KOVALTRY
BAYER
octocog alfa
Anti-hemofílico.
Apresentações.
Para as apresentações de 250 UI, 500 UI e 1000 UI: Cartucho com 1 frasco-ampola com pó liofilizado para solução injetável + 1 seringa preenchida com 2,5 mL de diluente (água para injetáveis) + 1 adaptador para frasco-ampola + 1 equipo.
Para as apresentações de 2000 UI e 3000 UI: Cartucho com 1 frasco-ampola com pó liofilizado para solução injetável + 1 seringa preenchida com 5 mL de diluente (água para injetáveis) + 1 adaptador para frasco-ampola + 1 equipo.
VIA INTRAVENOSA (I.V.)
USO ADULTO E PEDIÁTRICO
Composição.
Princípio ativo: fator VIII de coagulação recombinante (betaoctocogue).
Cada frasco-ampola contém 250, 500, 1000, 2000 ou 3000 UI de fator VIII de coagulação recombinante produzido por tecnologia de DNA recombinante. É um concentrado estéril, estável, purificado, não-pirogênico e seco. É produzido por células de rim de filhote de hamster (Baby Hamster Kidney - BHK) nas quais o gene do fator VIII humano foi introduzido.
A potência (UI) é determinada usando o ensaio cromogênico.
A atividade específica de Kovaltry® é de aproximadamente 4000 UI/mg de proteína.
Cada seringa preenchida com diluente contém: 2,5 mL de água para injetáveis (para as apresentações de 250 UI, 500 UI e 1000 UI) e 5 mL de água para injetáveis (para as apresentações de 2000 UI e 3000 UI).
Após reconstituição com água para injetáveis, cada mL do produto contém:
Kovaltry® 250 UI (Unidades Internacionais): 100 UI/mL de betaoctocogue.
Kovaltry® 500 UI (Unidades Internacionais): 200 UI/mL de betaoctocogue.
Kovaltry® 1000 UI (Unidades Internacionais): 400 UI/mL de betaoctocogue.
Kovaltry® 2000 UI (Unidades Internacionais): 400 UI/mL de betaoctocogue.
Kovaltry® 3000 UI (Unidades Internacionais): 600 UI/mL de betaoctocogue.
Excipientes: sacarose, histidina, glicina, cloreto de sódio, cloreto de cálcio e polissorbato 80.
Diluente: água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Kovaltry® é indicado para o tratamento e profilaxia de sangramento na hemofilia A em todos os grupos etários, manejo perioperatório na hemofilia A em todos os grupos etários, tratamento profilático para prevenir ou reduzir a frequência de episódios de sangramentos espontâneos em todos os grupos etários.
Kovaltry® não contém fator de von Willebrand e não é indicado para doença de von Willebrand.
2. RESULTADOS DE EFICÁCIA
-
- Controle e Prevenção de Sangramento
Foram conduzidos dois estudos multicêntricos, abertos, cruzados, não controlados, randomizados em pacientes tratados previamente (PTPs), adultos e adolescentes, com hemofilia A grave ( < 1%). Foi conduzido um estudo multicêntrico, aberto, não controlado em pacientes pediátricos tratados previamente (PTPs) < 12 anos de idade (Parte A) e pacientes não tratados previamente (PUPs)/ pacientes minimamente tratados (MTPs) < 6 anos de idade (Parte B) com hemofilia A grave.
LEOPOLD I (Estudo 1): O objetivo foi demonstrar a farmacocinética (PK), segurança e eficácia do tratamento profilático e hemostasia durante cirurgias. A duração do estudo foi de 1 ano com uma extensão opcional por mais 1 ano. Foram incluídos setenta e três (73) pacientes previamente tratados de forma profilática ou sob demanda (Parte A: 26 pacientes (PK), Parte B: 62 pacientes (segurança e eficácia) e Parte C: 5 pacientes apenas cirúrgicos ([mais 5 pacientes em extensão para 12 cirurgias]). Cinquenta e cinco (55) pacientes continuaram no estudo de extensão de um ano (para detalhes adicionais veja a Tabela 1).
LEOPOLD II (Estudo 2): O objetivo foi demonstrar a superioridade do tratamento profilático em relação ao tratamento sob demanda, durante um período de tratamento de 1 ano em 80 pacientes previamente tratados sob demanda. Os pacientes foram randomizados para serem tratados no regime sob demanda ou profilaxia (para detalhes adicionais veja a Tabela 1).
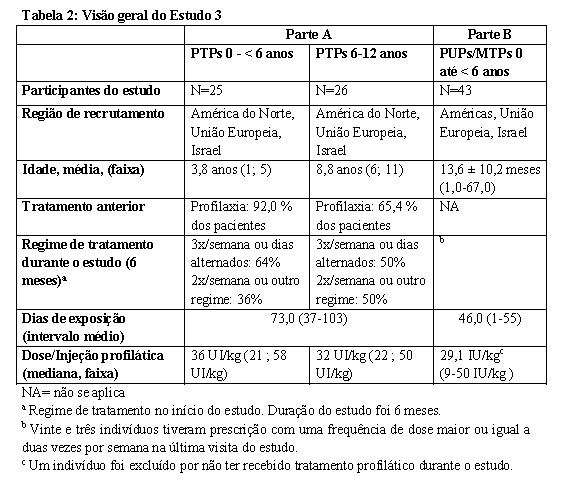
LEOPOLD Kids (Estudo 3): O objetivo foi avaliar a segurança, eficácia, farmacocinética e manejo perioperatório da hemostasia durante cirurgia em pacientes tratados previamente (PTPs: ≥ 50 DEs)
≤
12 anos (Parte A) e pacientes não tratados previamente (PUPs) e pacientes minimamente tratados (MTPs: ≤ 3 DEs) < 6 anos (Parte B).. Oitenta e dois pacientes (46 pacientes da Parte A, 36 pacientes da Parte B) continuaram no Estudo de extensão. Mais detalhes, consultar a Tabela 2,Tabela 4 e Tabela 6.
Um total de 247 pacientes (204 PTPs e 43 PUPs/MTPs) foram avaliados no programa de estudos, 153 pacientes ≥ 12 anos e 94 pacientes < 12 anos. Duzentos e oito (208) pacientes (174 PTPs, 34 PUPs/MTPs) foram tratados por pelo menos 12 meses, e 98 desses pacientes (78 PTPs, 20 PUPs/MTPs) por pelo menos 24 meses.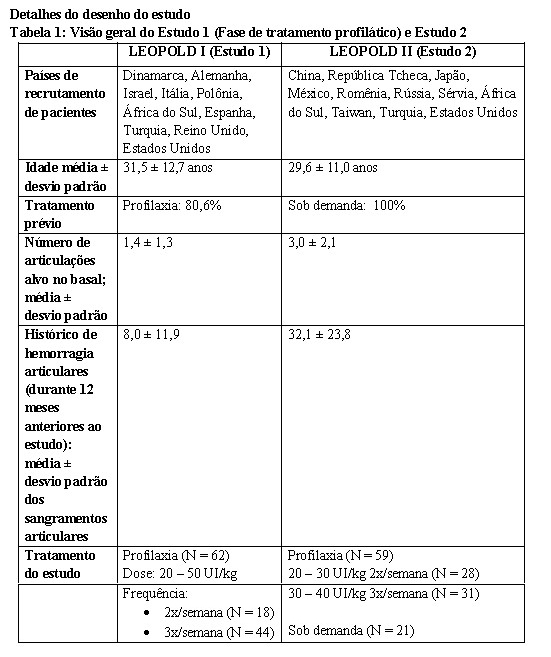
- Tratamento de sangramento (tratamento sob demanda) na hemofilia A em todos os grupos etários.
Adolescentes ( > 12 anos) e Adultos
Um total de 1892 episódios de sangramentos em 110 indivíduos foram tratados com Kovaltry® no Estudo 1 e no Estudo 2. A maioria dos episódios de sangramento foram espontâneos, localizados nas articulações e leve a moderado com relação à gravidade.
No Estudo 1 e no Estudo 2, em um total de 1859 sangramentos tratados, as respostas ao tratamento foram avaliadas pelos indivíduos comparando às suas experiências prévias de tratamento.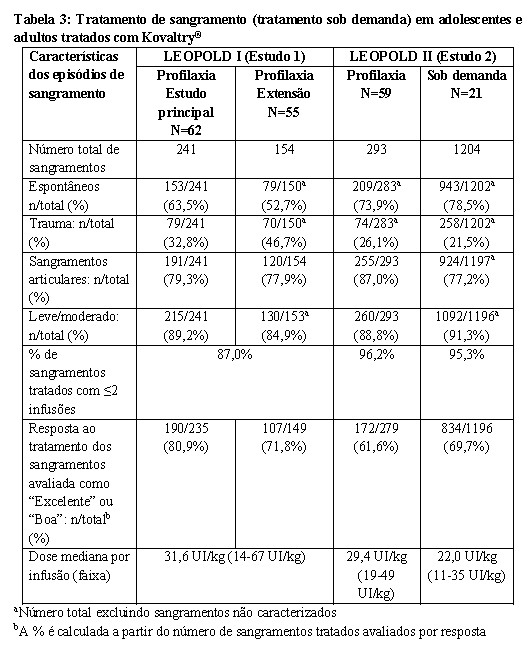
Crianças < 12 anos
Crianças de 0-12 anos previamente tratadas e PUPs/MTPs de 0-6 anos foram avaliadas para demonstrar segurança e eficácia do tratamento profilático. Para detalhes e resultados do estudo veja Tabela 2 e 4.
Um total de 97 episódios de sangramento em 28 indivíduos pediátricos tratados previamente e 105 episódios de sangramento em 37 PUPs/MTPs foram tratados com Kovaltry®. A maioria (96,9% em PTPs e 97,1% em PUPs/MTPs) dos sangramentos foram leves a moderados em relação à gravidade em ambos os grupos. Cinquenta e nove (72,8%) e 62 (59,0%) sangramentos foram relacionados a trauma para indivíduos previamente tratados e pacientes PUPs/MTPs, respectivamente. Durante o período de 6 meses de tratamento, a dose mediana de Kovaltry® para tratamento de episódios de sangramento em indivíduos previamente tratados foi 36,94 UI/kg por infusão (faixa 20,8-71,6 UI/kg).
A dose mediana por injeção para PUPs/MTPs foi 40,5 UI/kg (faixa de 21 a 112).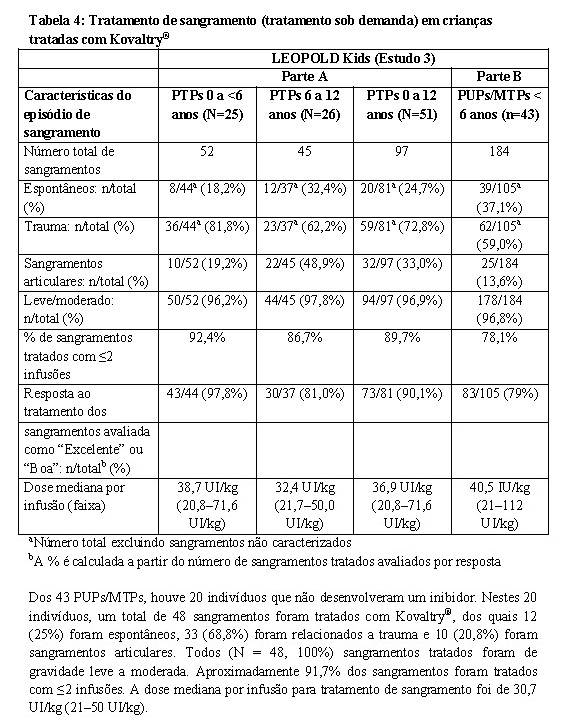
Dos 43 PUPs/MTPs, houve 20 indivíduos que não desenvolveram um inibidor. Nestes 20 indivíduos, um total de 48 sangramentos foram tratados com Kovaltry®, dos quais 12 (25%) foram espontâneos, 33 (68,8%) foram relacionados a trauma e 10 (20,8%) foram sangramentos articulares. Todos (N = 48, 100%) sangramentos tratados foram de gravidade leve a moderada. Aproximadamente 91,7% dos sangramentos foram tratados com ≤2 infusões. A dose mediana por infusão para tratamento de sangramento foi de 30,7 UI/kg (21-50 UI/kg).
- Manejo perioperatório na hemofilia A
Um total de 15 (PTPs: 14; PUP: 1) cirurgias de grande porte e 51 (PTPs: 46; PUPs: 5) cirurgias de pequeno porte foram realizadas em 50 indivíduos (PTPs:43 adultos e adolescentes e 1 criança menor de 12 anos de idade; PUP: 6 crianças menores de 6 anos de idade) com hemofilia A grave. Sete das 14 cirurgias de grande porte em PTPs foram procedimentos ortopédicos, incluindo artroplastias. Aproximadamente 51% das cirurgias de pequeno porte em PTPs foram extrações dentárias. Todos os indivíduos receberam Kovaltry® como injeção em bolus. Nos indivíduos adultos e adolescentes, as doses iniciais administradas de Kovaltry® estavam na faixa de 3000-5000 UI. A dose mediana total no dia da cirurgia foi 107,5 UI/kg (faixa 60 a 207 UI/kg). Em um único indivíduo menor que 12 anos de idade previamente tratado que foi submetido a cirurgia de grande porte, a dose de Kovaltry® inicial total administrada foi 2500 UI (108,7 UI/kg). O indivíduo não tratado previamente com < 6 anos de idade que foi submetido a uma cirurgia de grande porte tinha um inibidor do fator VIII de alto título e recebeu uma dose total de 5.000 UI de Kovaltry® no dia da cirurgia.
A perda de sangue, durante e após a cirurgia, ficou dentro dos limites esperados. O controle hemostático foi avaliado pelos cirurgiões como "bom" (sangramento perioperatório ligeiramente, mas não clinicamente aumentado significativamente acima das expectativas para o paciente não hemofílico; tratamento semelhante ao paciente não hemofílico) ou "excelente" (perda de sangue perioperatória semelhante ao não hemofílico paciente) para todas as cirurgias, exceto para uma cirurgia de pequeno porte em um PUP para o qual nenhuma avaliação foi fornecida.
- Tratamento profilático (profilaxia de sangramento) para prevenir ou reduzir a frequência de episódios de sangramentos espontâneos
Adolescentes ( > 12 anos) e Adultos
Um total de 140 indivíduos foram tratados com Kovaltry® por pelo menos 12 meses com mediana (faixa) de 157 dias de exposição (DEs) (25-178) no Estudo 1, [305 DEs (25-355) inclusive na fase de extensão], e 153 DEs (103-187) no Estudo 2. Em ambos Estudos, indivíduos na população Intenção-de-Tratar (ITT, do inglês Intent-To-Treat) receberam 95% a 100% do número de infusões profiláticas prescritas.
A Tabela 5 resume o consumo e as taxas gerais de sucesso com a profilaxia nos Estudos 1 e 2. A média e a mediana da taxa anualizada de sangramento (ABR, do inglês Annualized Bleeding Rate) para a população ITT no Estudo 1 foi de 3,8 ± 5,2 e 1 sangramento/ano, respectivamente. No Estudo 2, uma comparação das taxas de sangramento entre os indivíduos recebendo terapia sob demanda versus profilaxia (por ANOVA) demonstrou diferença estatisticamente significativa (p < 0.0001) na ABR mediana em indivíduos recebendo terapia sob demanda (60 sangramentos por ano) quando comparados com indivíduos recebendo profilaxia (2 sangramentos por ano). No Estudo 2, a ABR média em indivíduos recebendo terapia sob demanda foi 57,7 ± 24,6 versus 4,9 ± 6,8 em indivíduos recebendo profilaxia.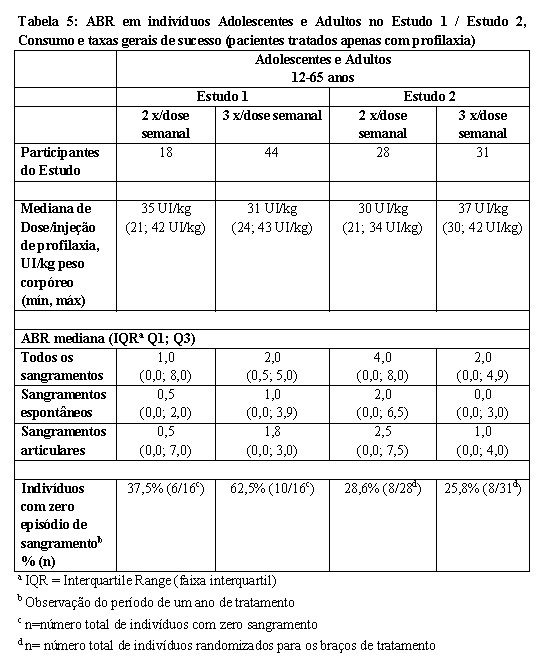
Crianças < 12 anos
Na Parte A, um total de 51 pacientes tratados previamente (PTPs) foram tratados com Kovaltry® por pelo menos 6 meses com mediana (faixa) de 73 DEs (37-103). Indivíduos receberam > 95% do número de infusões profiláticas prescritas. Para os 42 indivíduos que receberam tratamento profilático com Kovaltry® na Parte B, a duração mediana do tratamento profilático foi de aproximadamente 161,5 dias (intervalo de 13 a 656 dias), com uma mediana (intervalo) de 44,5 DEs (intervalo de 1 a 55 dias). Veja a Tabela 6.
Em crianças de 12 anos de idade ou mais novas (n=51), a mediana (IQR Q1; Q3) da ABR de 48 horas após infusão profilática foi 0 (0; 4) para todos os sangramentos, e 0 (0; 0) para sangramentos espontâneos e articulares. A mediana (IQR Q1; Q3) da ABR durante o tratamento profilático independente do tempo de infusão foi 1,9 (0; 6) para todos os sangramentos, 0 (0; 0) para sangramentos espontâneos e 0 (0; 2) para sangramentos articulares. A ABR média de 48 horas após infusão profilática foi 2,04 ± 2,91. A ABR média a qualquer tempo durante o regime profilático foi 3,75 ± 4,98.
Em ambos grupos etários (0 a < 6 anos e 6 a 12 anos), a ABR para sangramentos espontâneos e sangramentos articulares de 48 horas após tratamento profilático [mediana da ABR (IQR Q1; Q3)] foi 0 (0; 0). O número mediano (IQR Q1; Q3) anualizado de sangramentos espontâneos durante o tratamento profilático independente do tempo de infusão foi 0 (0; 0). A mediana (IQR Q1; Q3) do número anualizado de sangramentos articulares durante o tratamento profilático independente do tempo de infusão foi 0 (0; 1,9) no grupo de 0 a < 6 anos e 0 (0; 2,1) no grupo de 6 a 12 anos (ver Tabela 6).
A maioria (32/53) dos sangramentos que ocorreram dentro de 48 horas após uma infusão profilática anterior foram relacionados a trauma. Vinte e três (45,1%) indivíduos não reportaram nenhum sangramento duramente os 6 meses do período profilático.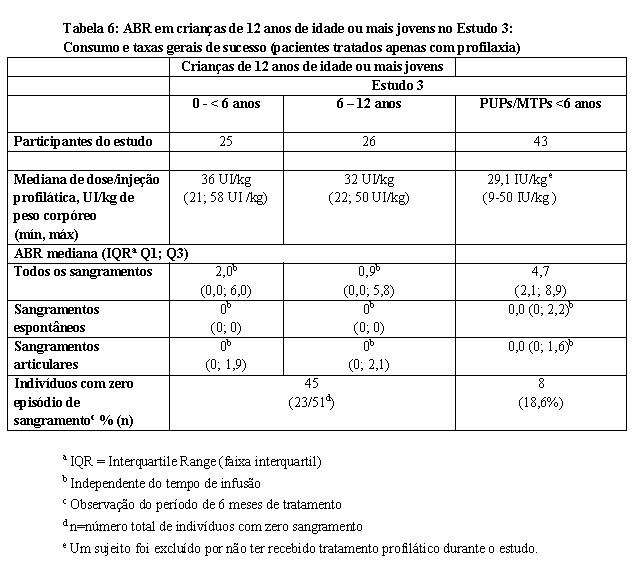
Em PUPs/MTPs, a ABR média para todos os sangramentos dentro de 48 horas após a infusão profilática foi de 1,9 ± 3,3. A ABR média para todos os sangramentos em qualquer momento durante o regime profilático foi de 7,1 ± 8,6.
Em PUPs/MTPs que não desenvolveram um inibidor (N=20), 12 indivíduos não experienciaram nenhum episódio de sangramento dentro de 48 horas após uma infusão profilática, 5 dos quais não tiveram episódios de sangramento durante todo o período de tratamento profilático. Para os 20 indivíduos sem inibidor do Fator VIII, a mediana (IQR Q1; Q3) ABR dentro de 48 horas após a infusão profilática foi 0 (0; 1,8) [média: 0,9 ± 1,4] para todos os sangramentos. A mediana (Q1; Q3) ABR independente do tempo de infusão foi de 2,9 (0,6; 7,4) [média; 4,6 ± 5,0] para todos os sangramentos.
Dos 94 indivíduos tratados nos estudos principais do LEOPOLD Kids, 82 indivíduos (46 indivíduos da Parte A, 36 indivíduos da Parte B) continuaram no estudo de extensão por um tempo médio de 3,1 anos (variação de 0,3 a 6,4 anos). Para estes 82 indivíduos, a mediana do tempo total em todo o estudo (principal e extensão) foi de 3,8 anos (intervalo de 0,8 a 6,7 anos). A maioria dos indivíduos (76 de 82, 92,7%) acumulou pelo menos 100 DEs cumulativos.
LEOPOLD Kids (Estudo 3) - Estudo de extensão - Resultados de eficácia
Durante o estudo de extensão, 67 indivíduos receberam Kovaltry® como tratamento profilático por uma mediana de 3,8 anos (intervalo de 0,5 a 6,4 anos). A dose média por infusão profilática foi de 34 UI/kg (intervalo de 16 - 57 UI/kg), e a maioria dos indivíduos (621 de 67; 92,591%) recebeu profilaxia com uma frequência de dosagem de pelo menos 2 vezes por semana durante o estudo de extensão.
A mediana (Q1: Q3) ABR para sangramentos totais ocorrendo dentro de 48 h após uma infusão profilática foi de 0,7 (0,0 a 1,9). A mediana (Q1; Q3) ABR independente do tempo de infusão foi de 1,9 (0,3 - 3,9).
Entre os 67 indivíduos, um total de 472 sangramentos foram tratados com Kovaltry®, exigindo 1-2 infusões para a maioria dos sangramentos (394 de 472; 83,5%), e a resposta ao tratamento foi boa ou excelente na maioria (415 de 472; 87,9%) dos casos.
3. CARACTERÍSTICAS FARMACOLÓGICAS
- Propriedades Farmacodinâmicas
- Mecanismo de ação
Kovaltry® fornece um meio de repor temporariamente o fator VIII de coagulação ausente para uma homeostase efetiva.
- Efeitos farmacodinâmicos
O tempo de tromboplastina parcial ativada (TTPa) é prolongado em pessoas com hemofilia. A determinação do TTPa é um ensaio convencional in vitro para avaliar a atividade biológica do fator VIII. O tratamento com rFVIII (FVIII recombinante) normaliza o TTPa de maneira semelhante ao obtido com o fator VIII plasmático.
- Propriedades Farmacocinéticas
As propriedades farmacocinéticas (PK) de Kovaltry® foram investigadas em 3 estudos em pacientes previamente tratados, adultos e crianças, em comparação com o Kogenate®. Para todas as avaliações farmacocinéticas, foram administrados 50 UI/kg de Kovaltry® ou Kogenate®. Foram coletadas amostras sanguíneas seriadas durante 48 horas em adultos e durante 24 horas em crianças < 12 anos de idade.
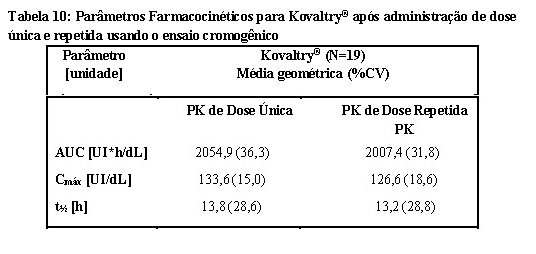
A farmacocinética foi avaliada em 26 pacientes previamente tratados (PTPs) (idade de 12 a 61 anos) com hemofilia A grave seguindo 50 UI/kg de Kogenate® ou Kovaltry® em um estudo randomizado, cruzado com pelo menos ≥ 3 dias de washout. Ambos os produtos foram avaliados usando ensaio cromogênico para essa análise farmacocinética.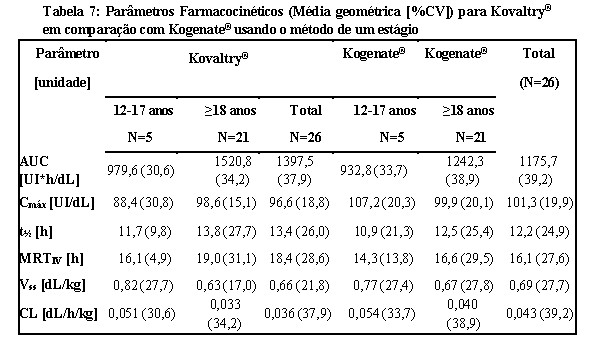
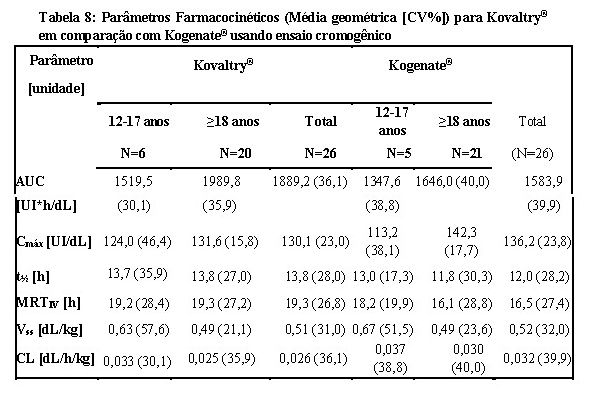
A AUC (Área sob a curva, do inglês Area Under the Curve) foi aproximadamente 19% maior, o CL (Clearance) 16% menor (p < 0,0005) e a meia vida (t½) cerca de 10% maior (p < 0,05) para Kovaltry® em comparação com Kogenate®. Em ambos os ensaios, os Intervalos de Confiança de 90% para a razão Kovaltry®/ Kogenate® de Cmáx estavam dentro dos critérios de bioequivalência de 0,80 a 1,25. A biodisponibilidade de Kovaltry® foi pelo menos não inferior à de Kogenate®. Para a AUC, o Intervalo de Confiança de 90% foi de 1,13 a 1,25 quando se utilizou o método de um estágio e de 1,11 a 1,28 quando se utilizou o ensaio cromogênico. No geral, os dados demonstram a não inferioridade da farmacocinética de Kovaltry® em comparação com Kogenate®.
A farmacocinética foi reavaliada em 19 pacientes após 6 a 12 meses.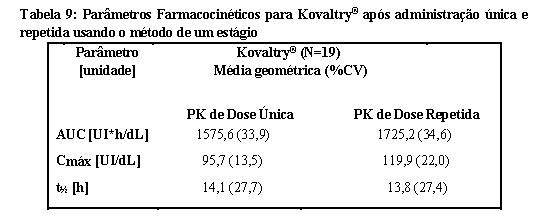
Medidas de farmacocinética repetidas após 6 a 12 meses de tratamento profilático com Kovaltry® não indicaram quaisquer alterações relevantes nas características farmacocinéticas após o tratamento em longo prazo.
Parâmetros farmacocinéticos calculados a partir de 19 pacientes com < 12 anos de idade estão disponíveis para 8 pacientes na faixa etária de 0 a < 6 anos e 11 pacientes na faixa etária de 6 a < 12 anos como mostrado na Tabela 11.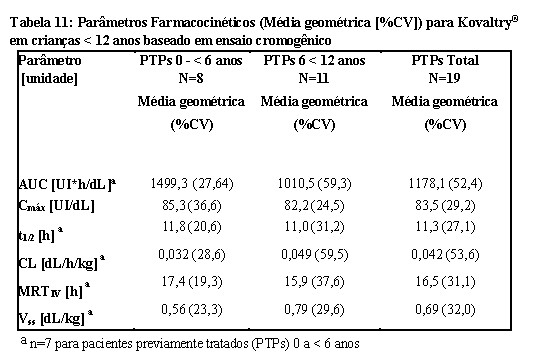
Crianças menores de 12 anos de idade têm concentrações plasmáticas mais baixas em comparação com pacientes previamente tratados (PTPs) > 12 anos de idade. A meia vida (t1/2) entre os grupos etários é semelhante.
O modelo farmacocinético populacional foi desenvolvido usando dados de farmacocinética e de recuperação de 183 pacientes que participaram dos estudos Leopold 1, Leopold 2 e Leopold Kids (≥ 18 anos N=109; de 12 a < 18 anos N=23; de 6 a < 12 anos N=26; < 6 anos N=25). A farmacocinética de Kovaltry® é melhor descrita utilizando um modelo de dois compartimentos. Idade, altura, peso, índice de massa corpórea (IMC), massa corpórea magra (LBW) e raça foram investigados como covariáveis, uma vez que foram considerados de interesse clínico. A massa corpórea magra explicou grande parte da variabilidade tanto do clearance quanto do volume de distribuição como esperado para um composto distribuído principalmente no plasma. Os parâmetros de farmacocinética previstos usando o modelo populacional de farmacocinética para os pacientes que participaram do estudo farmacocinético foram semelhantes aos observados utilizando métodos não compartimentais.
As análises de todas as recuperações registradas in vivo (RIV, do inglês Recorded In Vivo) em pacientes previamente tratados (PTPs), adultos/adolescentes, demonstrou um aumento mediano de FVIII:C > 2 UI/ dL por UI/kg de peso corpóreo de Kovaltry® administrado com ambos os ensaios. Este resultado é semelhante aos valores reportados para o fator VIII plasmático. Os valores de recuperação in vivo medianos foram 1,62 kg/dL para o grupo etário mais jovem (0 a < 6 anos) e 1,80 kg/dL para o grupo etário mais velho (6 a 12 anos). Não houve alteração relevante ao longo do período de tratamento de 6 a 12 meses.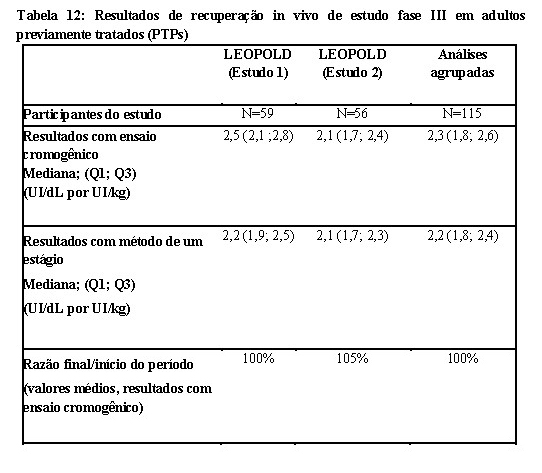
- Pacientes Pediátricos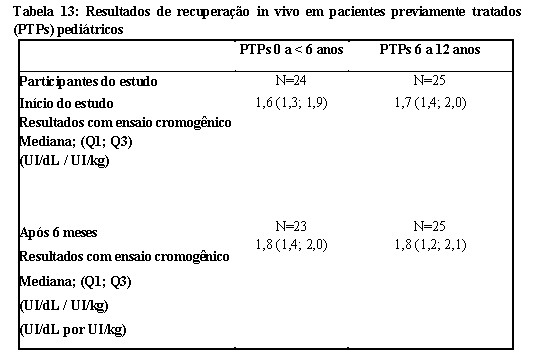
- Diferenças Étnicas
As diferenças étnicas na farmacocinética de Kovaltry® são improváveis porque a eliminação de Kovaltry®, uma preparação proteica, não é mediada pela depuração com enzimas que fazem o metabolismo de fármacos com polimorfismo genético. A análise farmacocinética da população contendo pacientes brancos (n = 132), asiáticos (n = 31), negros (n = 11) e hispânicos (n = 10) indicou que não houve diferenças étnicas significativas nas raças incluídas nos estudos.
- Dados pré-clínicos de segurança
Estudos não-clínicos avaliando Kovaltry® em modelos de eficácia em camundongos com hemofilia A demonstraram restauração da hemostasia. O programa não-clínico de segurança não identificou nenhuma preocupação para os seres humanos com base nos estudos de segurança farmacológica, toxicidade aguda, toxicidade de dose repetida e genotoxicidade.
- Embriotoxicidade/Teratogenicidade
O desenvolvimento embrio-fetal não foi avaliado em animais uma vez que o FVIII é uma proteína de reposição endógena; além disso a população de pacientes é principalmente masculina.
- Toxicidade reprodutiva
Nenhum efeito nos órgãos reprodutivos masculinos foi observado em estudos de toxicidade por administração repetida. O FVIII é uma proteína endógena, e nenhum efeito na fertilidade com esta proteína foi observado em seres humanos.
- Genotoxicidade e carcinogenicidade
Kovaltry® demonstrou ser não-genotóxico no ensaio de linfoma de camundongos.
Estudos carcinogênicos não foram realizados uma vez que o FVIII é uma proteína de reposição endógena e o rFVIII (FVIII recombinante) não demonstrou nenhum potencial genotóxico ou carcinogênico.
- Toxicidade de dose repetida
Doses várias vezes superiores à dose clínica recomendada (relacionada com o peso corpóreo) não demonstraram nenhuma toxicidade em estudos de dose única e multidose em ratos, coelhos e cães.
- Avaliação de risco ambiental
Não aplicável.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer um dos excipientes.
Reações alérgicas conhecidas à proteína de hamster ou camundongo.
5. ADVERTÊNCIAS E PRECAUÇÕES
- Hipersensibilidade
Hipersensibilidade à proteína de hamster ou camundongo. Reações de hipersensibilidade tipo alérgicas podem ocorrer com Kovaltry®. O produto pode conter traços de proteínas de hamster ou camundongos que em alguns pacientes podem causar reações alérgicas.
Os pacientes devem ser informados de que a potencial ocorrência de pressão no peito, tonturas, hipotensão leve e náuseas durante a infusão podem constituir uma advertência antecipada para hipersensibilidade e reações anafiláticas. Tratamento sintomático e terapia para hipersensibilidade deverão ser instituídos conforme apropriado. Se ocorrerem reações alérgicas ou anafiláticas, a injeção/infusão deve ser interrompida imediatamente. Em caso de anafilaxia, os padrões médicos vigentes para o tratamento devem ser observados.
- Inibidores
A formação de anticorpos neutralizantes (inibidores) contra o fator VIII é uma complicação conhecida no manejo de pacientes com hemofilia A. Esses inibidores são geralmente imunoglobulinas IgG dirigidas contra a atividade pró-coagulante do fator VIII, as quais são quantificadas em Unidades Bethesda (UB) por mL de plasma usando o ensaio modificado. O risco de desenvolver inibidores está correlacionado à exposição ao fator VIII, sendo este risco mais elevado nos primeiros 20 dias de exposição e a outros fatores genéticos e ambientais. Raramente, os inibidores podem se desenvolver após os primeiros 100 dias de exposição.
Em geral, todos os pacientes tratados com produtos de fator VIII de coagulação devem ser cuidadosamente monitorados para o desenvolvimento de inibidores através de observações clínicas e testes laboratoriais apropriados.
- Infecções relacionadas a cateter
Infecções relacionadas a cateter podem ser observadas quando Kovaltry® é administrado através de equipos de acesso venoso central (CVADs, do inglês Central Venous Access Devices). Estas infecções não foram associadas ao produto propriamente dito.
- Distúrbios cardiovasculares
Pacientes com hemofilia com doenças ou fatores de risco cardiovasculares podem ter o mesmo risco de desenvolver eventos cardiovasculares que pacientes não hemofílicos quando a coagulação tiver sido normalizada pelo tratamento com fator VIII. Consequentemente os pacientes devem ser avaliados quanto aos fatores de risco cardíaco.
- Fertilidade, gravidez e lactação
Não foram conduzidos estudos em reprodução animal com fator VIII. Com base na ocorrência rara de hemofilia A em mulheres, a experiência em relação ao uso do fator VIII durante a gravidez e lactação não está disponível. Portanto, o fator VIII deve ser utilizado durante a gravidez e lactação somente se for claramente indicado.
- Efeitos sobre a habilidade de dirigir veículos ou operar máquinas
Kovaltry® não influencia sobre a habilidade de dirigir veículos ou operar máquinas.
"Este medicamento não deve ser utilizado em mulheres grávidas sem orientação médica ou do cirurgião-dentista." (Categoria C)
"Atenção diabéticos: contém açúcar."
6. INTERAÇÕES MEDICAMENTOSAS
Nenhuma interação de produtos do fator VIII de coagulação humana com outros medicamentos foi reportada.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Kovaltry® deve ser armazenado sob refrigeração (temperatura entre 2°C e 8°C). Não congelar.
Manter o frasco-ampola e a seringa preenchida dentro da embalagem para proteger da luz.
Não utilizar o produto fora do prazo de validade indicado nos rótulos e no cartucho do produto.
Dentro do seu prazo de validade, o armazenamento do pó liofilizado para solução injetável pode ser feito à temperatura até 25°C por até 12 meses ou à temperatura até 30°C por até 6 meses, como em situações de armazenamento doméstico.
Se produto for armazenado fora do refrigerador, adicione a data de sua retirada da refrigeração e anote o novo prazo de validade no cartucho e no frasco-ampola. A nova data de validade deve ser 12 meses (25°C) ou 6 meses (30°C) a partir da data da retirada do produto do refrigerador, ou a data de validade previamente descrita na embalagem, a que ocorrer mais cedo. Uma vez que o produto é retirado da refrigeração, ele não pode ser devolvido ao refrigerador.
Após reconstituído, Kovaltry® deve ser administrado o mais rapidamente possível e não mais do que 3 horas após a reconstituição. Este produto é para uso único. Qualquer solução não utilizada deve ser descartada.
O prazo de validade de Kovaltry® é de 30 meses a partir da data de sua fabricação.
"Número de lote e datas de fabricação e validade: vide embalagem."
"Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original."
"Após o preparo, administrar em até 3 horas."
- Características organolépticas
Pó liofilizado para solução injetável:
- branco a levemente amarelado (antes da reconstituição).
- líquido límpido e incolor (após reconstituição com água para injetáveis).
Diluente (água para injetáveis): líquido límpido.
"Antes de usar, observe o aspecto do medicamento."
"Todo medicamento deve ser mantido fora do alcance das crianças."
8. POSOLOGIA E MODO DE USAR
- Método de administração
Uso intravenoso.
- Dosagem
O número de unidades de fator VIII administrado é expresso em Unidades Internacionais (UI), de acordo com o padrão atualmente vigente da Organização Mundial de Saúde (OMS) para produtos de fator VIII. A atividade de fator VIII no plasma é expressa em porcentagem (relativa ao plasma humano normal) ou em Unidades Internacionais (relativa ao Padrão Internacional para fator VIII plasmático). Uma Unidade Internacional (UI) de atividade de fator VIII é equivalente a essa quantidade de fator VIII em 1 mL de plasma humano normal. O cálculo da dose necessária de fator VIII é baseado na constatação empírica de que 1 (uma) Unidade Internacional (UI) de fator VIII por kg de peso corpóreo aumenta a atividade do fator VIII plasmático em 1,5% a 2,5% da atividade normal.
A dose e a duração da terapia de substituição para alcançar a hemostasia devem ser individualizadas de acordo com as necessidades do paciente (peso, gravidade da deficiência na função hemostática, local e extensão/gravidade do sangramento, nível de inibidores e nível de fator VIII desejado).
O efeito clínico do fator VIII é o elemento mais importante na avaliação da eficácia do tratamento. Pode ser necessário administrar mais Kovaltry® do que se estimava a fim de atingir resultados clínicos satisfatórios. Se a dose calculada não atingir os níveis esperados de fator VIII ou se o sangramento não for controlado após a administração da dose calculada, deve-se suspeitar da presença de inibidor circulante no paciente. Sua presença deve ser constatada e o nível do inibidor quantificado por exames laboratoriais apropriados. Quando um inibidor está presente, a dose requerida de Kovaltry® é extremamente variável e a dose pode somente ser determinada pela resposta clínica.
Tratamento sob demanda
A dose necessária é determinada usando a seguinte fórmula:
Unidades requeridas = peso corpóreo (kg) x aumento do fator VIII desejado (% ou UI/dL) x recíproco da recuperação observada
A dose individual usual é de 10-30 UI/kg de peso corpóreo. Doses mais altas são recomendadas para hemorragias maiores ou com risco para a vida.
A dose necessária para alcançar a hemostasia depende do tipo e da gravidade do episódio de sangramento.
Para pacientes com uma recuperação abaixo de 2 (que pode ocorrer, por exemplo, em crianças pequenas), doses maiores podem ser necessárias.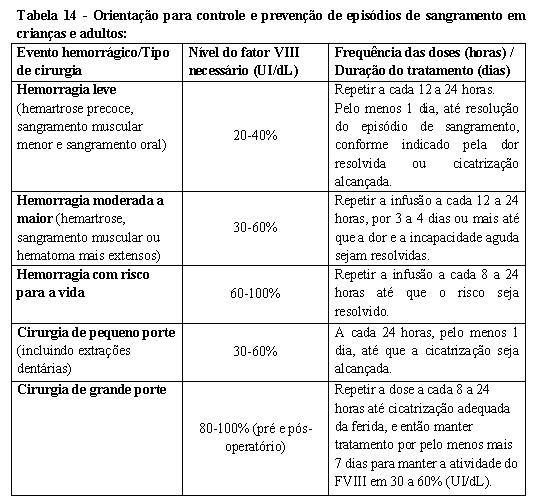
Tratamento profilático para pacientes adolescentes e adultos
Para profilaxia em longo prazo contra sangramentos em pacientes com hemofilia A grave, as doses recomendadas são 20 a 40 UI de fator VIII por kg de peso corpóreo duas ou três vezes por semana. Em alguns casos, especialmente em pacientes mais jovens, podem ser utilizados intervalos de dose mais curtos ou doses maiores.
- Taxa de Administração
A taxa de administração deve ser adaptada à resposta individual de cada paciente.
- Informações adicionais para populações especiais
- Pacientes pediátricos
Kovaltry® é apropriado para o uso em pacientes pediátricos. Estudos de segurança e eficácia foram realizados com crianças entre 0 a 12 anos. As doses profiláticas recomendadas são de 20 a 50 UI/kg duas vezes por semana, três vezes por semana ou em dias alternados, de acordo com as necessidades individuais. Para pacientes pediátricos acima de 12 anos de idade a dose recomendada é a mesma que aquela para adultos.
- Pacientes geriátricos
Os estudos clínicos não incluíram pacientes com 65 anos de idade ou mais para que fosse possível determinar se respondem de maneira diferente que os pacientes mais jovens. Entretanto, a experiência clínica com outros produtos de fator VIII não identificaram diferenças entre os pacientes idosos e mais jovens. Assim como para qualquer paciente que receba fator VIII de coagulação recombinante (rFVIII), a seleção de dose para um paciente idoso deve ser individualizada.
- Pacientes com insuficiência hepática
O ajuste de dose para pacientes com insuficiência hepática não foi estudado nos estudos clínicos.
- Pacientes com insuficiência renal
O ajuste de dose para pacientes com insuficiência renal não foi estudado nos estudos clínicos.
- Gênero
Não há dados disponíveis para mulheres, como a hemofilia raramente ocorre em mulheres, mulheres não foram incluídas nos estudos clínicos.
- Dose máxima diária
Ver no item "Dosagem" a dose necessária para obtenção da hemostasia.
- Incompatibilidades
Este medicamento não deve ser misturado a outros produtos medicinais ou solventes.
- Instruções de uso
Para infusão, o produto deve ser preparado sob condições assépticas. Se algum componente da embalagem estiver aberto ou danificado, não utilize este componente. Medicamentos parenterais devem ser inspecionados visualmente quanto à presença de material particulado e alteração da cor antes da administração. Não utilize Kovaltry® se notar alguma partícula ou turbidez na solução.
Kovaltry® deve ser reconstituído e administrado utilizando os componentes fornecidos em cada embalagem.
É recomendado que toda vez que Kovaltry® for administrado a um paciente, que o nome do paciente e o número de lote do produto seja anotado para se manter um vínculo entre o paciente e o lote do produto.
Adaptador para frasco-ampola e diluente (seringa preenchida)
O produto reconstituído deve ser filtrado antes da administração para remover possível material particulado da solução. A filtração é feita utilizando o adaptador para frasco-ampola.
1. Aqueça com suas mãos tanto o frasco-ampola como a seringa fechados até uma temperatura confortável (não exceder 37°C).
2. Remova o lacre protetor do frasco-ampola (Fig. A). Limpe assepticamente a tampa de borracha com álcool, tendo o cuidado de não manusear a tampa de borracha.
3. Coloque o frasco-ampola com produto em uma superfície firme, não escorregadia. Retire a cobertura de papel da embalagem plástica do adaptador para frasco-ampola. Não remova o adaptador da embalagem plástica. Segurando a embalagem do adaptador, coloque-o sobre o frasco-ampola e pressione firmemente para baixo (Fig. B). O adaptador irá se acoplar sobre a tampa do frasco-ampola. Não remova a embalagem do adaptador nesta etapa.
4. Segurando a seringa pelo corpo, retire a tampa da ponta da seringa (Fig. C). Não toque a ponta da seringa com as mãos ou qualquer superfície. Coloque a seringa de lado para seu uso posterior.
5. Agora remova e descarte a embalagem do adaptador (Fig. D).
6. Conecte a seringa preenchida na parte rosqueada do adaptador para frasco-ampola girando no sentido horário (Fig. E).
7. Segure o êmbolo pela parte superior e retire-o da embalagem. Evite tocar a lateral e a parte rosqueada do êmbolo. Imediatamente conecte o êmbolo girando-o firmemente no sentido horário na tampa de borracha da seringa com rosca. (Fig. F).
8. Injete o diluente empurrando lentamente o êmbolo para baixo (Fig. G).
9. Gire em círculos delicadamente o frasco-ampola até completa dissolução (Fig. H). Não agite o frasco. Assegure-se de que o pó esteja completamente dissolvido. Não use soluções que contenham partículas visíveis ou que estejam turvas.
10. Transfira a solução para a seringa segurando o frasco-ampola na extremidade acima do adaptador para frasco-ampola e seringa (Fig. I) e em seguida puxe o êmbolo lenta e suavemente. Assegure-se de que o conteúdo total do frasco-ampola foi passado para seringa. Remova o máximo de ar possível antes de remover a seringa do frasco-ampola, empurrando o ar lenta e cuidadosamente de volta para dentro do frasco-ampola.
11. Com o êmbolo no lugar, remova a seringa do adaptador para frasco-ampola (o adaptador para frasco-ampola deve permanecer acoplado ao frasco-ampola). Conecte a seringa ao equipo fornecido dentro da embalagem de Kovaltry® e injete por via intravenosa (Fig. J).
12. Se o mesmo paciente for receber a administração de mais de um frasco-ampola, faça a reconstituição de cada frasco-ampola com a respectiva seringa preenchida com diluente e, então, junte as soluções em uma seringa maior (não fornecida na embalagem de Kovaltry®) e administre da maneira usual.
Medicamentos de uso parenteral devem ser inspecionados visualmente quanto à presença de material particulado ou alteração na cor antes da administração, sempre que a solução e o recipiente permitirem.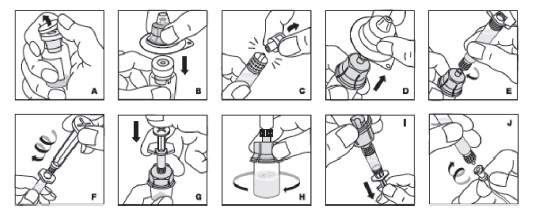
9. REAÇÕES ADVERSAS:
- Resumo do perfil de segurança
Um total de 236 pacientes (193 PTPs, 43 PUPs/MTPs) constituíram a população de segurança agrupada nos três estudos de fase III em pacientes tratados previamente (PTP), pacientes não tratados previamente (PUPs) e pacientes minimamente tratados (MTPs); LEOPOLD I (Estudo 1), LEOPOLD II (Estudo 2) e LEOPOLD kids (Estudo 3). O tempo médio no estudo clínico para a população de segurança agrupada foi de 558 dias (intervalo de 14 a 2.436 dias) com uma mediana de 183 dias de exposição (DEs) (intervalo de 1 a 1230 DEs). A maioria dos pacientes (n = 201 de 236; 85,2%) acumulou ≥ 100 DEs.
O número total de dias de exposição para todos os tratamentos sob demanda, manejo perioperatório, profilático, estudos farmacocinéticos (PK) e indução de tolerância imunológica (ITI)) foi 65029 DEs, dos quais 59585 DEs foram para tratamento profilático.
Pacientes que receberam Kovaltry® para manejo perioperatório (n = 5) com período de tratamento de 2 a 3 semanas e aqueles que receberam doses únicas de Kovaltry® para estudos de PK (n = 6) foram excluídos da análise de segurança agrupada.
As reações adversas reportadas com mais frequência na população agrupada foram pirexia (9,3%), dor de cabeça (8,5%) e erupção cutânea (rash) (5,5%). As reações adversas reportadas com mais frequência em PUPs/PMTPs foram a inibição do FVIII (ver item "Imunogenicidade).
Lista tabulada das reações adversas
A tabela apresentada abaixo está de acordo com Classificação por Sistema de Órgão MedDRA (SOC e Nível de Termo Preferido).
As reações adversas baseadas nas experiências dos estudos clínicos com Kovaltry® estão apresentadas na tabela abaixo, ordenadas por Classificação por Sistema de Órgão (SOC). As frequências foram avaliadas de acordo com a seguinte padronização: muito comum (≥ 1/10 pacientes), comum (≥ 1/100 a < 1/10 pacientes), incomum ( > 1/1.000 a < 1/100 pacientes), rara ( > 1/10.000 a < 1/1.000 pacientes), muito rara ( < 1/10.000).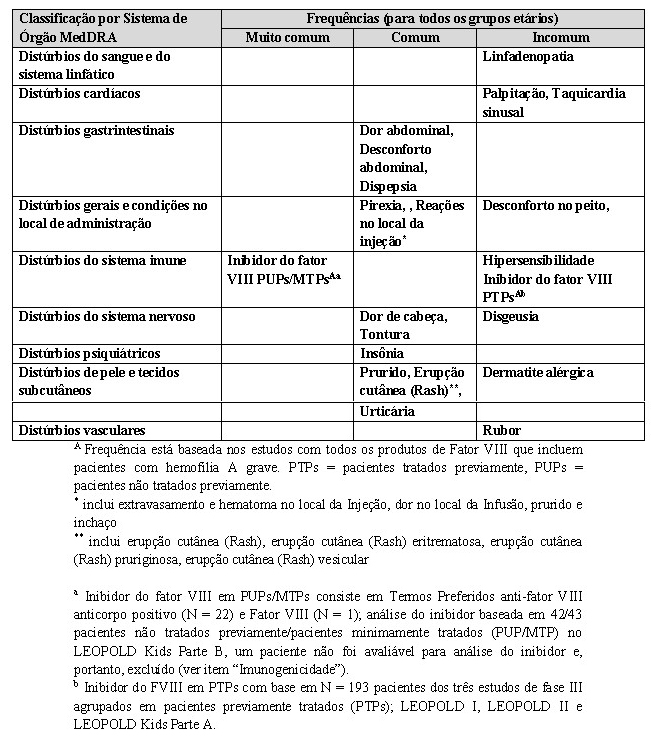
Descrição de reações adversas selecionadas
- Imunogenicidade
A imunogenicidade de Kovaltry® foi avaliada em pacientes tratados previamente (PTPs) e pacientes não tratados previamente/pacientes minimamente tratados (PUPs/MTPs). Durante os estudos clínicos com Kovaltry® em aproximadamente 200 pacientes pediátricos e adultos diagnosticados com hemofilia A grave (FVIII < 1%) com exposição prévia à concentrados do fator VIII ≥ 50 dias de exposição (DE), houve um caso de ocorrência de inibidor transitório de baixo título (pico do título: 1,0 Unidades Bethesda [UB]/mL) ocorreu em um PTP de 13 anos de idade após 549 DEs concomitantes com uma infecção aguda e anticorpos anticardiolipina IgG positivos. A recuperação do Fator VIII foi normal (2,7 UI/dL por UI/kg).
No estudo clínico que incluiu pacientes não tratados previamente (PUPs) e pacientes minimamente tratados (MTP
s) (definido como tendo até ≤ 3
dias de exposição [DEs] para um produto de fator VIII no momento do recrutamento), inibidores de fator VIII foram detectados em 23 de 42 pacientes com uma mediana (intervalo) de 9 (4 - 42) DEs no momento do primeiro teste positivo de inibidor. Destes, 6 pacientes tiveram inibidores de baixo título (≤ 5,0 Unidades Bethesda [UB]) e 17 pacientes tiveram inibidores de alto título ( > 5,0 Unidades Bethesda [UB]). Em pacientes que desenvolveram altos títulos de inibidores, mutações de alto risco para o desenvolvimento de inibidores foram identificadas em 12 dos 14 pacientes com dados de mutação de FVIII disponíveis.
Informações adicionais para populações especiais
-Pacientes pediátricos
Em estudos clínicos com 51 pacientes pediátricos previamente tratados (PTPs), afrequência, o tipo e a grav