manifestações depois de mais de um ano de tratamento. Muitas vezes, os pacientes com HAP relatada durante o tratamento com dasatinibe estavam tomando medicações concomitantes ou tinham comorbidades em complemento à malignidade subjacente.
Pacientes devem ser avaliados quanto à presença de sinais e sintomas de doença cardiopulmonar subjacente antes de iniciar a terapia com LADIZAC®. Pacientes que desenvolvem dispneia e fadiga após o início da terapia devem ser avaliados quanto às etiologias mais comuns, incluindo derrame pleural, edema pulmonar, anemia, ou infiltração no pulmão. Durante esta avaliação, as diretrizes para o controle de reações adversas não-hematológicas devem ser seguidas (vide 8. POSOLOGIA E MODO DE USAR). Caso a reação adversa seja grave, o tratamento deverá ser suspenso até que o evento tenha evoluído para a resolução ou melhorado. Caso não seja encontrado diagnóstico alternativo, o diagnóstico de HAP deverá ser considerado. Caso a HAP seja confirmada, LADIZAC® deve ser permanentemente descontinuado. Acompanhamento deve ser realizado de acordo com as diretrizes de prática padrão. Melhoras nos parâmetros clínicos e hemodinâmicos foram observadas em pacientes tratados com dasatinibe com HAP após a interrupção da terapia de dasatinibe.
Lactose
O LADIZAC® contém 20 mg de lactose em uma dose de 100 mg ao dia e 28 mg de lactose monoidratada em uma dose de 140 mg ao dia.
Uso Pediátrico
A segurança e eficácia do dasatinibe em pacientes com menos de 18 anos de idade não foram estabelecidas.
Uso Geriátrico
Nenhuma diferença na RCC e RMM confirmadas foram observadas entre pacientes jovens e idosos. Dos 2712 pacientes nos estudos clínicos de dasatinibe, 617 (23%) tinham 65 anos de idade ou mais e 123 (5%) tinham 75 anos ou mais. Enquanto o perfil de segurança de dasatinibe na população geriátrica foi similar à população mais jovem, pacientes com 65 anos de idade ou mais foram mais propensos a apresentar as reações adversas mais comumente relatadas: fadiga, derrame pleural, dispneia, tosse, hemorragia gastrointestinal baixa, e distúrbio de apetite, e são mais prováveis de experienciar as reações adversas menos frequentemente reportadas: distensão abdominal, tontura, efusão do pericárido, insuficiência cardíaca congestiva e perda de peso, e devem ser cuidadosamente monitorados.
Insuficiência hepática
O efeito da insuficiência hepática na farmacocinética do dasatinibe foi avaliado em voluntários saudáveis com função normal do fígado e pacientes com moderada (Child-Pugh Classe B) e grave (Child-Pugh Classe C) insuficiência hepática. Comparado aos voluntários saudáveis com função hepática normal, os parâmetros farmacocinéticos de normalização da dose foram diminuídos nos pacientes com insuficiência hepática.
Nenhum ajuste de dose é necessário em pacientes com insuficiência hepática (vide 3. CARACTERÍSTICAS FARMACOLÓGICAS - Farmacocinética). Recomenda-se cuidado ao administrar LADIZAC® a pacientes com insuficiência hepática.
Insuficiência Renal
No momento não existem estudos clínicos com dasatinibe em pacientes com insuficiência renal. Menos de 4% do dasatinibe e seus
metabólitos são excretados pelos rins.
Carcinogênese, Mutagênese, Comprometimento da Fertilidade
- Carcinogênese
Em um estudo de carcinogenicidade de 2 anos, foram administradas a ratos doses orais de dasatinibe de 0,3; 1 e 3 mg/kg/dia. A maior dose resultou em níveis de exposição plasmática da droga (AUC) de aproximadamente 60% da exposição humana com 100 mg uma vez ao dia. Foi observado um aumento estatiscamente significativo na incidência combinada de carcinoma de células escamosas e papilomas no útero e colo de doses elevadas em fêmeas e de adenoma de próstata de baixas doses em machos. A relevância desses achados do estudo de carcinogenicidade em ratos para humanos não é conhecida.
- Mutagênese
O dasatinibe foi clastogênico quando testado in vitro em células de ovário de hamster chinês, com ou sem ativação metabólica. O dasatinibe não foi mutagênico quando testado em um ensaio de células bacterianas in vitro (teste de Ames) e não foi genotóxico em um estudo em células de micronúcleo de ratos in vivo.
- Comprometimento da Fertilidade
O dasatinibe não afetou a fertilidade masculina ou feminina em um estudo de fertilidade convencional e de desenvolvimento embrionário inicial em ratos, mas induziu a letalidade embrionária em exposição à droga plasmática (AUC) similar às exposições em humanos na dose de 100 mg uma vez ao dia. Em estudos de desenvolvimento embriofetal, o dasatinibe também induziu a letalidade embrionária com diminuições associadas no tamanho das ninhadas em ratos, bem como alterações esqueléticas fetais em ratos e coelhos. Estes efeitos ocorreram em doses que não produziram toxicidade materna, indicando que o dasatinibe é uma substância tóxica seletiva para reprodução a partir da implantação até a conclusão da organogênese. Em um estudo exploratório de desenvolvimento pré e pós natal, a exposição indireta de filhotes de ratos ao dasatinibe (no útero ou durante o aleitamento) iniciando a partir do final da organogênese até o início da lactação era incompatível com a sobrevivência dos filhotes, mesmo com exposições maternas que são subterapêutica.
Os efeitos potenciais de dasatinibe no esperma foram avaliados em um estudo oral de fertilidade e desenvolvimento embrionário inicial em ratos. O dasatinibe não é uma substancia tóxica para a reprodução em ratos machos em exposições clinicamente relevantes. No entanto, os dados da avaliação da toxicidade reprodutiva em pacientes do sexo masculino em tratamento com dasatinibe são limitados.
Homens e mulheres com vida sexual ativa e em idade fértil devem usar meios de contracepção adequados durante a terapia com LADIZAC®.
Gravidez
O uso de LADIZAC® não é recomendado em mulheres grávidas ou que planejam engravidar. Se LADIZAC® for usado durante a gestação ou caso a paciente engravide durante a terapia com LADIZAC®, a paciente deve ser informada sobre o risco potencial ao feto. Deve-se aconselhar as mulheres com potencial reprodutivo a evitar a gravidez, o que pode incluir o uso de contracepção, durante o tratamento com LADIZAC®.
O dasatinibe pode causar danos ao feto quando administrado a mulheres grávidas. Desfechos adversos fetais e infantis foram relatados em mulheres que tomaram dasatinibe durante a gravidez. Nos estudos de reprodução em animais, foram observadas toxicidades embrio-fetal, incluindo malformações ósseas, em ratos e coelhos em concentrações plasmáticas abaixo daquelas em humanos recebendo doses terapêuticas de dasatinibe. Houveram relatos pós-comercialização de aborto espontâneo e anomalias fetais e infantis de mulheres que utilizaram dasatinibe durante a gravidez.
Em estudos pré-clínicos, em concentrações plasmáticas abaixo daquelas observadas em humanos recebendo doses terapêuticas de dasatinibe, foi observada toxicidade fetal em ratos e coelhos. Morte dos fetos foi observada em ratos.
Categoria D
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico
em caso de suspeita de gravidez.
Lactação
N