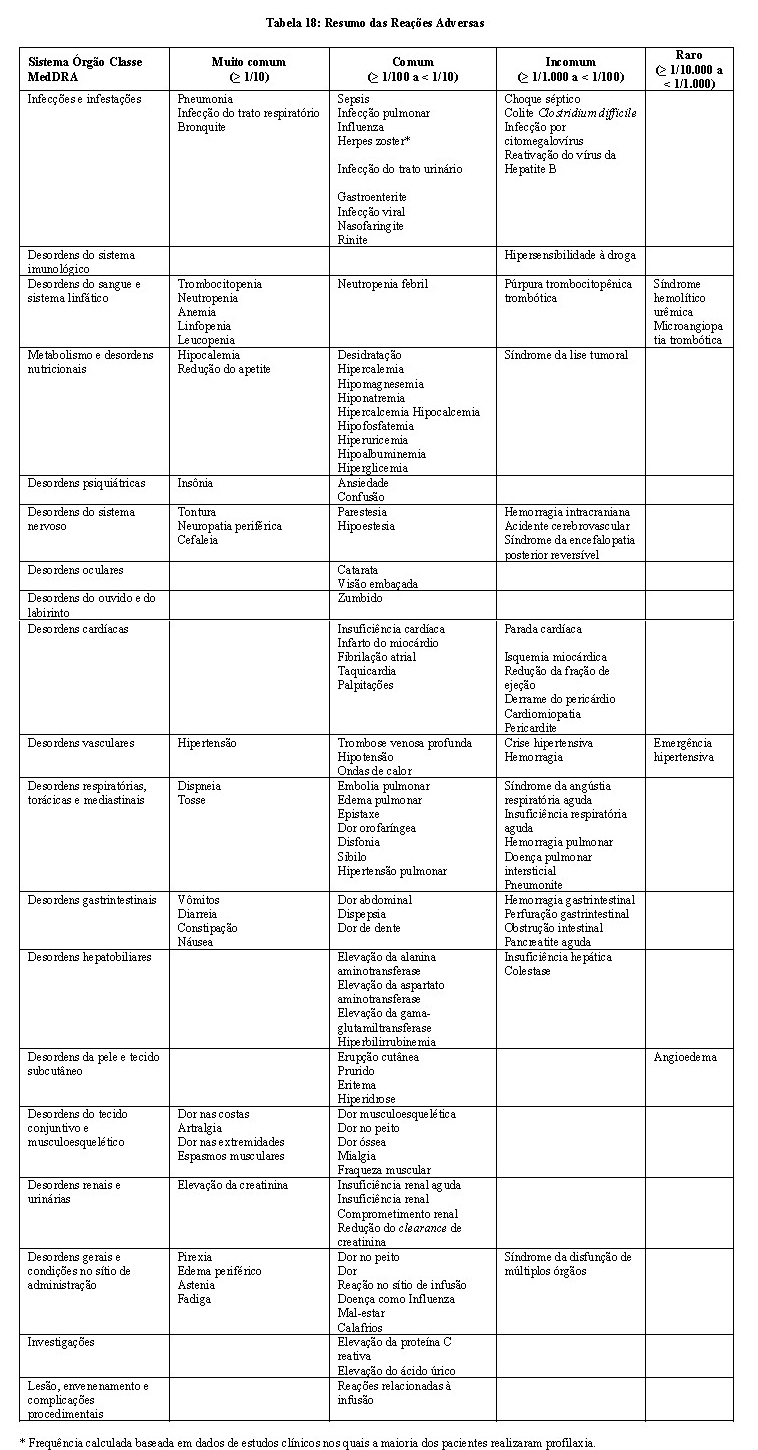
KYPROLIS
AMGEN
carfilzomibe
Inibidor proteassomal.
Apresentações.
60 mg de pó liofilizado para solução injetável em embalagem com 1 frasco-ampola.
USO INTRAVENOSO
USO ADULTO
Composição.

Informações técnicas.
1. INDICAÇÕES
KYPROLIS em combinação com daratumumabe e dexametasona, lenalidomida e dexametasona, ou com dexametasona isolada é indicado para o tratamento de pacientes com mieloma múltiplo recidivado que receberam de uma a três terapias prévias.
KYPROLIS, como um agente isolado, está indicado para o tratamento de pacientes com mieloma múltiplo recidivado ou refratário que tenham recebido pelo menos duas terapias prévias que incluíram bortezomibe e um agente imunomodulador.
2. RESULTADOS DE EFICÁCIA
Resumo das informações de eficácia clínica
Em combinação com lenalidomida e dexametasona para o tratamento de pacientes com mieloma múltiplo (recidivado)
Estudo PX-171-009
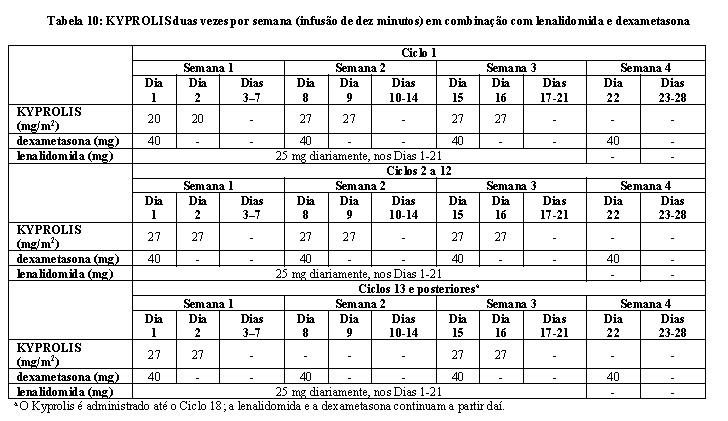
A segurança e a eficácia de KYPROLIS foram avaliadas em um estudo randomizado, em regime aberto e multicêntrico que incluiu 792 pacientes com mieloma múltiplo recidivado que haviam recebido de 1 a 3 terapias prévias (mediana de 2), o estudo avaliou a combinação de KYPROLIS com lenalidomida e dexametasona versus lenalidomida e dexametasona isoladamente, randomizados 1:1. Os pacientes que tinham as seguintes características foram excluídos do estudo: clearance de creatinina < 50 mL/min, insuficiência cardíaca congestiva Classe funcional III ou IV da New York Heart Association ou infarto do miocárdio nos 4 meses anteriores ao estudo. O tratamento com KYPROLIS foi administrado por um máximo de 18 ciclos, a menos que fosse descontinuado anteriormente por progressão da doença ou toxicidade inaceitável. A administração de lenalidomida e dexametasona poderia continuar até progressão da doença ou toxicidade inaceitável.
O estudo incluiu uma população representativa de mieloma múltiplo recidivado; as características basais, inclusive aquelas relacionadas à doença, foram bem balanceadas entre os dois braços de tratamento.
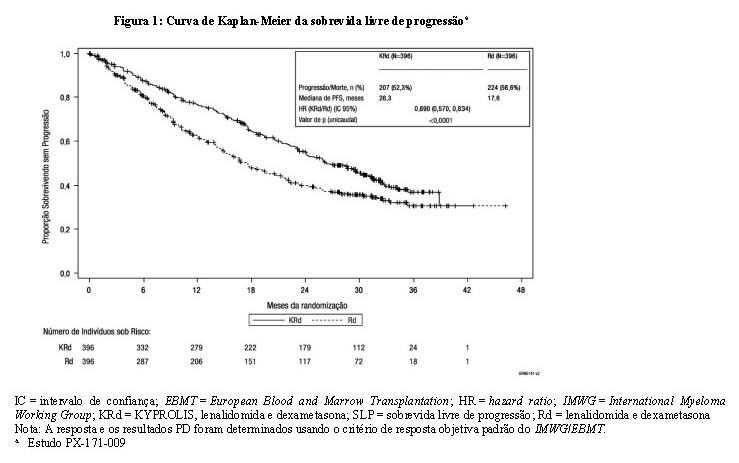
Os pacientes no braço KYPROLIS, lenalidomida e dexametasona (KRd) apresentaram melhor sobrevida livre de progressão (SLP) em comparação aos pacientes no braço lenalidomida e dexametasona (Rd) (Hazard Ratio [HR] = 0,69; com valor de p unicaudal < 0,0001). Este achado representa uma melhora de 45% na SLP ou uma redução de 31% no risco de evento, conforme determinado pelo uso do critério de resposta objetiva padrão do International Myeloma Working Group (IMWG)/European Blood and Marrow Transplantation (EBMT) por um Comitê de Revisão Independente (Independent Review Committee, IRC).
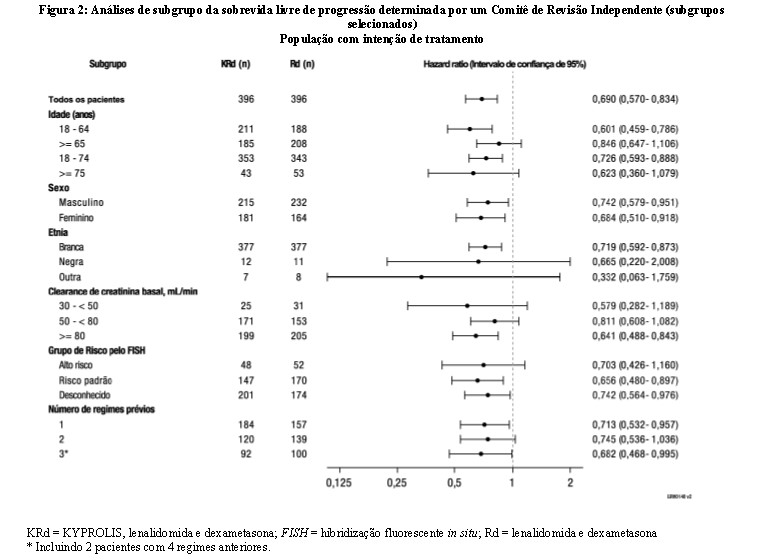
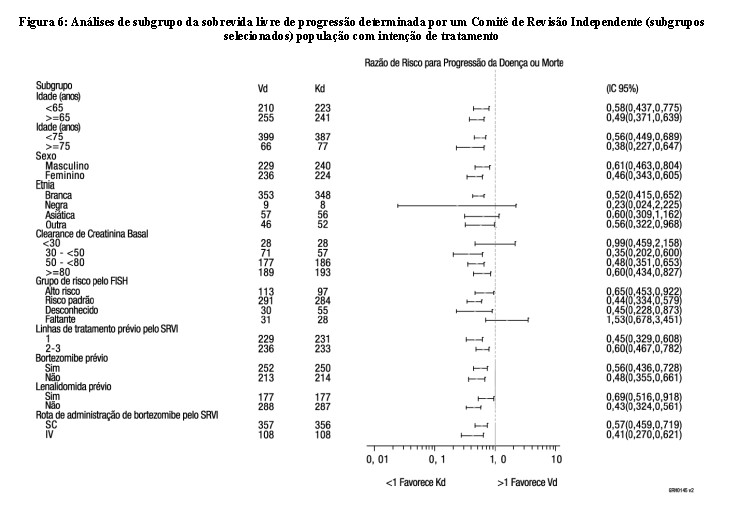
A SLP mediana foi de 26,3 meses (IC 95%: 23,3 a 30,5 meses) no braço KRd versus 17,6 meses (IC 95%: 15,0 a 20,6 meses) no braço Rd, uma diferença na mediana de 8,7 meses (vide Figura 1). O benefício do KRd na SLP foi consistentemente observado em todos os subgrupos (vide Figura 2).
Uma análise prévia de sobrevida global (SG) foi realizada após 246 mortes no braço KRd e 267 mortes no braço Rd. A mediana de acompanhamento foi de aproximadamente 67 meses. Uma vantagem estatisticamente significante na SG foi observada em pacientes no braço KRd em comparação com pacientes no braço Rd. Os pacientes no braço KRd tiveram uma redução de 21% no risco de morte em comparação com aqueles no braço Rd (HR = 0,79; IC 95%: 0,67, 0,95; valor p = 0,0045). A mediana da SG melhorou em 7,9 meses em pacientes do braço KRd em comparação com os do braço Rd (vide Tabela 1 e Figura 3).
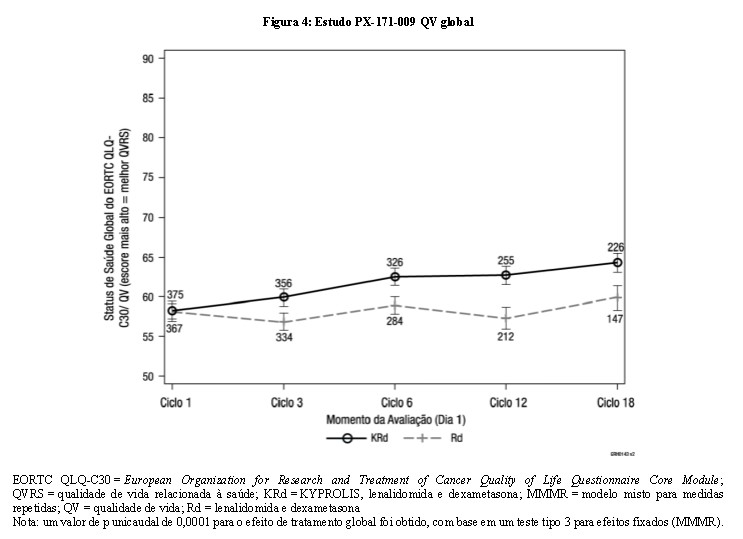
A taxa de resposta global (TRG) foi mais alta no braço KRd versus o braço Rd (87,1% versus 66,7%; valor de p unicaudal < 0,0001). A taxa e a profundidade da resposta foram maiores no braço KRd versus o braço Rd, resposta completa (RC) ou maior foi de 31,8% no braço KRd (incluindo 14,1% de resposta completa rigorosa [RCr]) versus 9,3% de RC ou maior no braço Rd (incluindo 4,3% de RCr). Os pacientes tratados com KRd relataram status de saúde global melhorado, com escores mais elevados de Status de Saúde Global/Qualidade de Vida (QV) comparados com os pacientes tratados com Rd ao longo de 18 ciclos de tratamento (valor de p unicaudal = 0,0001) medidos pelo EORTC QLQ-C30, um instrumento validado em mieloma múltiplo (vide Figura 4).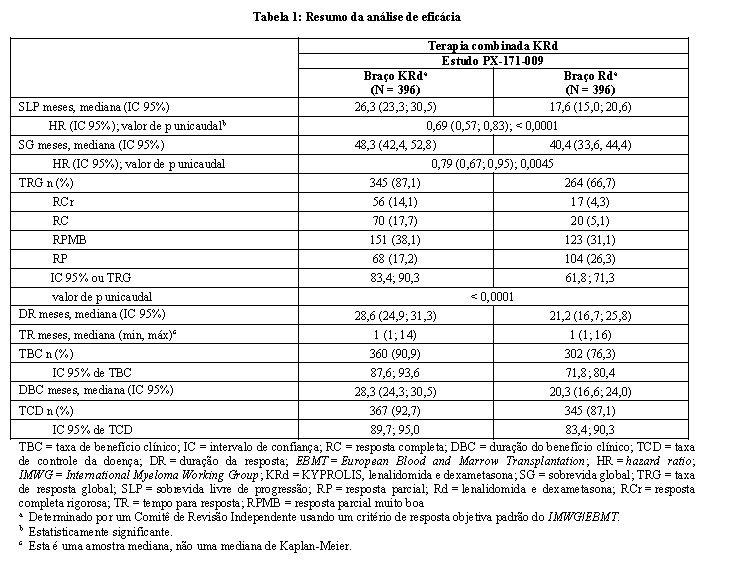

KYPROLIS em combinação com dexametasona para o tratamento de pacientes com mieloma múltiplo (recidivado)
Estudo 2011-003
A segurança e a eficácia de KYPROLIS 56 mg/m2 duas vezes por semana foram avaliadas em um estudo de Fase 3, randomizado, aberto e multicêntrico de KYPROLIS mais dexametasona (Kd) versus bortezomibe mais dexametasona (Vd) em pacientes com múltiplo mieloma refratário ou recidivado que receberam de 1 a 3 linhas prévias de tratamento. Um total de 929 pacientes foi incluído e randomizado (464 no braço Kd; 465 no braço Vd). Pacientes foram excluídos se apresentaram menos que RP em todos os regimes anteriores: clearance de creatinina < 15 mL/min; transaminase hepática ≥ 3 x limite superior normal (ULN); ou fração de ejeção ventricular esquerda < 40% ou outras condições cardíacas significativas. Este estudo avaliou KYPROLIS em uma dose inicial de 20 mg/m2, que foi aumentada para 56 mg/m2 no Ciclo 1, Dia 8, administrada duas vezes por semana como uma infusão de 30 minutos até progressão da doença ou toxicidade inaceitável.
O desfecho primário do estudo foi a SLP determinada por um Comitê de Revisão Independente (IRC) usando os critérios de resposta do International Myeloma Working Group (IMWG). O estudo mostrou melhora significativa na SLP para os pacientes do braço Kd em relação aos do braço Vd (HR: 0,53, IC 95%: 0,44; 0,65 [valor de p < 0,0001]), com uma diferença na mediana da SLP de 9,3 meses (18,7 meses no braço Kd [IC 95%: 15,6; NE] versus 9,4 meses no braço Vd [IC 95%: 8,4; 10,4]) (vide Figura 5).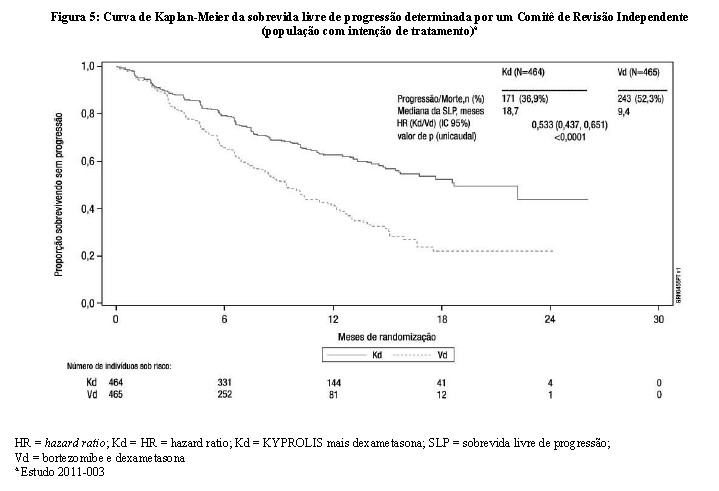
Os desfechos secundários chaves foram a SG, TRG e incidência de eventos de neuropatia periférica (Grau ≥ 2).
A análise de SG pré-planejada foi realizada após 189 mortes no braço Kd e 209 mortes no braço Vd. A mediana do acompanhamento foi de aproximadamente 37 meses. Uma vantagem estatisticamente significativa na SG foi observada em pacientes no braço Kd em comparação aos pacientes no braço Vd (HR = 0,791; IC 95%: 0,648, 0,964; valor de p = 0,010) (vide Tabela 2 e Figura 7).
A TRG foi de 76,9% (IC 95%: 72,8; 80,7) para os pacientes no braço Kd e 62,6% (IC 95%: 58,0; 67,0) para os pacientes no braço Vd (odds ratio = 2,032; IC 95%: 1,519: 2,718); (valor de p < 0,0001) (vide Tabela 2).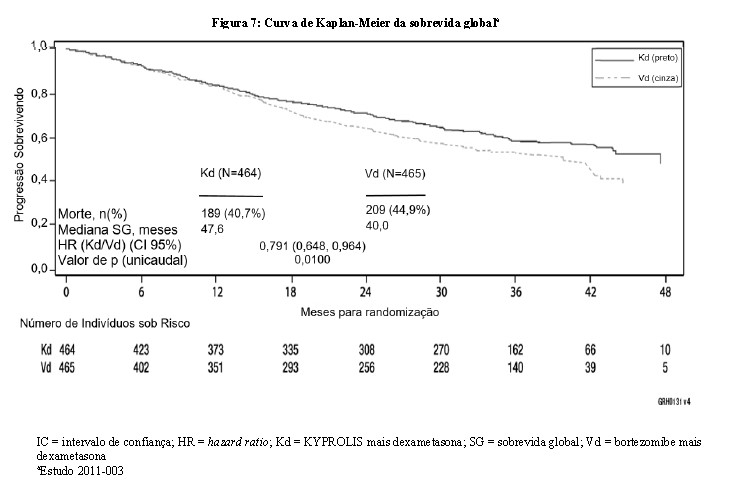
No momento da análise de SG pré-planejada, a incidência de eventos de neuropatia periférica Grau ≥ 2 no braço Kd (taxa de evento de 6,9% [IC 95%: 4,6; 9,2]) foi aproximadamente 5 vezes menor que no braço Vd (taxa de evento de 34,9% [IC 95%: 30,5; 39,2]) (odds ratio = 0,139; IC 95%: 0,092; 0,208; valor de p < 0,0001).
Estudo 20140355
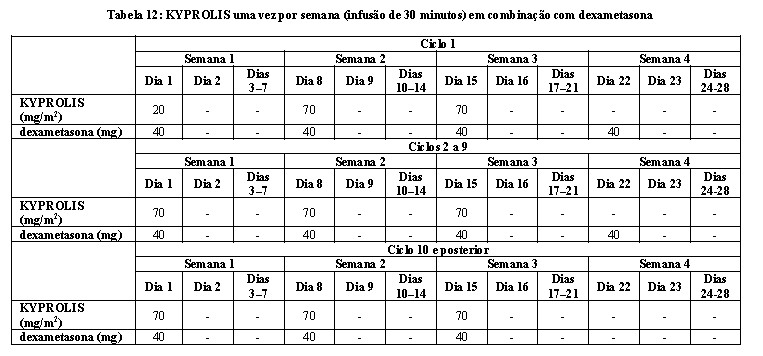
A segurança e a eficácia de KYPROLIS 70 mg/m2 uma vez por semana foram avaliadas em um estudo Fase 3 randomizado, aberto, multicêntrico de Kd 70 mg/m2 uma vez por semana versus Kd 27 mg/m2 duas vezes por semana em pacientes com mieloma múltiplo refratário e recidivado que receberam de 2 a 3 linhas prévias de tratamento. Um total de 478 pacientes foi incluído e randomizado (240 no braço Kd 70mg/m2; 238 no braço Kd 27 mg/m2). Este estudo avaliou KYPROLIS em uma dose inicial de 20 mg/m2 que foi aumentada para 70 mg/m2 no Ciclo 1, Dia 8, administrado uma vez por semana com uma infusão de 30 minutos até a progressão da doença ou toxicidade inaceitável.
O desfecho primário do estudo foi a SLP. O estudo mostrou uma maior duração da SLP para os pacientes tratados com Kd 70 mg/m2 uma vez por semana em relação aos tratados com Kd 27 mg/m2 duas vezes por semana (HR: 0,68, IC 95%: 0,54,0,87 [valor de p < 0,00010]), com uma diferença na mediana da SLP de 3,7 meses (11,3 meses no braço Kd 70 mg/m2 uma vez por semana versus 7,6 meses no braço Kd 20/27 mg/m2 duas vezes por semana) (vide Figura 8).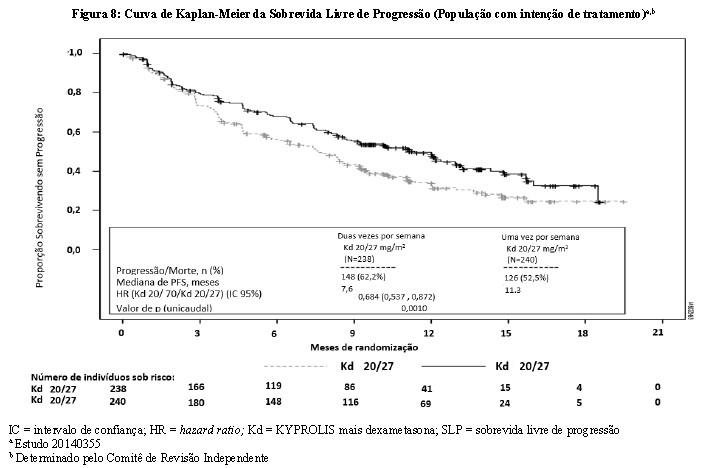
Os desfechos-chaves secundários foram TRG e SG. A TRG foi de 63,8% (IC 95%: 57,3, 69,8) para pacientes no braço Kd 70 mg/m2 uma vez por semana e 41,2% (IC 95%: 34,9, 47,7) para pacientes no braço Kd 27 mg/m2 duas vezes por semana (odds ratio = 2,53; IC 95% 1,75, 3,66; valor p < 0,0001) (vide Tabela 3). No momento da análise primária da SLP, o HR para SLP foi de 0,80 (IC 95%: 0,56, 1,14; p unicaudal = 0,1070).
Estudo 20130403
A segurança e eficácia de Kyprolis 70 mg/m2 uma vez por semana foram avaliadas num estudo clínico de fase 1/2, multicêntrico e aberto. A dose máxima tolerada (DMT) de Kd 70 mg/m2 uma vez por semana foi determinada na Fase 1, e a Fase 2 incluiu pacientes com mieloma múltiplo recidivado ou refratário que receberam 1 a 3 linhas prévias de tratamento. Houve um total de 104 pacientes no grupo Kd 70 mg/m2 uma vez por semana em ambas as fases combinadas. Os pacientes foram excluídos do estudo quando apresentavam: taxas de depuração de creatinina < 30 mL/min, insuficiência cardíaca congestiva classe III a IV da segundo a New York Heart Association ou infarto do miocárdio nos últimos 6 meses.
O desfecho primário foi a TRG definida como a proporção de indivíduos que obtiveram uma resposta parcial confirmada [RP] ou melhor. A TRG (resposta completa [RC] + muito boa resposta parcial RPMB] + resposta parcial [RP]) foi de 76,9% (95% CI: 67,6, 84,6) (N = 104). A duração mediana da resposta (DOR) foi de 18 meses (IC 95%: 14,5, 21,9). A eficácia de Kd uma vez por semana é resumida na Tabela 3.
KYPROLIS como um agente isolado no mieloma múltiplo
KYPROLIS em monoterapia foi avaliado em dois estudos de mieloma múltiplo recidivado e refratário.
Estudo PX-171-003 A1
A segurança e a eficácia de KYPROLIS foram avaliadas em um estudo clínico de braço único e multicêntrico. Um total de 266 pacientes com mieloma múltiplo recidivado e refratário que haviam recebido pelo menos duas terapias prévias (incluindo bortezomibe e talidomida e/ou lenalidomida) foram incluídos. Os pacientes que tinham as seguintes características foram excluídos do estudo: taxas de clearance de creatinina < 30 mL/min, insuficiência cardíaca congestiva Classe funcional III ou IV da New York Heart Association ou infarto do miocárdio nos 6 meses anteriores ao estudo.
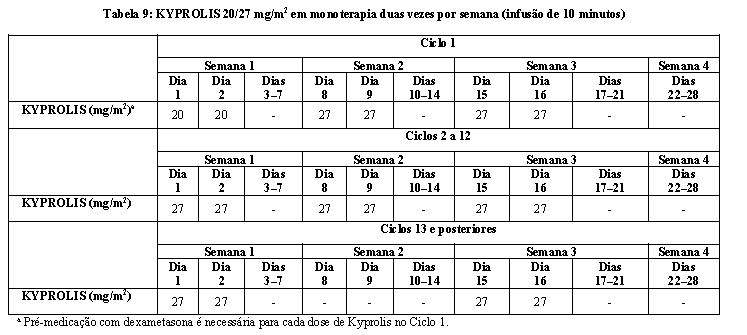
KYPROLIS foi administrado por via intravenosa (IV) em um período de 2 a 10 minutos em dois dias consecutivos a cada semana, durante três semanas, seguidas por um período de descanso de 12 dias (ciclo de tratamento de 28 dias), até a progressão da doença, toxicidade inaceitável ou por um máximo de 12 ciclos. Os pacientes receberam 20 mg/m2 em cada dose no Ciclo 1 e 27 mg/m2 nos ciclos subsequentes. Para reduzir a incidência e a gravidade da febre, tremores, calafrios, dispneia, mialgia e artralgia, dexametasona 4 mg por via oral ou por infusão IV foi administrada antes de cada dose de KYPROLIS durante o primeiro ciclo e antes de todas as doses de KYPROLIS durante o primeiro ciclo de escalonamento da dose (27 mg/m2). A pré-medicação com dexametasona (4 mg por via oral ou IV) era reiniciada se esses sintomas reaparecessem durante os ciclos subsequentes.
O desfecho primário foi a taxa de resposta global (TRG) determinada pela avaliação de um Comitê de Revisão Independente usando os critérios do IMWG/EBMT. A TRG (resposta completa rigorosa [RCr] + resposta completa [RC] + resposta parcial muito boa [RPMB] + resposta parcial [RP]) foi de 22,9% (IC 95%: 18,0; 28,5) (N = 266). A mediana da duração da resposta (DR) foi de 7,8 meses (IC 95%: 5,6; 9,2).
Estudo PX-171-011
O estudo PX-171-011 foi um estudo de Fase 3 com 315 pacientes que haviam recebido pelo menos três terapias prévias, que avaliou a monoterapia com KYPROLIS versus um braço de comparador ativo (referido no protocolo como Melhor Tratamento de Suporte [MTS]) com um protocolo metronômico com corticosteroides em baixa dose (84 mg/ciclo de 28 dias) e ciclofosfamida opcional (1.400 mg/ciclo de 28 dias, que foi usado por 91,8% dos pacientes randomizados para o braço comparador). Os pacientes que tinham as seguintes características foram excluídos do estudo: taxas de clearance de creatinina < 15 mL/min, insuficiência cardíaca congestiva Classe funcional III ou IV da New York Heart Association ou infarto do miocárdio nos 3 meses anteriores ao estudo.
Os pacientes incluídos no Estudo PX-171-011 haviam sido previamente tratados de maneira mais agressiva e tinham pior função de órgãos e da medula em comparação àqueles incluídos no Estudo PX-171-003A1.
O estudo não atingiu seu desfecho primário de eficácia de prolongamento da SG com a monoterapia com KYPROLIS em comparação ao controle. O HR foi de 0,975 (IC 95%: 0,760; 1,249); com um valor de p unicaudal de 0,4172. A SG mediana foi de 10,2 meses (IC 95%: 8,4; 14,4 meses) no braço de KYPROLIS versus 10,0 meses (IC 95%: 7,7; 12,0 meses) no braço comparador.
A SLP mediana foi de 3,7 meses no braço de KYPROLIS versus 3,3 meses no braço comparador. O HR foi de 1,091 [IC 95%: 0,843; 1,410]; com valor de p unicaudal de 0,2479. A TRG no braço KYPROLIS foi de 19,1% em comparação a 11,4% no braço comparador.
KYPROLIS em combinação com dexametasona e daratumumabe em mieloma múltiplo
A segurança da administração de 20/56 mg/m2 de KYPROLIS duas vezes por semana em combinação com daratumumabe e dexametasona (KdD) foi avaliada em um estudo fase 3, randomizado, aberto (CANDOR). Além disso, a segurança da administração de 20/70 mg/m2 de KYPROLIS uma vez por semana em combinação com daratumumabe e dexametasona foi avaliada em um estudo não randomizado, aberto, multi coorte (EQUULEUS). CANDOR foi um estudo randomizado, aberto, multicêntrico e de superioridade de KYPROLIS com daratumumabe e dexametasona (KdD) duas vezes por semana (20/56 mg/m2) comparado à administração de KYPROLIS mais dexametasona (Kd) duas vezes por semana (20/56 mg/m2) em pacientes com mieloma múltiplo recidivado ou refratário que receberam de 1 a 3 linhas de terapia prévia. Os pacientes foram excluídos caso apresentassem asma persistente moderada ou severa conhecida nos últimos 2 anos, doença pulmonar obstrutiva crônica (DPOC) conhecida com FEV1 < 50% de insuficiência cardíaca congestiva ativa e normal prevista. Um total de 466 pacientes foram incluídos e randomizados em uma proporção 2:1 (312 no grupo KdD e 154 no grupo Kd). A randomização foi estratificada pelo ISS na triagem (estágio 1 ou 2 vs estágio 3), exposição anterior a inibidor de proteassoma (sim vs não), número de linhas prévias de terapia (1 vs ≥ 2) e terapia prévia com anticorpo CD38 (antígeno de diferenciação de cluster 38) (sim vs não).
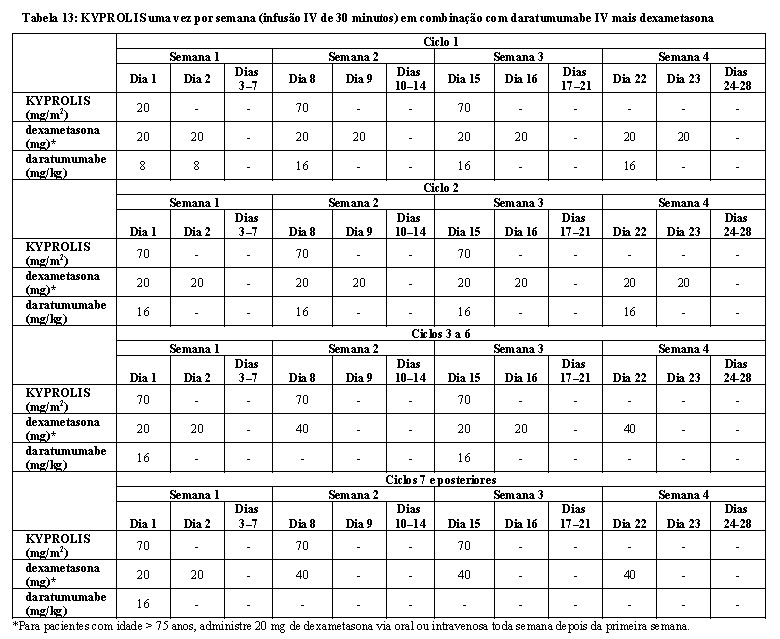
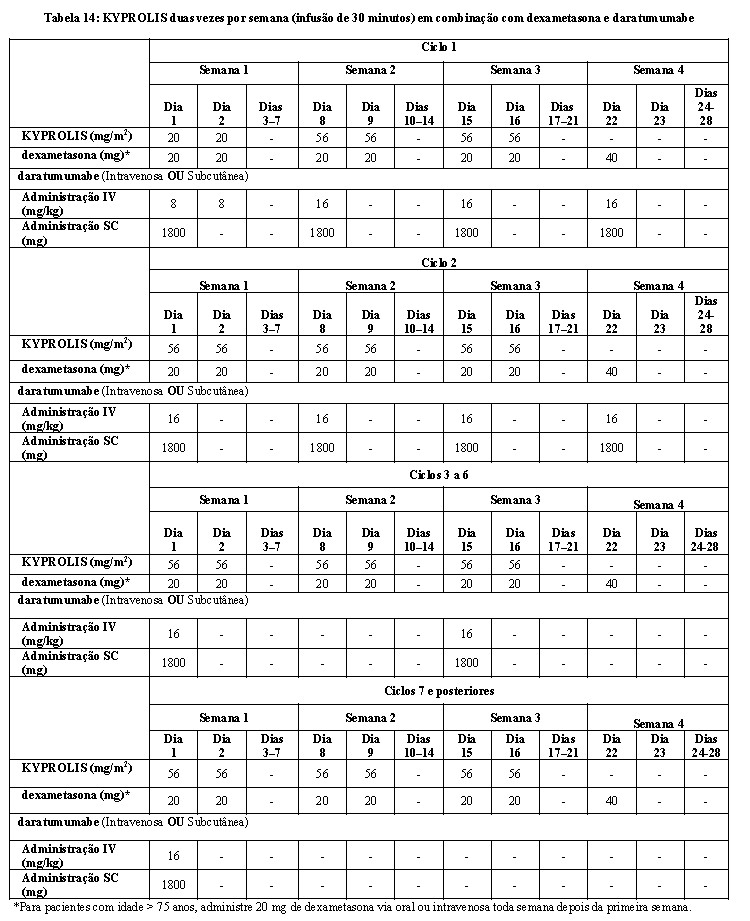
Nos grupos KdD e Kd, o KYPROLIS foi administrado a uma dose inicial de 20 mg/m2, que foi aumentada para 56 mg/m2 no Ciclo 1, Dia 8 em diante. O KYPROLIS foi administrado duas vezes por semana como uma infusão de 30 minutos nos Dias 1, 2, 8, 9, 15 e 16 de cada ciclo de 28 dias. No grupo KdD, o daratumumabe foi administrado em uma dose de 16 mg/kg no Ciclo 1 dividida em 8 mg/kg nos Dias 1 e 2. Em seguida, o daratumumabe foi administrado a 16 mg/kg uma vez por semana nos Dias 8, 15 e 22 do Ciclo 1 e nos Dias 1, 8, 15 e 22 do Ciclo 2 e depois a cada duas semanas por 4 ciclos (ciclos 3 a 6) e, então, a cada 4 semanas nos ciclos restantes ou até a progressão da doença. Nos dois grupos, a dexametasona 20 mg foi administrada nos Dias 1, 2, 8, 9, 15 e 16 e depois 40 mg via oral ou intravenosa no Dia 22 de cada ciclo de 28 dias. Os dados demográficos e as características basais estão resumidos na Tabela 4.
A eficácia do KYPROLIS foi avaliada pela SLP usando os critérios de resposta do IMWG. No momento da análise primária, o estudo demonstrou melhora na SLP no grupo KdD, comparado ao grupo Kd. A SLP mediana não foi ainda atingida no grupo KdD, versus 15,8 meses no grupo Kd (razão de risco [HR]=0,630; IC 95%: 0,464, 0,854; p=0,0014) representando uma redução de 37% no risco de progressão da doença ou morte nos pacientes tratados com KdD.
Outros desfechos incluíram TRG. A TRG foi de 84,3% para pacientes no grupo KdD e de 74,7% no grupo Kd (consulte a Tabela 5). A duração mediana da resposta não pode ser estimada para o grupo KdD e foi de 16,6 meses (13,9, NE) para o braço Kd. O tempo mediano de resposta foi de 1,0 (1, 14) mes para o grupo KdD e 1,0 (1,10) mes para o grupo Kd.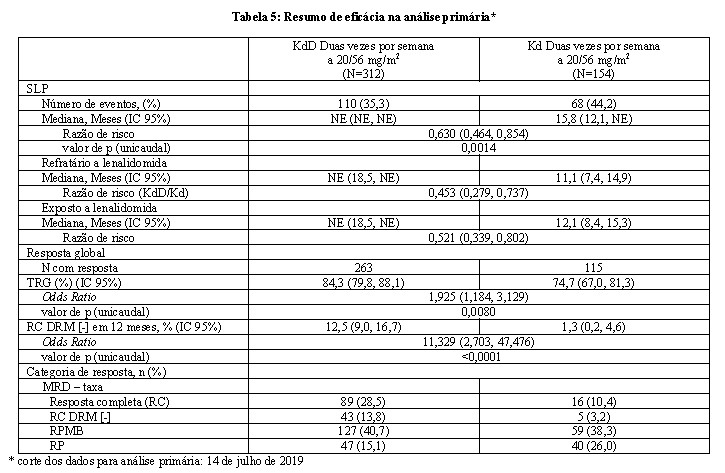
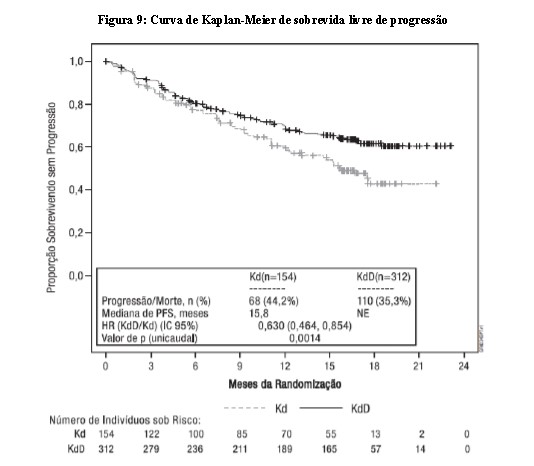
Uma análise final pré-planejada de sobrevida global (SG) foi realizada quando os pacientes tiveram a oportunidade de serem tratados por um período máximo de aproximadamente 5 anos. No momento da análise final, 148 participantes (47,4%) no grupo KdD e 80 participantes (51,9%) no grupo Kd tinham morrido. A SG mediana (IC 95%) foi de 50,8 (44,7, NE) meses para o grupo KdD e 43,6 (35,3, NE) meses para o grupo Kd, com uma HR (KdD/Kd) de 0,784 (IC 95%: 0,595, 1,033; p unicaudal = 0,0417). Esse valor de p unicaudal não atendeu o nível de significância estatística de 0,021 para a análise final. O tempo mediano de acompanhamento foi de 50,6 meses no grupo KdD e 50,1 meses para o grupo Kd.
O EQUULEUS foi um estudo aberto, de vários grupos de daratumumabe em combinação com terapia de mieloma múltiplo. O grupo que avalia semanalmente o KYPROLIS com daratumumabe mais dexametasona (KdD) incluiu 85 pacientes com mieloma múltiplo recidivado ou refratário. Os pacientes foram excluídos caso apresentassem asma persistente moderada ou severa conhecida nos últimos 2 anos, doença pulmonar obstrutiva crônica (DPOC) conhecida com FEV1 < 50% de insuficiência cardíaca congestiva ativa e normal prevista. O KYPROLIS foi administrado a uma dose inicial de 20 mg/m2, que foi aumentada para 70 mg/m2 no Ciclo 1, Dia 8 e sucessivamente. O KYPROLIS foi administrado uma vez por semana como uma infusão de 30 minutos nos Dias 1, 8 e 15 de cada ciclo de 28 dias. Dez pacientes receberam uma única primeira dose de daratumumabe a 16 mg/kg no Ciclo 1, Dia 1. Os 75 pacientes restantes receberam uma primeira dose dividida de daratumumabe a 8mg/kg por dia no Ciclo 1, Dias 1 e 2. A partir daí, uma única dose de 16 mg/kg foi administrada nos Dias 8, 15 e 22 do Ciclo 1 e nos Dias 1, 8, 15 e 22 do Ciclo 2, a cada 2 semanas, durante 4 ciclos (ciclos 3 a 6) e, depois a cada 4 semanas durante os ciclos restantes de cada ciclo de 28 dias. A dexametasona é administrada via oral ou intravenosa a uma dose 20 mg nos Ciclos 1 e 2 nos Dias 1, 2, 8, 9, 15, 16, 22 e 23. Nos ciclos 3-6, a dexametasona é administrada a uma dose de 20 mg nos Dias 1, 2, 15 e 16 e a uma dose de 40 mg no Dia 22. Nos ciclos 7 e a partir dele, a dexametasona é administrada a uma dose de 20 mg nos Dias 1 e 2 e a uma dose de 40 mg nos Dias 8, 15 e 22. Para pacientes com idade > 75 anos, administre 20 mg de dexametasona via oral ou intravenosa semanalmente depois da primeira semana. Administre a dexametasona de 30 minutos a 4 horas antes do KYPROLIS. A dexametasona 40 mg foi administrada nos Dias 1, 8, 15 e 22 de cada ciclo de 28 dias. O tratamento deve ser continuado, até a progressão da doença ou toxicidade inaceitável.
A eficácia do KYPROLIS foi avaliada por SLP usando os critérios de resposta do IMWG. A SLP mediana foi de 26 meses (IC 95%: 14,8, NE), depois de uma mediana de acompanhamento de 24 meses. Outros desfechos incluíram TRG. A TRG foi de 81%. A mediana de sobrevida global não foi atingida. A taxa de sobrevida em 12 meses foi de 82% e a taxa de sobrevida em 24 meses foi de 71%. O tempo mediano de resposta foi de 0,95 meses (intervalo: 0,9, 14,3). A duração mediana da resposta foi de 28 meses (IC 95%: 20,5, NE). O tempo mediano para terapia antimieloma posterior foi de 29 meses (IC 95%: 19,5, NA).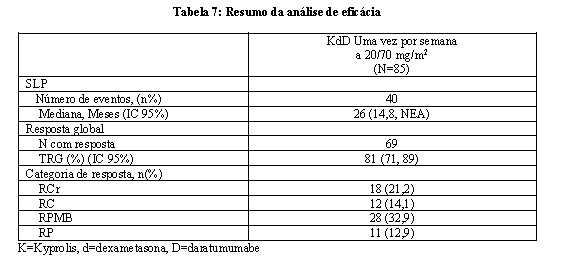
Referências bibliográficas
1.Siegel D et al., A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012; 120:2817. 2.Dimopoulos M et al., Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomized, phase 3, open-label, multicentre study. The Lancet; published online December 5, 2015, http://dx.doi.org/10.1016/S1470-2045(15)00464-7. 3.Stewart A.K., Carfilzomib, Lenalidomide and Dexamethasone for Relapsed Multiple Myeloma, The New England Journal of Medicine, January 8, 2015; 372;2
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O carfilzomibe é um inibidor de proteassoma epoxicetona tetrapeptídeo que se liga seletiva e irreversivelmente à treonina N terminal nos sítios ativos do proteassoma 20S, a partícula central proteolítica dentro do proteassoma 26S; ele apresenta pouca ou nenhuma atividade contra outras classes de proteases. O carfilzomibe tem atividades antiproliferativas e pró-apoptóticas em modelos pré-clínicos de tumores sólidos e hematológicos. Em animais, carfilzomibe inibiu a atividade proteassomal em sangue e em tecidos e retardou o crescimento tumoral em modelos de mieloma múltiplo e de tumores sólidos e hematológicos. In vitro, carfilzomibe demonstrou neurotoxicidade mínima e mínima reação a proteases não proteassomais.
Efeitos farmacodinâmicos
O carfilzomibe administrado por via IV resultou em supressão da atividade do proteassoma semelhante à quimiotripsina (chymotrypsin-like [CT-L]) medida no sangue uma hora após aprimeira dose. Doses ≥ 15 mg/m2 consistentemente induziram uma inibição (≥ 80%) da atividade do proteassoma CT-L. Além disso, a administração de carfilzomibe na dose de 20 mg/m2 resultou em inibição do polipeptídeo 2 de baixa massa molecular (latent membrane protein 2 [LMP2]) e das subunidades da multicatalytic endopeptidase complex-like 1 (MECL1) do imunoproteassoma, que variou de 26% a 32% e 41% a 49%, respectivamente. A inibição do proteassoma foi mantida por ≥ 48 horas após a primeira dose de carfilzomibe para cada semana de administração. A administração combinada com lenalidomida e dexametasona não afetou a inibição do proteassoma.
Propriedades farmacocinéticas
Nas doses entre 20 e 70 mg/m2, o carfilzomibe administrado como uma infusão de 30 minutos resultou em um aumento dose-dependente nas concentrações plasmáticas máximas (Cmáx) e área sob a curva (AUC). Após doses repetidas de 70 mg/m2 de carfilzomibe, a exposição sistêmica (AUC) e a meia-vida foram similares às do Dia 15 dos Ciclos 1 e 2, sugerindo que não houve acúmulo sistêmico de carfilzomibe. Uma infusão de 30 minutos resultou em uma meia-vida e em uma AUC semelhantes, mas em uma Cmáx 2 a 3 vezes menor em comparação àquela observada com uma infusão de 2 a 10 minutos na mesma dose.
Distribuição - O volume de distribuição médio no estado de equilíbrio dinâmico com a dose de 20 mg/m2 de carfilzomibe foi de 28 L. Quando testada in vitro, a ligação de carfilzomibe às proteínas plasmáticas foi de aproximadamente 97%, nas concentrações de 0,4 a 4 micromolar.
Metabolismo - O carfilzomibe foi rapidamente e extensivamente metabolizado. Os metabólitos predominantes medidos no plasma humano e na urina e gerados in vitro por hepatócitos humanos, foram fragmentos peptídicos e o diol de carfilzomibe, sugerindo que a clivagem por peptidase e epóxido hidrolase foram as vias principais do metabolismo. Mecanismos mediados por citocromo P450 desempenharam um papel pouco importante no metabolismo global do carfilzomibe. Os metabólitos não têm atividade biológica conhecida.
Eliminação - Após a administração IV de doses ≥ 15 mg/m2, carfilzomibe foi rapidamente eliminado da circulação sistêmica com uma meiavida ≤ 1 hora no Dia 1 do Ciclo 1. O clearance sistêmico variou de 151 a 263 L/hora e excedeu o fluxo sanguíneo hepático, sugerindo que o carfilzomibe foi amplamente eliminado extrahepaticamente. O carfilzomibe é eliminado primariamente via metabolismo com excreção subsequente na urina.
Populações especiais - Análises farmacocinéticas populacionais indicam que não houve efeitos da idade, sexo ou etnia sobre a farmacocinética de carfilzomibe.
Insuficiência hepática - A farmacocinética do carfilzomibe foi estudada em pacientes com neoplasias em estágio avançado em progressão ou recidivadas com insuficiência hepática crônica moderada ou leve comparada com àqueles com função hepática normal.
Nenhuma diferença significativa nas exposições (AUC e Cmáx) foram observadas entre pacientes com função hepática normal e àqueles com insuficiência hepática leve ou moderada. Não é necessário ajuste de dose inicial em pacientes com insuficiência hepática basal leve ou moderada. A farmacocinética de carfilzomibe não foi estudada em pacientes com insuficiência hepática grave (vide "ADVERTÊNCIAS E PRECAUÇÕES - Insuficiência hepática").
Insuficiência renal - A farmacocinética de carfilzomibe foi estudada em pacientes com mieloma múltiplo recidivado com função renal normal; insuficiência renal leve, moderada ou grave; e em pacientes com doença renal em estágio avançado com necessidade de hemodiálise. Exposições ao carfilzomibe (AUC e Cmáx) em pacientes com insuficiência renal foram similares àqueles com função renal normal.
Nenhum ajuste de dose inicial è necessário para pacientes com insuficiência renal basal (vide "ADVERTÊNCIAS E PRECAUÇÕES - Insuficiência renal").
Citocromo P450 - Com base em dados in vitro e in vivo, não se espera que carfilzomibe iniba as atividades do CYP3A4/5 e/ou afete a exposição a substratos do CYP3A4/5. Um estudo clínico usando midazolam oral como um marcador do CYP3A mostrou que a farmacocinética de midazolam não foi afetada pela administração concomitante de carfilzomibe.
P-gp - O carfilzomibe é um substrato da glicoproteína P (P-gp). No entanto, considerando que o carfilzomibe é administrado por via IV e é extensivamente metabolizado, o perfil farmacocinético de carfilzomibe muito provavelmente não é afetado por inibidores ou indutores da P-gp.
4. CONTRAINDICAÇÕES
KYPROLIS está contraindicado em pacientes com hipersensibilidade conhecida ao carfilzomibe ou a qualquer componente da formulação do produto.
Categoria D para gravidez: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Desordens cardíacas
Piora da insuficiência cardíaca ou diagnóstico desta condição (por exemplo: insuficiência cardíaca congestiva, edema pulmonar, redução da fração de ejeção), isquemia e infarto do miocárdio ocorreram após a administração de KYPROLIS. Morte devido à parada cardíaca ocorreu dentro de um dia da administração de KYPROLIS e resultados fatais foram relatados com insuficiência cardíaca e infarto do miocárdio.
Embora hidratação adequada seja necessária antes do Ciclo 1, todos os pacientes devem ser monitorados para evidências de sobrecarga de volume, especialmente pacientes em risco de insuficiência cardíaca. O volume total de fluidos pode ser ajustado conforme indicação clínica em pacientes
com insuficiência cardíaca basal ou que estão em risco de insuficiência cardíaca (vide "POSOLOGIA E MODO DE USAR").
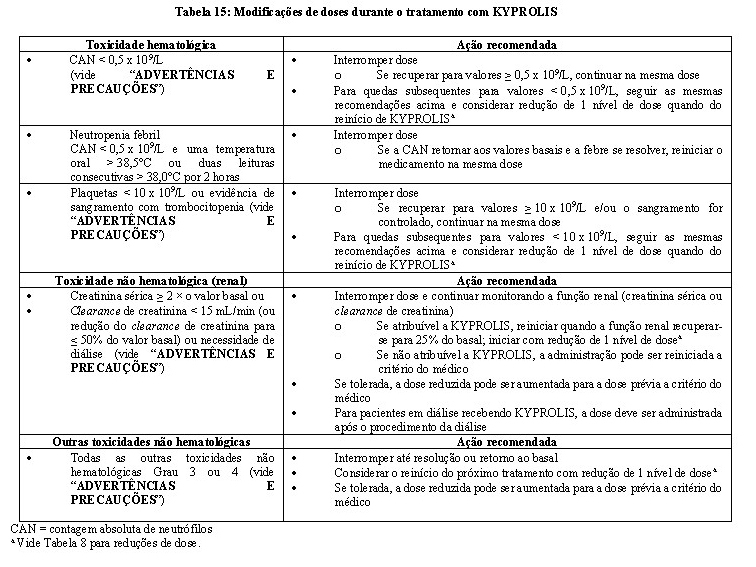
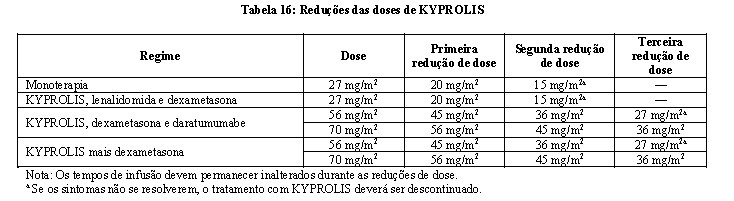
KYPROLIS deve ser interrompido em casos de eventos cardíacos Grau 3 ou 4 até a recuperação; deve-se considerar o reinício de KYPROLIS com 1 nível de redução de dose com base na avaliação do risco/benefício.
Orisco de insuficiência cardíaca è maiorem pacientes idosos (≥ 75 anos). O risco de insuficiência cardíaca é aumentado em pacientes asiáticos.
Pacientes com insuficiência cardíaca em Classe funcional III ou IV da New York Heart Association, infarto do miocárdio recente, anormalidades de condução, angina ou arritmias não controladas com medicamentos não foram elegíveis para os estudos clínicos. Devido ao maior risco de complicações cardíacas, a terapia com KYPROLIS nestes pacientes é recomendada apenas quando houver uma avaliação criteriosa baseada no perfil risco/benefício. Antes de iniciar o tratamento, estes pacientes devem ser submetidos a uma avaliação médica completa (em particular, o controle da pressão arterial e gestão de fluídos) e então permanecer sob acompanhamento cuidadoso.
Alterações eletrocardiográficas
Foram relatados casos de prolongamento do intervalo QT em estudos clínicos. Um efeito de KYPROLIS no intervalo QT não pode ser excluído. Uma avaliação dos possíveis efeitos de carfilzomibe na função cardíaca foi realizada através da análise, com leitura de avaliação central cega, ECG triplicado em 154 pacientes com malignidades avançadas, incluindo mieloma múltiplo, o efeito de carfilzomibe na repolarização cardíaca utilizando o intervalo QT com a fórmula de correção Fridericia (intervalo QTcF) e análise da relação da concentração-QTc não demonstra sinal claro de qualquer efeito relacionado a dose. O limite superior do intervalo de confiança (IC) 95% unilateral para efeito previsto no QTcF no Cmáx foi de 4,8 ms. Com a formula de correção Bazett (intervalo QTcB), o limite superior do intervalo de confiança (IC) 95% unilateral para o efeito previsto no QTcB no Cmáx foi de 5.9 ms.
Este medicamento pode potencializar o prolongamento do intervalo QT, o que aumenta o risco de ataque de arritmias ventriculares graves, como "torsades de pointes", que é potencialmente fatal (morte súbita).
Toxicidade pulmonar
Síndrome da Angústia Respiratória Aguda (SARA), insuficiência respiratória aguda e doença pulmonar infiltrativa difusa aguda, como pneumonite e doença pulmonar intersticial, ocorreram em pacientes recebendo KYPROLIS e alguns destes eventos foram fatais. Deve-se avaliar e interromper KYPROLIS até que essas condições se resolvam e deve-se considerar o reinício de KYPROLIS com base na avaliação do risco/benefício.
Hipertensão pulmonar
Hipertensão pulmonar tem sido relatada em pacientes tratados com KYPROLIS. Alguns desses eventos foram fatais. Deve-se avaliar e interromper KYPROLIS até a resolução do quadro e deve-se considerar o reinício de KYPROLIS com base na avaliação do risco/benefício.
Dispneia
Dispneia foi comumente relatada em pacientes tratados com KYPROLIS. Deve-se avaliar a dispneia, para que se possam excluir condições cardiopulmonares, incluindo insuficiência cardíaca e síndromes pulmonares. Deve-se interromper KYPROLIS em casos de dispneia Grau 3 e 4 até a resolução do quadro e deve-se considerar o reinício de KYPROLIS com base na avaliação do risco/benefício.
Hipertensão
Hipertensão, incluindo crises e emergências hipertensivas, foram observadas durante o tratamento com KYPROLIS. Alguns desses eventos foram fatais. É recomendado controlar a hipertensão antes de iniciar KYPROLIS. Todos os pacientes devem ser rotineiramente avaliados para a hipertensão arterial durante o tratamento com KYPROLIS e tratados conforme a necessidade. Caso a hipertensão não possa ser controlada, a dose de KYPROLIS deve ser reduzida. Em caso de crise hipertensiva, KYPROLIS deve ser interrompido até a resolução do quadro e deve-se considerar
o reinício de KYPROLIS com base na avaliação do risco/benefício.
Insuficiência renal aguda
Casos de insuficiência renal aguda foram relatados em pacientes que receberam KYPROLIS. Alguns desses eventos foram fatais. Insuficiência renal aguda foi relatada mais frequentemente em pacientes com mieloma múltiplo recidivado e refratário avançado que receberam KYPROLIS em monoterapia. A incidência foi aumentada em pacientes com um clearance de creatinina estimado diminuído, calculado com a equação de Cockcroft e Gault, antes do início da terapia com KYPROLIS. Deve-se monitorar a função renal com medidas regulares da creatinina sérica e/ou do clearance de creatinina estimado. Deve-se reduzir ou interromper o tratamento, conforme se julgar apropriado.
Síndrome de lise tumoral
Casos de Síndrome da lise tumoral (SLT), incluindo resultados fatais, foram relatados em pacientes que receberam KYPROLIS. Pacientes com uma alta carga tumoral devem ser considerados como de maior risco para a SLT. Deve-se assegurar que os pacientes estejam bem hidratados antes da administração de KYPROLIS no Ciclo 1, e nos ciclos subsequentes, conforme necessário. Reduções de medicamentos que diminuem o ácido úrico devem ser consideradas em pacientes de alto risco para SLT. Deve-se monitorar os pacientes para sinais de SLT durante o tratamento, incluindo mensurações regulares dos eletrólitos séricos e com a adoção de medidas rápidas, se necessário. Deve-se interromper o uso de KYPROLIS até a resolução da SLT.
Reações infusionais
Reações infusionais, incluindo reações que ameaçam a vida, foram relatadas em pacientes que receberam KYPROLIS. Sinais e sintomas incluem febre, calafrios, artralgia, mialgia, rubor facial, edema facial, vômitos, fraqueza, dispneia, hipotensão, síncope, aperto torácico ou angina. Essas reações podem ocorrer imediatamente ou até 24 horas após a administração de KYPROLIS. Deve-se administrar dexametasona antes do uso de KYPROLIS, seja como pré-medicação ou como parte da terapia combinada, para reduzir a incidência e a gravidade das reações (vide
"POSOLOGIA E MODO DE USAR").
Hemorragia e trombocitopenia
Casos de hemorragia (por exemplo: gastrintestinal, pulmonar e hemorragia intracraniana) têm sido relatados em pacientes tratados com
KYPROLIS, frequentemente associados com trombocitopenia. Alguns destes eventos foram fatais (vide "REAÇÕES ADVERSAS").
KYPROLIS causa trombocitopenia com nadirs de plaquetas observados no Dia 8 ou Dia 15 de cada ciclo de 28 dias, com recuperação para os valores basais de contagem de plaquetas até o início do próximo ciclo (vide "REAÇÕES ADVERSAS"). Deve-se monitorar as contagens plaquetárias frequentemente durante o tratamento com KYPROLIS. Deve-se reduzir ou interromper as doses, conforme apropriado.
Trombose venosa
Casos de eventos tromboembólicos venosos, incluindo trombose venosa profunda e embolia pulmonar, com resultados fatais, foram relatados em pacientes que receberam KYPROLIS.
Tromboprofilaxia deve ser considerada com base na avaliação individual de risco/benefício.
Toxicidade hepática
Casos de insuficiência hepática, incluindo casos fatais, foram relatados. KYPROLIS pode causar elevações das transaminases séricas (vide
"REAÇÕES ADVERSAS").As enzimas hepáticasebilirrubinadevemseravaliadasnoiníciodotratamentoemonitoradas mensalmentedurante
o tratamento com KYPROLIS, independentemente dos valores iniciais, e devem ser feitas modificações de dose adequadas com base na toxicidade. Deve-se reduzir ou interromper as doses, conforme apropriado.
Este medicamento pode causar hepatotoxicidade. Por isso, requer uso cuidadoso, sob vigilância médica estrita e acompanhado por controles períodicos da função hepática mensalmente.
Microangiopatia trombótica
Casos de microangiopatia trombótica, incluindo púrpura trombocitopênica trombótica e síndrome hemolítica urêmica (PTT/SHU) foram relatados em pacientes que receberam KYPROLIS; alguns desses eventos foram fatais. Deve-se monitorar os pacientes para sinais e sintomas de PTT/SHU. Caso se suspeite desse diagnóstico, deve-se interromper KYPROLIS e avaliar os pacientes para PTT/SHU; se esses diagnósticos forem excluídos, KYPROLIS pode ser reiniciado. A segurança do reinício da terapia com KYPROLIS em pacientes que experimentaram previamente PTT/SHU não é conhecida.
Síndrome da encefalopatia posterior reversível
A síndrome da encefalopatia posterior reversível (SEPR), previamente conhecida como síndrome da leucoencefalopatia posterior reversível (SLPR), é uma desordem neurológica que pode se apresentar com convulsões, cefaleia, letargia, confusão, cegueira, alteração do estado de consciência e outros distúrbios visuais e neurológicos, juntamente com hipertensão, e o diagnóstico é confirmado por imagens neurorradiológicas. Casos de SEPR foram relatados em pacientes recebendo KYPROLIS. KYPROLIS deve ser descontinuado em casos de suspeita de SEPR. A segurança do reinício da terapia com KYPROLIS em pacientes que experimentaram previamente SEPR não é conhecida.
Reativação do vírus da Hepatite B (HBV)
Casos de reativação do vírus da Hepatite B (HBV) foram reportados em pacientes em tratamento com KYPROLIS. Os pacientes devem fazer exames para HBV antes de iniciar o tratamento. Para os pacientes que são portadores do HBV, deve ser considerada a profilaxia com antivirais. Portadores do HBV que precisam de tratamento com KYPROLIS devem ser cuidadosamente monitorados quanto a sinais e sintomas de uma infecção ativa causada pelo HBV durante todo o tratamento e depois do fim do tratamento. Os pacientes com testes positivos para HBV devem considerar procurar um especialista antes ou durante o tratamento.
A segurança de prosseguir com o tratamento com KYPROLIS após a reativação do HBV ser controlada de forma adequada não é conhecida. Portanto, o prescritor deverá ponderar os riscos e os benefícios ao considerar prosseguir com o tratamento em face a esta situação.
Leucoencefalopatia multifocal progressiva
Casos de Leucoencefalopatia Multifocal Progressiva (PML) foram reportados em pacientes tratados com KYPROLIS com terapia imunossupressora prévia ou concomitante. A relação causal com o KYPROLIS não é conhecida. Os pacientes devem ser monitorados quanto a sinais ou sintomas neurológicos, cognitivos ou comportamentais novos ou agravantes que possam sugerir PML como parte do diagnóstico de distúrbios do SNC. Se houver suspeita de PML, suspenda a administração de KYPROLIS; os pacientes devem ser encaminhados prontamente para um especialista e os testes para o diagnóstico devem ser iniciados. Descontinue o uso do KYPROLIS, caso o diagnóstico de PML seja confirmado.
Incidência elevada de eventos fatais e eventos adversos sérios em combinação com melfalano e prednisona em pacientes inelegíveis para transplante recém diagnosticados com mieloma múltiplo
Em um estudo clínico de 955 pacientes com mieloma múltiplo recém-diagnosticado, não elegíveis para transplante, randomizados para KYPROLIS (20/36 mg/m2 por infusão de 30 minutos duas vezes por semana durante quatro seman